Mitophagy in Cancer: A Tale of Adaptation

1
Laboratory of Cell Death Research and Therapy, Department for Cellular and Molecular Medicine, Campus Gasthuisberg, University of Leuven (KU Leuven), Herestraat 49, B-3000 Leuven, Belgium
2
Laboratory of Cell Death Research and Therapy, VIB-KU Leuven Center for Cancer Biology, 3000 Leuven, Belgium
*
Author to whom correspondence should be addressed.
Cells 2019, 8(5), 493; https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050493
Submission received: 2 May 2019
/
Revised: 18 May 2019
/
Accepted: 21 May 2019
/
Published: 22 May 2019
(This article belongs to the Special Issue Tumor Microenvironment: Interaction and Metabolism)
Abstract
:In the past years, we have learnt that tumors co-evolve with their microenvironment, and that the active interaction between cancer cells and stromal cells plays a pivotal role in cancer initiation, progression and treatment response. Among the players involved, the pathways regulating mitochondrial functions have been shown to be crucial for both cancer and stromal cells. This is perhaps not surprising, considering that mitochondria in both cancerous and non-cancerous cells are decisive for vital metabolic and bioenergetic functions and to elicit cell death. The central part played by mitochondria also implies the existence of stringent mitochondrial quality control mechanisms, where a specialized autophagy pathway (mitophagy) ensures the selective removal of damaged or dysfunctional mitochondria. Although the molecular underpinnings of mitophagy regulation in mammalian cells remain incomplete, it is becoming clear that mitophagy pathways are intricately linked to the metabolic rewiring of cancer cells to support the high bioenergetic demand of the tumor. In this review, after a brief introduction of the main mitophagy regulators operating in mammalian cells, we discuss emerging cell autonomous roles of mitochondria quality control in cancer onset and progression. We also discuss the relevance of mitophagy in the cellular crosstalk with the tumor microenvironment and in anti-cancer therapy responses.
1. Introduction
Mitochondria are double-membrane organelles deputed at cell energy supply; defects in mitochondrial functions not only affect cell homeostasis, bioenergetics and redox control but also are decisive for cell death. In the particular case of cancer cells, mitochondrial-harbored metabolic pathways are rewired to meet the increased bioenergetics and biosynthetic needs of the cancer cells and to handle oxidative stress. Therefore, a tight control of the mitochondrial network homeostasis is essential for cancer cells.
Several highly interrelated mechanisms, including mitochondrial dynamics (fusion and fission) as well as macroautophagy (mitophagy), operate in mammalian cells as key mitochondrial quality control processes, and their implication in tumor development and progression has recently been elucidated. In particular, the selective removal of mitochondria through the process of mitophagy has been recently implicated in reshaping the metabolic landscape within cancer cells and the interaction between cancer cells and other key components of the tumor microenvironment (TME), to foster the adaptive and survival ability of cancer cells. Moreover, and considering the tight relationship between mitochondrial homeostasis and susceptibility to cell death, mitochondria quality control and mitophagy in primis are critical in anti-cancer therapeutic response as well as cancer-related off target effects.
In this review, after a brief introduction of the main mitophagy pathways, we discuss the interplay of mitophagy with the key pathways involved in tumorigenesis, its coordination of the TME and its implication in the success (or not) of current anti-cancer therapies.
2. Molecular Mechanisms Leading to Mitophagy
Macroautophagy (hereafter referred to as autophagy) is a self-degradation process which is typically stimulated under conditions of nutrient deprivation or cellular stress. During autophagy, proteins, macromolecules and/or organelles are engulfed in a double-membrane vesicle called autophagosome, which eventually fuses with the lysosome where cargo degradation takes place (for recent reviews on mechanisms of autophagy, see [1,2]). The breakdown of intracellular material allows the recycling of essential building blocks to occur for metabolic and biosynthetic pathways. In mammalian cells, ubiquitylation operates as a prominent—albeit not unique—mechanism to selectively tag cytoplasmic cargoes destined for degradation by the autophagic machinery. Ubiquitylated targets are then recognized by specific autophagy receptors (such as p62/SQSTM1 and optineurin (OPTN); for a review on the topic, please see [3]) which are capable of binding both ubiquitin and the lipidated members of the ATG8 family of pro-autophagic proteins (LC3A/LC3B/LC3C/GABARAP/GABARAPL1/GABARAPL2, reviewed in [4]) via their LC3-interacting domain (LIR).
Mitophagy is a specialized form of autophagy in which damaged, dysfunctional or obsolete mitochondria are recognized by the autophagy machinery and eventually degraded by the lysosome. Damaged mitochondria are, in general, those mitochondria which are not able to execute oxidative phosphorylation (OXPHOS) efficiently. This is mainly because of the dissipation of their transmembrane potential and consequent accumulation of reactive oxygen species (ROS) causing an increase in the overall cellular oxidative stress levels, precipitating mitochondria-mediated cell death [5]. Since mitochondria are not found as isolated organelles but as a highly dynamic network, the dysfunctional mitochondrion needs to be separated from the healthy network, requiring the tight coordination between fusion, fission and mitophagy machineries (see Box 1 for a summary of the fusion and fission mechanisms). In particular, depolarized mitochondria will be either not able to fuse with the healthy mitochondrial network or isolated from the network by fission, resulting in isolated mitochondria ready to be degraded by mitophagy (for extensive reviews on the topic, see [6,7]). Instead, elongated mitochondria are spared from degradation and remain bioenergetically functional [8,9]. Isolated and damaged mitochondria are then recognized by specific mitophagy receptors whose identity depends on the specific trigger causing mitochondrial clearance, and which function as molecular bridges for the interaction with the autophagy machinery [10].
Box 1. Mitochondrial dynamics.

Fusion is the union of two separate mitochondria into a single entity to guarantee at least one copy of mitochondrial DNA (mtDNA) per mitochondrion as well as efficient OXPHOS [11]. Fusion is a highly coordinated process chiefly controlled by the mitofusins 1 and 2 (MFN1 and MFN2) anchored to the outer mitochondrial membrane (OMM) which, by forming homo- or heterodimers, coordinate fusion of the two OMMs, whereas Opa1 and cardiolipin (CL) orchestrate the fusion of the inner mitochondrial membranes (IMMs) [11]. MFN1 and MFN2 are required in both OMMs to assist membrane fusion in contrast to Opa1, whose presence in IMM is sufficient to enable the fusion [12]. Opa1 interacts with MFN1 and disruption of this interaction results in mitochondrial fragmentation [13]. Moreover, Opa1 cleavage by the mitochondrial metalloproteases OMA1 and YME1L results in one long isoform (L-Opa1) that remains anchored to the IMM and one short isoform (S-Opa1) that is released in the inter-membrane space (IMS) [14,15]. Although increased Opa1 cleavage is often associated with mitochondrial dysfunction [16], Opa1 proteolytic activation is required for mitochondrial fusion [17] since L-Opa1 is essential for IMM tethering [18] and S-Opa1 enhances the interaction of L-Opa1 and CL during IMM fusion [19].
In contrast, fission refers to the excision of one part of the mitochondrion from the main organelle. This separation can occur either symmetrically, where the two resulting mitochondria have similar respiratory capacity and membrane potential, or asymmetrically, where fission will result in one healthy mitochondrion and one damaged, the latter being targeted for degradation by mitophagy [20]. Fission is coordinated by the dynamin-related protein 1 (DRP1). DRP1 is a cytosolic protein that will translocate to the OMM and induces mitochondrial excision by interacting with OMM-anchored receptors, including the mitochondrial dynamics proteins of 49 and 51 kDa (MiD51, MiD49) [21]. Once at the OMM, DRP1 self-assembles to form a constricting ring around the portion of the mitochondrion to be excised [22]. The self-assembly of the different DRP1 isoforms into the constriction ring is assisted by other fission elements like Mff [23] and CL [24]. The role of FIS1 in mitochondrial fission is controversial, since it was initially described an OMM-anchored receptor assisting the mitochondrial localization of DRP1 [25] but recent data showed that FIS1 can induce mitochondrial fragmentation by inhibiting the GTP-ase activity of the fusion machinery (MFN1, MFN2 and Opa1) [26]. Phosphorylation of different serine residues within DRP1’s GTP-ase domain will enhance (serine 616) or diminish (serine 637) its affinity for the OMM and its receptor molecules [27]. The division site for the recruitment of DRP1 at the mitochondria is marked by the endoplasmic reticulum (ER). The ER wraps mitochondria and enables mitochondria constriction at the ER-mitochondria contact sites [28,29]. A pivotal role is played by the ER-bound protein inverted formin 2 (INF2) which by controlling actin assembly induces constrictions, before DRP1 recruitment to the mitochondria ([29]; for extensive reviews on this subject, see [30,31]).
Below we provide an overview of some of the best characterized (canonical) or emerging mitophagy pathways described to date in mammalian cells.
2.1. Canonical Mitophagy Pathways
2.1.1. PINK1/Parkin-Mediated Mitophagy
The PINK1/Parkin pathway is the most studied pathway of mitophagy (Figure 1) [32,33]. The serine/threonine PTEN-induced putative kinase 1 (PINK1) is the initiator of this pathway. Under normal conditions, PINK1 is imported to the IMM via the Translocase of the Outer Membrane (TOM) and Translocase of the Inner Membrane (TIM) complexes, where PINK1 is cleaved by the presenilin-associated rhomboid-like (PARL), a mitochondrial resident rhomboid serine protease, and subsequently degraded by the (mitochondrial) proteasome, keeping PINK1 levels low under basal conditions [34,35]. The translocation of PINK1 towards the IMM is membrane-voltage-dependent; therefore, mitochondrial depolarization abolishes PINK1 translocation [34]. Moreover, and in response to mitochondrial depolarization, PDK2 phosphorylates PARL, rendering it unable to cleave PINK1 [34,36]. Both events result in PINK1 accumulation at the OMM, where it recruits the U3 ubiquitin ligase Parkin [37]. PINK1 phosphorylates serine 65 of Parkin’s ubiquitin-like domain, promoting the E3 ubiquitin ligase activity of Parkin. Once activated, Parkin will ubiquitinate key mitochondrial proteins (such as MFN1, MFN2, VDAC1 or Miro1), a process that contributes to the isolation of the damaged mitochondria. On the one hand, Parkin mediated ubiquitylation of MFN1 and MFN2 prevents them to engage in fusion [38,39]. On the other hand, upon ubiquitylation Miro1, a protein that attaches mitochondria to the microtubules, will no longer bind the microtubules but the PINK1-Parkin complex, supporting the isolation the damaged mitochondrion [40,41]. Furthermore, these ubiquitin chains are phosphorylated by PINK1, which triggers further cycles of Parkin recruitment and activation, leading to the amplification of the mitophagy signal [42]. Finally, polyubiquitylation of mitochondrial proteins will be recognized by the autophagy cargo adaptors p62 and OPTN [43] and interaction with LC3, forming a complex that is degraded by the autophagic machinery [44].
2.1.2. BNIP3/NIX-Mediated Mitophagy
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L/NIX) are LIR-containing proteins located at the OMM which are able to directly bind LC3/GABARAP and trigger mitophagy without requiring protein ubiquitination or additional adaptors (Figure 1) [45,46,47]. They belong to the BH3-only group of the BCL-2 family of proteins, although their ability to bind BCL-2 remains context dependent [48]. Both BNIP3 and NIX are under the transcriptional regulation of Hypoxia-Inducible Factor 1 alpha (HIF1α), and are therefore thought to mediate hypoxia-triggered mitophagy [49,50]. However, in particular conditions, BNIP3 and NIX are also under the transcriptional regulation of FOXOa3 [51,52] or NF-kB [53], suggesting their participation in signaling pathways beyond hypoxia. Regarding their interactors, BNIP3 and NIX have been shown to form homodimers and heterodimers [54,55], as well as to separately interact with Mieap (a p53-regulated mitochondrial protein associated with the removal of oxidized species [56]) and cadherin6 (CDH6, protein modulator of mitophagy and DRP1-mediated fission [57]) to guarantee mitochondrial homeostasis. Moreover, Opa1 has been shown to interact with BNIP3 to promote mitochondrial fragmentation [58] and phosphorylation of BNIP3 in its C-terminal domain disrupts Opa1-BNIP3 interaction promoting mitophagy and reducing cell death [59]. Phosphorylation of BNIP3 and NIX within their LIR domain (serines 17 and 24 for BNIP3 and serines 34 and 35 for NIX) increases their affinity for LC3 binding [60,61], suggesting that phosphorylation is the main molecular switch controlling their pro-mitophagy or pro-death activity.
2.1.3. FUNDC1-Mediated Mitophagy
FUN14 Domain Containing 1 (FUNDC1) is another OMM protein capable to bind directly LC3 and trigger mitophagy (Figure 1) [62,63]. Alike BNIP3 and NIX, also FUNDC1 can trigger hypoxia-induced mitophagy [62]—even though it is not a HIF1α target [64]—and its phosphorylation in the LIR domain (serine 17) can increase its affinity for LC3 [65]. Interestingly, FUNDC1 can interact with both Opa1 and DRP1 and this interaction is modulated by FUNDC1 phosphorylation on serine 13. Dephosphorylation of FUNDC1 by phosphoglycerate mutase 5 (PGAM5), a mitochondrial serine/threonine protein phosphatase activated by PARL cleavage under condition of loss of mitochondria membrane potential [66], reduces Opa1-pFUNDC1 complex, which in turn increases FUNDC1′s affinity for DRP1 and promotes mitochondrial fragmentation and mitophagy [67,68]. Alternatively, the E3 ubiquitin ligase MARCH5 ubiquitylates FUNDC1 and DRP1 to fine-tune the mitophagic upregulation in response to hypoxia [69].
2.2. Non-Canonical Mitophagy Pathways
2.2.1. Lipid-Mediated Mitophagy
CL is a phospholipid which is particularly enriched in the IMM, which is able to directly interact with GABARAP upon translocation from the IMM to the OMM [70,71] in response to loss of mitochondrial transmembrane potential mediated by the hexameric intermembrane space protein NDPK-D [72]. Prohibitin 2 (PHB2) is an IMM mitophagy receptor which can bind LC3 upon membrane depolarization and proteasomal rupture favoring Parkin-mediated mitophagy [73]. Mechanisms of mitophagy induced by PHB2 have been associated with CL redistribution within the mitochondrial membranes [74], although PHB2′s contribution to CL-mediated mitophagy has not yet been elucidated. Additionally, ceramides -sphingolipids present in the OMM- can bind LC3B-II to target the mitochondria for degradation upon DRP1-mediated mitochondrial fragmentation [75]. Notably, ceramide 18 (Cer18) binding to LC3B-II inhibits mitochondrial function and oxygen consumption, induces autophagic cell death in vitro and suppress tumor growth in vivo [75], suggesting that lipid binding to LC3 may regulate the functional outcome of mitophagy.
2.2.2. AMBRA1-Mediated Mitophagy
Another mitophagy receptor is Autophagy And Beclin 1 Regulator 1 (AMBRA1), a Parkin interactor [76] that directly binds LC3 and triggers mitophagy [77] to protect the cells from apoptotic cell death caused by oxidative stress [78]. Mechanistically, IKKα phosphorylates AMBRA1 at serine 1014, enhancing its affinity for the ATG8 members GABARAP/GABARAPL1/GABARAPL2 and IKKα inhibition impairs AMBRA1-mediated mitophagy [79]. Furthermore, after membrane depolarization AMBRA1 promotes the mitochondrial localization of the E3 ligase HUWE1, which by ubiquitylating MFN2 promotes mitophagy [79]. Of note, HUWE1 absence also abolishes AMBRA1-mediated mitophagy, suggesting that HUWE1 could also modulate AMBRA1 serine 1014 phosphorylation [79], although the precise mechanism has not yet been elucidated. Overexpression of AMBRA1ActA (a fusion protein specifically expressed at the mitochondria) exacerbates mitophagy and suppresses oxidative stress and apoptosis induced by mitochondrial poisons [78], indicating the cytoprotective role of this pro-mitophagic protein.
2.2.3. BCL2L13-Mediated Mitophagy
The mammalian ortholog of the only mitophagy receptor in yeast (atg32) is BCL2L13/BCL-RAMBO, an OMM protein able to bind LC3 and able to induce DRP1-independent mitochondrial fragmentation [80]. Not only BCL2L13 induces Parkin-independent mitophagy [80] and has been associated with NIX/FUNDC1-mediated mitophagy [81], but it is also involved in autophagy-independent mitochondrial quality control [82].
2.2.4. FKBP8-Mediated Mitophagy
The recently described mitophagy receptor FK506-binding protein 8 (FKBP8/FKBP38), a known inhibitor of the mammalian target of rapamycin (mTOR), is a protein anchored to the OMM which is able to bind lipidated LC3A/GABARAP; its overexpression promotes mitochondrial fission in a similar fashion as BNIP3 or NIX overexpression [83]. However, FKBP38 operates as a preferential LC3A recruiter, suggesting that depending on the expression of LC3 members, a different panel of mitophagy receptors is engaged during mitophagy. Strikingly, FKBP8 is not degraded in the autophagosome during mitophagy, but once the mitochondria have been targeted for degradation, FKBP8 escapes from degradation by relocating to the ER, where it exerts an antiapoptotic effect due to its ability to bind Bcl2 [83,84].
2.2.5. Rab-Mediated Mitophagy
Endosomal trafficking and recycling governed by the Rab family of small GTPases contributes to the mitophagic removal of mitochondria. Recent studies have highlighted mechanisms for the removal of mitochondria that are dependent on the activity of several Rab-GTPases. A recently described pathway, independent of the classical ATG5/ATG7-LC3A/B/C autophagosome axis but regulated by the Beclin1 and ULK1 complex, involves a Rab9-mediated vesicular mechanism where autophagosomes are generated by the fusion of isolation membrane with vesicles derived from the trans-Golgi and late endosomes [85]. Recent work shows that another regulator of Rab activity, RABGEF1, a guanine nucleotide exchange factor (GEF) of endosomal proteins, is recruited in a Parkin-mediated fashion to damaged mitochondria. Mitochondria associated RABGEF1 enables the targeting of Rab5 and Rab7a to the damaged mitochondria and promotes ATG9-mediated vesicle assembly and the subsequent autophagosomal encapsulation [86].
Mitochondria can also be sequestered into Rab5-decorated endosomes through the endosomal sorting complex (ESCRT) and delivered to the lysosomes for degradation [87]. For this pathway, Parkin-mediated ubiquitylation of mitochondria is necessary while the canonical autophagy machinery is dispensable. The reason why mitochondria would be cleared preferentially by the endocytic pathway instead of using the autophagy machinery is not completely clear, even if it is apparent that redundant pathways of mitochondria degradation exist in mammalian cells. A possibility is that endosomes act as a first line of defense, before autophagy is stimulated, to rapidly remove potentially damaging dysfunctional mitochondria [87].
3. Mitophagy and Cancer
Most of the above described proteins involved in the mitophagic processes have been shown to be dysregulated in cancer patients (Table 1), but whether they behave as tumor promoter or tumor suppressor seems to be highly dependent on the cancer subtype and context [32,88]. For example, the pro-mitophagic receptor BNIP3 has tumor suppressor functions in breast cancer (Table 1), whereas it is thought to have a tumor promoter role in melanoma, renal cell carcinoma and pancreatic cancer (Table 1). To discuss how mitophagy contributes to tumor progression, we will first review the interplay between mitophagy with the key pathways involved in tumorigenesis in terms of metabolic modulation. Later, we will discuss their impact on the modulation of the TME.
3.1. Mitophagy Modulators and Cancer Metabolism
Metabolic reprogramming is one of the hallmarks of cancer [199]. Cancer cells need to plastically rewire their metabolism to fulfill the three basic needs of dividing cells: rapid ATP generation to maintain energy status, metabolic precursor supply to meet the high rates of macromolecule biosynthesis and maintenance of an appropriate cellular redox status [200]. To do so, cancer cells have acquired the ability to use a variety of fuel sources to adapt their metabolism according to their needs and to cope with metabolic and nutrient stresses. Growing evidence indicates that autophagy supports the metabolic plasticity of cancer cells, by providing virtually all essential components of carbon metabolism through the degradation of carbohydrates, proteins, lipids and nucleotides (recently reviewed in [201]). Several genetic studies support the current view that both glucose-dependent metabolic pathways and mitochondria metabolism are pivotal in tumorigenesis [199,200,202,203].
In the following subsections, we will discuss emerging links connecting oncogene-driven metabolic pathways and key modulators of the mitophagic machinery.
3.1.1. Mitophagy and Its Contribution to the Warburg Effect
A main metabolic phenotype observed in cancer cells is driven by the Warburg effect, which consists in the shift from ATP generation through OXPHOS to ATP generation through glycolysis, even in the presence of oxygen [204], being the transcription factor HIF-1α one of its major drivers. Stabilization of HIF-1α subunit under hypoxia activates the expression of the glycolytic program by encoding glucose transporters and glycolytic enzymes, as well as by promoting the conversion of pyruvate into lactate instead of its incorporation in the tricarboxylic acid (TCA) cycle [205]. Notably, HIF-1α transcriptional program also comprises the pro-mitophagic receptors BNIP3 and NIX, which, by instigating mitophagy, would diminish the mitochondrial mass thereby reducing the overall oxygen consumption of the cell and promoting its survival under low oxygen conditions [49,205] (Figure 2a). Cancer cells have also developed O2-independent mechanisms to stabilize HIF-1α under normoxia and drive tumorigenesis, highlighting the essential pro-glycolytic role of this transcription factor. Moreover, changes in the expression levels of the mitophagy receptors BNIP3 and NIX can feedback on HIF-1α stability. For instance, loss of BNIP3 in a mouse model of mammary tumorigenesis reduces mitophagy and increases mitochondrial ROS levels, which results into increased normoxic HIF-1α stabilization, eventually promoting the Warburg effect and subsequently tumor progression [206] (Figure 2b). A similar effect has also been observed in the human breast cancer cell line MCF-7 in vitro, where insulin-like growth factor 1 (IGF-1) induces BNIP3 expression in a HIF-1α dependent-manner. However, MCF-7 cells with acquired resistance to an IGF-1 receptor kinase inhibitor show reduced BNIP3 levels, impaired mitophagy, accumulation of dysfunctional mitochondria and increased ROS production, leading to increased ATP production through glycolysis [207]. Likewise, in glioblastoma cells, loss of PINK1 promotes the Warburg effect by the ROS-dependent stabilization of HIF-1α and reduced pyruvate kinase muscle isozyme 2 (PKM2) activity, both key regulators of aerobic glycolysis [208].
Glycolysis is also modulated by the interplay between the tumor suppressor p53 and mitophagy. A p53-BNIP3 axis modulates the glycolytic flux in radioresistant head and neck squamous cell carcinoma cell lines [209]. In these radioresistant cancer cells, BNIP3-dependent clearance of abnormal mitochondria reduces the glycolytic shift while maintaining oxygen consumption only in the presence of p53 (Figure 2c). Although the molecular mechanism linking p53 to BNIP3 is still unclear, BNIP3 is the dominant mitophagy receptor since loss of Parkin, is ineffective [209]. This is interesting, since Parkin is a p53-regulated gene mediating the effects of p53 on mitochondria energy metabolism, antioxidant defense and irradiation-induced tumorigenesis [210,211]. Hence, while these studies suggest that mitophagy contributes to p53-mediated effects on cancer metabolism and tumorigenesis they also highlight that, depending on specific cancer context, the nature of the mitophagy receptor involved could be critical. Additionally, other mitophagy-unrelated functions of BNIP3 [212] and Parkin [210] could play a role, depending on the cancer and type of stress considered.
High glycolytic rates in cancer cells are controlled by other key oncogenes, such as c-Myc and K-RAS. Besides coordinating with HIF-1α the expression of several glucose transporters and glycolytic enzymes [213,214], c-Myc also modulates mitophagy by regulating choline metabolism. In B-lymphoma cells, c-Myc activates the transcription of the key enzyme phosphate cytidylyltransferase 1 choline-α (PCYT1A) and PCYT1A upregulation prevents lymphoma cells to undergo a mitophagy-dependent necroptosis [215].
3.1.2. Mitophagy and OXPHOS
As mentioned, while cancer cells engage in aerobic glycolysis, and some tumors rely mostly on this pathway to meet their bioenergetic demands, they also strive to maintain pools of respiring mitochondria to adjust their metabolic and biosynthetic requirements [216]. This cancer cell autonomous plasticity requires that pathways controlling mitochondria clearance and biogenesis are intricately linked. Clear examples are the key regulator of mitochondrial biogenesis, peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) and c-Myc. The transcriptional coactivator PGC-1α is downregulated by HIF-1α to support the glycolytic switch in low oxygen condition (reviewed in [217]). Oncogenic c-Myc coordinates a vast array of genes involved in cell cycle control and glycolysis, but it is also a key promoter of mitochondrial biogenesis and overall mitochondria metabolism. Mitochondrial biogenesis strengthens c-Myc’s effects on cell-cycle progression and glycolytic metabolism, enabling cancer cells with the metabolic flexibility that supports growth (Figure 2d). Another important pathway involved in the clearance of damaged mitochondria is the MAPK pathway, which stabilizes PINK1 and subsequently promotes mitophagy in response to ROS-induced stress [218].
Inefficient OXPHOS caused by a leaky or defective electron transport chain can lead to ROS production [219,220]. Interestingly, high OXPHOS activity induced by feeding HeLa cells with glutamine is also coupled to enhanced mitophagy, through a mechanism involving the translocation of the small GTPase Ras homolog enriched in brain protein (Rheb) to the mitochondria and its binding to NIX. Although Rheb can interact with both BNIP3 and NIX, resulting in the blockade of Rheb-mediated activation of mTORC1, mitophagy coupled to increased OXPHOS is both mTORC1- and BNIP3-independent [221]. Thus, although mainly operating in concert, BNIP3 and NIX may affect mitochondrial degradation through independent mechanisms or in a cancer-subtype specific fashion. In line with this, silencing of BNIP3 in melanoma cells blunts glutamine-mediated effects on melanoma cell growth, migration and bioenergetics [222], suggesting that BNIP3 is vital to maintain mitochondria fitness required for glutamine-driven melanoma aggressiveness.
Hence, mitophagy may prevent the accumulation of damage that is inherently associated with elevated mitochondrial metabolism, thereby maintaining the (re)generation of mitochondria that are metabolically adapted to cope with the metabolic and nutrient stress from the TME. An interesting conjecture proposes that the coordinated induction of mitochondria biogenesis and mitophagy may be used to generate pools of mitochondria that are better suited to catabolize fatty acids through fatty acid oxidation (FAO) [33]. Given that RAS-driven tumors require elevated autophagy to preserve mitochondrial function and proficient FAO ([223] and reviewed in [201]), this conjecture seems indeed plausible (Figure 2d). This is particularly important, considering that FAO is emerging as crucial fuel for aggressive cancer types, like breast cancer cells [224], and that acetyl-CoA from oxidized fatty acids is a key regulator of epigenetic remodeling of chromatin [225], which may further support metabolic rewiring in cancer cells. In this scenario, PGC1α could be a key player, as specifically regulates FAO [226]. Actually, upregulation of PGC1α in nasopharyngeal carcinoma provides resistance to radiation by promoting FAO [227]. In addition, PGC1α regulates mitophagy during myogenesis by buffering ROS production, which can cause mitophagy at high levels [228]. Considering this, it could be possible that PGC1α contributes to the aggressiveness of tumors by connecting lipid metabolism, mitophagy and mitochondrial homeostasis.
The role of mitophagy, as for autophagy more in general, might be also modulated during the various phases of tumorigenesis [229]. Notably, AMBRA1 may also influence cancer metabolism and tumor progression by regulating the degradation of c-Myc. Mechanistically, AMBRA1 favors the interaction between c-Myc and its phosphatase PP2A, which leads to the dephosphorylation and degradation of c-Myc. This interaction is enhanced when mTOR is inhibited, reducing the cell division rate [230]. In addition, HUWE1, the E3 ubiquitin ligase involved in AMBRA1-mediated mitophagy, also participates in c-Myc degradation, suppressing RAS-driven tumorigenesis by preventing c-Myc/Miz1 accumulation [231]. AMBRA1 mutant mice develop spontaneous tumors [232], suggesting a tumor suppressor role for AMBRA1. However, considering that besides regulating c-Myc degradation and mitophagy, AMBRA1 plays additional functions in cancer cells [78], it remains challenging to pinpoint the exact contribution of AMBRA1-mediated mitophagy in tumorigenesis. In contrast, previous studies have shown that autophagy favors RAS-mediated transformation by supporting glycolysis [233] and mitochondria metabolism [234]. Additionally, loss of autophagy in K-RAS [223] or in mutant B-RAF-driven [235] lung cancer models impairs tumor growth and switches tumor fate from carcinomas to benign tumors, called oncocytomas. Interestingly, in both cases this inhibition of tumorigenesis is associated with the accumulation of defective mitochondria, suggesting mitophagy impairment.
Transformation mediated by c-Myc or RAS-v12 overexpression increases AMP-activated protein kinase (AMPK) phosphorylation, favoring the activation of FoxO3, which upregulates the expression of genes such as BNIP3 and LC3 [236]. In addition, Hepatitis B virus X protein (HBx), a leading factor in Hepatitis B virus-related hepatocellular carcinoma, promotes PINK1-Parkin mediated mitophagy through the activation of the mitochondrial peptidase LON under starvation, which could be a determinant event in the development of hepatocellular carcinoma [237].
Altogether, these studies reveal that mitophagy either directly or indirectly impacts the metabolism of cancer cell, but if tumor progression is favored by mitophagy or not, will depend on the mitophagy pathway involved, type of tumor and possibly on the stage of tumor development.
3.1.3. Mitophagy and Iron Metabolism
Iron homeostasis is essential for numerous cellular processes, and either too much or too little iron can be detrimental for cell survival. Iron is fundamental for cell growth, but excessive iron accumulation induces the production of ROS and oxidative injury [240]. Iron trafficking is regulated at both the systemic and organellar level, being mitochondrial functions crucial in maintaining cellular iron homeostasis. Iron is transported to the mitochondrion for the biosynthesis of heme and iron-sulfur clusters through mitochondrial iron importers such as SLC25A37/mitoferrin-1 and SLC25A28/mitoferrin-2 [241,242]. Dysfunction of mitochondrial iron trafficking plays an important role in mitochondrial diseases as well as cancer. Recently, it has been described that PINK1 and Parkin regulate mitochondrial iron accumulation in pancreatic cancer [127,243]. Depletion of PINK1 and Parkin in mice accelerates K-RAS-driven pancreatic tumorigenesis due to mitochondrial iron accumulation. Since PINK1 and Parkin mediate autophagic degradation of SLC25A37 and SLC25A28, PINK1- or Parkin--deficient mice show increased SLC25A37 and SLC25A28 levels, leading to mitochondrial iron accumulation, activation of the HIF1α glycolytic program, ultimately promoting the Warburg effect. This effect was rescued by genetic depletion of HIF1α or by deferiprone treatment, a mitochondrial iron chelator. Notably, K-RAS-driven pancreatic tumorigenesis was also inhibited, suggesting that HIF-1α metabolic reprogramming induced by mitochondrial iron accumulation contributed to pancreatic tumorigenesis in Pink1- or Park2-deficient mice. Furthermore, in a STAT3 deficient model of colorectal cancer, elevated mitophagy in intestinal epithelial cells (IECs) caused an accumulation of iron (II) in lysosomes, provoking lysosomal membrane permeabilization. This enabled antigen processing and stimulation of dendritic cells-mediated CD8+ T cells-induced anti-tumor immunity [244].
Hence, these studies support the view that mitophagy or mitophagy players take part in the control of iron trafficking at organellar level in cancer cells and stromal cells, affecting tumor progression at different molecular and cellular levels; from metabolic reprogramming to anti-tumor immunity regulation. Considering the emerging role played by mitochondria during iron-dependent ferroptosis—a regulated form of necrosis to which drug-resistant cancer cells are particularly vulnerable [245]—it is tempting to assume that mitophagic-control of iron metabolism in cancer cells may become a druggable target in cancer therapy.
3.2. Mitophagy and Cancer Stem Cells
Tumors are complex cellular systems where different subpopulations of cells coexist. Cancer stem cells (CSCs) constitute one of these subpopulations, which is characterized by their ability of self-renewal, dedifferentiation, generating the bulk tumor cells, and metastatic potential. CSCs are not a fixed entity and phenomena of dedifferentiation of mature tumor cells to CSCs can occur [246]. Therefore, the interaction of CSCs and tumor cells with the TME is important to decide cancer cell fate. Mitophagy also plays a role in the regulation of CSC subpopulation, since it is not only involved in the promotion of the stemness, but also in the acquisition of chemoresistance. Mitophagy regulates hepatic CSC subpopulation by suppressing p53 activity [247]. Increased Parkin-mediated mitophagy has been shown to promote p53 co-localization with mitochondria, resulting into simultaneous p53 and mitochondria degradation in a mitophagy-dependent manner [247]. Mitophagy inhibition leads to PINK-mediated p53 phosphorylation at serine 392, provoking p53 translocation into the nucleus and binding to the NANOG promoter, which prevents the activation of NANOG expression by the OCT4 and SOX2 transcription factors [247]. Since NANOG is an essential transcription factor for maintaining the stemness of CSCs [248], this mechanism would reduce the hepatic CSC subpopulation. Exacerbated mitophagy also promotes stemness in esophageal squamous cell carcinoma cells, as Parkin-dependent mitophagy was found to increase the expression of the stem cell marker CD44 in cancer cells undergoing epithelial–mesenchymal transition (EMT) [249]. Metabolic regulation is also a key determinant of the stem phenotype in cancer cells and mitophagy could play an important role in this context too. In line with this, in lung cancer and nasopharyngeal carcinoma, CSCs exhibit reduced mitochondrial mass compared to non-CSCs [250,251].
Hence, it seems that the role of mitophagy in CSCs may largely depend on the affected regulators and signaling pathways that control the differentiation of mature tumor cells to CSCs or the maintenance of stemness.
3.3. Non-Autonomous Effects of Mitophagy: Mitochondrial Transfer
Although mitophagy is crucial to preserve cell homeostasis, under certain conditions, either healthy or damaged mitochondria can be exchanged between cells to improve the OXPHOS capacity of the receiving cells or be degraded by the mitophagy machinery of the receiving cell, in a process called transmitophagy [252,253]. For instance, cancer associated fibroblasts (CAFs) in contact with breast cancer cells have been shown to have a radically different mitochondrial network than that of non-cancerous fibroblasts, suggesting that a mitochondrial exchange is orchestrated by the cancer cells [254]. In this scenario, CAFs are the highly-mitophagic donor cells and the cancer cells the recipients wanting to maintain their high OXPHOS status. In another cancer model, B16 murine melanoma cells depleted from mitochondria (ρ0) and injected in syngeneic mice have been shown to incorporate mitochondria from the host to maintain their “OXPHOS addiction” as well as tumor growth [255]. Similarly, mitochondrial transfer between cells within astrocytomas promotes tumor growth [256]. Additionally, leukemic cells have been shown to accept intact mitochondria from stromal cells to increase their OXPHOS capacity and resist the loss of membrane potential induced by different chemotherapeutic treatments [257]. In fact, there is a general consensus that stromal cells, mainly mesenchymal stem cells (MSCs) or fibroblasts, are the source of healthy mitochondria for cancer cells [253,258,259,260], although endothelial cells [261,262] or macrophages [257] have also been reported as stromal cells involved in the mitochondrial exchange.
Although several studies suggest that mitochondrial transfer is an active process, the mechanistic underpinnings of this process remain highly controversial. Mitochondrial transfer could require direct cell-cell contact between the donor and acceptor cell [257], mainly in the form of nanotubes. Nanotubes are F-actin based membrane tubes that can transport mitochondria, among other intercellular structures, to relieve mitochondrial stress in the acceptor cell [263] which have been observed within tumoral structures [264]. Most of the mitochondrial transfers using nanotubules in a cancer context mainly refer to either MSCs as donor cells and a cancer cell as acceptor [265,266] or cancer cell donor to cancer cell acceptor [264,267]. On the other hand, other studies suggest that contact between donor and acceptor is not necessary, as (fragments of) mitochondria could be trafficked and released via extracellular vesicles [253,258,259]. For example, pro-inflammatory myeloid-derived regulatory cells secrete extracellular vesicles containing depolarized mitochondria, which can be incorporated by T-cells [268]. T-cells and prostate cancer cells-derived extracellular vesicles have also been shown to contain mitochondrial fragments marked by the presence of various mitophagy regulators (e.g., PINK1, MFN1, MFN2…) and mtDNA [91,269]. It has also been claimed that the functionality of the mitochondria is irrelevant for their transfer, while intact mtDNA is indispensable for mitochondria to be transferred to another cell [270]. Furthermore, mtDNA transfer alone (not the complete mitochondrion) from the host to ρ0 cancer cells was shown to be sufficient to recover the respiratory capacity of mitochondria-deficient cancer cells [271]; however, the transfer mechanisms remain elusive. Interestingly, few reports suggest that extracellular vesicles could also travel within nanotubes and thus, these two mechanisms are perhaps not mutually exclusive [272,273]). What is clear is that both mechanisms are highly dependent on cytoskeletal dynamics and in line with this, cytoskeletal disruption by vincristine [257] or cytochalasin D [270] treatment hampers mitochondrial transfer. In this regard, Miro1 overexpression in MSCs favor the donation of healthy mitochondria to relief cellular stress in the acceptor cell [259,274,275]
Together, these studies highlight that intercellular mitochondrial transfer is an emerging mechanism of mitochondrial quality control as well as a crucial mechanism of interaction between cancer and stromal cells. However, the mechanistic underpinnings of this process are still largely elusive and more studies are needed to fully appreciate how and when mitochondrial transfer between cancer cells and their stroma contributes to cancer progression.
3.4. Mitophagy, Innate Immunity and Inflammation
Mitochondria are emerging as key modulators of cellular danger signaling and systemic immunity responses aiming to restore cellular or tissue homeostasis. This is perhaps not surprising since mitochondria are ancestral remnants of a-proteobacteria (endosymbiotic theory) and, as such, the mitochondrial genome (mtDNA) harbors CpG DNA repeats and encodes for formylated peptides (for a recent review on the topic, see [276]. Upon cellular injury or death, various mitochondrial factors or products (including but not limited to mtDNA, CL, ROS, ATP, cytochrome c or N-formyl-peptides) are released in the extracellular environment and circulation and operate as damage-associated molecular pattern (DAMPs), which activate immune responses by binding to pattern recognition receptors (PRRs) of innate immune cells [277]. Intracellularly and upon mitochondrial damage, mtDNA and ROS released in the cytosol can engage either the NLRP3 inflammasome or AIM2 inflammasome, which will mediate the proteolytic maturation and secretion of potent pro-inflammatory cytokines (such as IL-1β and IL-18) through the activation of caspase-1, thus eliciting pro-inflammatory responses [278]. Cytosolic mtDNA can also be sensed by cGAS, which upon stimulation will engage the ER-resident protein STING into activating the Type-I interferon transcriptional program [279].
Dysfunctional mitochondria not only provide potent DAMPs, but also sense and decode danger signals by operating as a signaling platform for the recruitment and modulation of the molecular machinery detecting incoming cellular damage. Typically, during viral infection, mitochondria stimulate antiviral signaling through the recruitment of RIG-I and MDA5, two members of the NOD-like receptor family of PRRs, via the mitochondria-associated adaptor protein MAVS (for an extensive review on this subject, please see [280]. Interestingly, MAVS interacts with essential mediators of mitochondrial dynamics (MFN1 and TBC1D15) and this interaction would modulate STING and NLPR3 inflammasome signaling as well [281]. Moreover, NLRP3 and STING are particularly enriched at the ER-mitochondria contact sites (MAMs; [282,283]), ER microdomains that have also been shown to be critical for PINK1/Parkin-mediated mitophagy initiation [284,285,286]. These observations together suggest that mitophagy is intimately linked to mitochondrial sensing and decoding of intracellular danger signaling. In fact, in PINK1- or Parkin-deficient mice the increased oxidative stress caused by mitochondrial iron accumulation in pancreatic cells resulted in AIM2 inflammasome activation, ultimately leading to the release of HMGB1, a non-histone nuclear protein that once released in the extracellular environment upon cell death operates as a DAMP [127]. In this particular case, HMGB1 was released in its oxidized form, which can bind the AGER receptor on innate immune cells and trigger PD-L1 expression, hereby exerting an immunosuppressive role [127]. The concept of PINK1/Parkin- mediated mitophagy as an inflammasome dampener is supported by recent compelling evidence regarding the cargo receptor p62. Upon activation of NF-κB signaling by the NLRP3 inflammasome, parallel mitochondrial damage is induced to trigger Parkin/p62-mediated mitophagy in order to prevent excessive IL-1β-dependent inflammation [287,288]. The activation of the NLPR3 inflammasome signaling is critically dependent on the SESN2-mediated recruitment of p62 to the ubiquitylated mitochondrial membrane [289].
Thus, mitophagy operates as a self-limiting system to protect cells from exacerbated inflammation by removing bona fide activators of the NLRP3 inflammasome (ROS, iron and mtDNA). Given the pleiotropic effects in inflammation, immunosurveillance and therapy responses of IL-1β and IL-18, the activation of the inflammasome in cancer may have contextual pro-tumorigenic or anti-tumorigenic roles (for a review on the topic, see [290]).
On the other side, mitophagy has also been shown to modulate the adaptive immune response in terms of dendritic cell-T cell synapse [291], activation of both CD8+ T cell [244] and memory NK cells [292], suggesting that additional fine-tuning of the immune responses by mitophagy may occur through mechanisms that do not involve the inflammasome. However, more studies are required to fully appreciate the role of mitophagy in cancer inflammation and anti-tumor immunity.
4. Mitophagy and Anti-Cancer Therapies
Historically, mitophagy has been considered a protective mechanism used by the cancer cells against the onset of mitochondrial apoptosis, the main cell death pathway driven by the metabolic stress of the TME in cancer cells. Considering that several classes of anti-cancer drugs or treatments either directly or indirectly cause mitochondrial dysfunction, ROS production and cytochrome c release launching caspase activation, targeting these dysfunctional mitochondria by mitophagy could hamper the initiation of the apoptotic cascade. Therefore, the induction of mitophagy by anti-cancer therapies may modulate their cytotoxic ability and contribute to therapy resistance.
This assumption is supported by several examples where the genetic inhibition of mitophagy pathways sensitize cancer cells towards cell death induced by anticancer treatments [293,294]. In fact, autophagy (and mitophagy by extension) is highly sensitive to microtubule-dynamics modulators since microtubules are essential for the autophagosome fusion with the lysosome [295,296]. In particular, downregulation of PINK1/Parkin- or Rab9a-mediated mitophagy contributes to radiosensitizing cancer cells [297,298], and genetic downregulation of key mitophagy receptors such as PINK1, FUNDC1 or AMBRA1 also chemosensitizes cancer cells [170,299,300], supporting the pro-survival role of mitophagy in cancer cells in response to chemo/radiotherapy.
Considering this pro-survival role of mitophagy after anti-cancer treatment, it could be expected that therapy-resistant cells would display higher mitophagy levels. Indeed, enhanced mitophagy contributes to cisplatin and etoposide resistance in cancer cells [301] and mitophagy impairment resensitizes drug-resistant cancer cells [302,303].
However, different mitophagic pathways may be engaged to counteract therapy-induced mitochondrial damage and contribute to the therapy resistance of cancer cells. For example, in docetaxel-treated prostate cancer cells, trehalose induces ULK1-independent mitophagy and reduces their sensitivity to their treatment [304]. Moreover, Opa-interacting protein 5 (OIP5) overexpression prevented docetaxel-induced cell death in gastric cancer cells by activating MFN2/PINK1-mediated mitophagy [305]. Instead, NIX-mediated mitophagy drives doxorubicin-resistance [306], whereas in the case of cisplatin/etoposide, the E3 ubiquitin-ligase ARIH1 -rather than Parkin- mediates PINK1-induced mitophagy of damaged mitochondria in response to cisplatin/etoposide [301]. Together, these studies suggest that the expression of certain receptors or mediators of mitophagy in specific cancer subtypes is a decisive factor in eliciting therapy resistance by mitochondrial clearance. However, although high levels of mitophagy have been correlated with therapeutic resistance, ULK1-driven mitophagy activation has also been shown to have an antiproliferative effect in therapy-resistant colon cancer cell lines [307], implying that mitophagy could play a dual role in therapy resistance.
Mitochondrial clearance (or alternative pathways of mitochondrial removal) could also be involved in modulating cancer cell non-autonomous response to therapy-mediated cell death. Mitochondrial transfer from surrounding cells via microtubules could also contribute to cancer cell therapy resistance, since mitochondria from endothelial cells or stromal cells have been reported to increase the chemoresistance of breast cancer cells [261] or leukemic cells [257], respectively.
Although the involvement of mitophagy has not been formally proven, blocking the release of the mitochondrial DAMP N-formyl peptides during immunogenic cancer cell death elicited in response to anthracyclines and reduces anti-tumor immunity [308], suggesting a potential regulatory role for mitochondrial clearance, an aspect that deserves further investigation.
When talking about therapy resistance, CSCs deserve special mention. CSCs are a subpopulation within cancer cells which is particularly resistant to therapy and which is responsible for the tumor repopulation after treatment (for a recent review on the topic, see [309]). Interestingly, the ability of CSCs to resist therapy seems to correlate to their mitophagic capacity, since higher mitophagic levels were detected in cisplatin-resistant oral squamous cell carcinomas’ CSCs [310], as well as in oxaliplatin-resistant or doxorubicin-resistant human colorectal CSCs [306,311]. In fact, blockade of mitophagy using a nanomedicine (188Re-Liposome) in chemotherapy-resistant ovarian cancer (stem) cells restored their sensitivity to the treatment not only in xenograft mice but also in two proof-of-concept therapy-resistant cancer patients [312]. Regarding the role of mitophagy in CSC-chemoresistance, it has been pointed out that higher mitophagic levels contribute to a higher OXPHOS state within the CSC population to facilitate its proliferation [313]; although in the case of therapy-resistance, it is more likely that mitophagy, by removing the damaged mitochondria, tightly controls ROS production and mitochondrial transmembrane potential, eventually reducing apoptotic cell death [314].
Altogether, preserving mitochondrial fitness by mitophagy provides cancer cells a mechanism to resist anti-cancer therapy (Figure 3); nevertheless, further research is necessary to understand which mitophagy players are instrumental for cancer drug- or radio-resistance in order to develop novel therapeutic strategies aiming to proficiently harness mitophagy in anti-cancer therapy.
Mitophagy and Cancer-Related Side Effects
Lastly, mitophagy is also relevant for off-target toxicities derived from anti-cancer therapies as well as from cancer itself. In terms of chemotherapeutics, one of the major side-effects related to doxorubicin treatment is its cardiotoxicity caused by enhanced Parkin-mediated or BNIP3-mediated mitophagy in cardiomyocytes [315,316,317].
Nonetheless, the most life-threatening side effect of cancer is cancer-associated cachexia: it occurs in more than 80% of the late-stage patients, it is directly associated to 20% of cancer-related deaths [318], and it constitutes a major prognostic factor independently of the cancer type [319]. Cachexia is multifactorial syndrome characterized by muscular mass and adipose tissue loss together with anorexia and weakness [320] and in cancer not only develops particularly fast [321] but it can also be induced by the chemotherapeutic treatment [322]. Cachectic muscle waste and loss have been associated to hypercatabolic breakdown of the muscle by autophagy: LC3B-II, ATG5 and Beclin1 have been shown to accumulate in skeletal muscle tissue from cachectic cancer patients [323,324,325] and in cancer mouse models [326,327]. Recently, it has been pointed out that excessive mitophagy in skeletal muscle cells would promote cachexia development. Alterations in BNIP3 or PINK1 [325] transcript levels are found in cachectic muscle of cancer patients [323,325,328] as well as in mouse models [329,330], but whether and how they regulate mitophagy-induced cachexia remains to be further studied.
In different cancer mouse models (and independently of chemotherapeutic treatment), cachectic muscle displayed alterations in mitochondrial homeostasis [331] (such as mitochondrial uncoupling, aberrant mitochondrial expression of CL [332,333], as well as giant mitochondria unable to fuse proficiently [334]), which would be then targeted for mitophagic degradation. Of note, UCP3, a mitochondrial protein that has been shown to accumulate in cancer-induced cachexia [334], is under the transcriptional control of Tumor Necrosis Factor alpha (TNFα) [335] and certain pro-inflammatory cytokines seem to be highly pro-autophagic/pro-mitophagic in cachectic environments. For example, TNFα and IL-6 trans-signaling induced by the cancer cells accelerates autophagy/mitophagy in skeletal muscle, hereby promoting cachexia [326,336]. Intriguingly, AMBRA1 has been associated with muscular atrophy [337], and has been recently shown to modulate the IL6-STAT3 axis [338], although no direct link to cancer-associated cachexia has been drawn yet.
In contrast to the putative detrimental effect of mitophagy in cancer patients, there are few cases where mitophagy induction in healthy tissue would be beneficial instead: renal failure is a side effect of cisplatin treatment and higher DRP1 levels and mitophagic levels in the renal tubular cells protect them from such a damage [339]. In fact, rapamycin administration reduces cisplatin-mediated nephrotoxicity in C57Bl6 mice by stimulating PINK1/Parkin-mediated mitophagy in tubular renal cells [340]. This suggests that rapamycin, possibly by enhancing mitophagy, could diminish chemotherapy-driven tissue damage.
Altogether, concomitant mitophagy modulation during chemotherapeutic treatment might contribute not only to overcoming cancer-cell resistance, but also to diminishing side toxicities derived from the treatment or the cancer itself. However, more mechanistic studies are necessary to understand the extent of the relationship between cancer-associated mitophagy and healthy tissue as well as the mitophagic pathways involved.
5. Conclusions and Perspectives
In the past years, the world of selective autophagy has grown tremendously, as has our understanding of the molecular underpinnings and physio-pathological implications of selective autophagy pathways. Among various selective degradation pathways operating in mammalian cells, mitophagy is emerging as a crucial determinant of cancer cell plasticity and interface with the TME. Reflecting the dynamic and plastic role of autophagy in carcinogenesis, mitochondrial clearance in cancer appears to operate as a mechanism recruited on-demand by the developing cancer cells to modulate key malignant features during cancer initiation and development. Growing evidence shows that mitophagy pathways act as key regulators of cancer cell mitochondrial mass, dynamics, redox homeostasis, bioenergetics, oncogene-driven metabolic reprogramming and cell death signals. This is perhaps not surprising, considering how mitochondrial biology and metabolic plasticity are central to cancer growth and response to anticancer therapies. The emerging view is that mitophagy represents a flexible mechanism supporting the metabolic adaptation and survival of cancer cells within the harsh TME.
However, from a therapeutic perspective, the redundancy in mitophagy receptors and alternative pathways for mitochondria clearance, which highlights the vital relevance of this process to maintain homeostasis, poses also key challenges when considering new therapeutic avenues harnessing pro-survival mitophagy in cancer treatment. Likewise, our knowledge of additional (and perhaps mitophagy-unrelated) functions of known mitophagy modulators and their link with oncogenic signals must be deepened in order to be able to advance more effective cancer treatments.
Funding
M.V.P. is the recipient of a FWO Doctoral Fellowship from the Flemish Research Foundation (FWO-Vlaanderen, 1186017N), Belgium. B.F.A. is supported by the EOS consortium (30837538). P.A. is supported by grants from the Flemish Research Foundation (FWO-Vlaanderen; G076617N, G049817N, G070115N), the EOS consortium (30837538), KU Leuven (C16/15/073) and Stichting tegen Kanker (FAF-F/2018/1252).
Acknowledgments
We apologize to the authors of several high-quality scientific articles that contributed significantly to the development of the field, which could not be cited due to space limits.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AGER | Advanced Glycosylation End-Product Specific Receptor |
| AIM2 | Absent In Melanoma 2 |
| AMBRA1 | Autophagy and Beclin 1 Regulator 1 |
| AMBRA1ActA | AMBRA1 fusion protein that localizes in the mitochondria |
| AMPK | AMP-activated protein kinase |
| ARIH1 | Ariadne RBR E3 Ubiquitin Protein Ligase 1 |
| Atg32 | autophagy-related protein 32 |
| ATG5 | autophagy-related protein 5 |
| ATG8 | autophagy-related protein 8 |
| ATG9 | autophagy-related protein 9 |
| ATP | adenosin triphosphate |
| B16 | murine melanoma cell line |
| BCL-2 | B-Cell CLL/Lymphoma 2 |
| BCL2L13/BCL-RAMBO | BCL2 Like 13 |
| Beclin1 | Coiled-Coil Myosin-Like BCL2-Interacting Protein |
| BNIP3 | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 |
| BNIP3L/NIX | BNIP3-like |
| B-RAF | V-Raf Murine Sarcoma Viral Oncogene Homolog B |
| C57Bl6 | mouse strain |
| CAFs | cancer associated fibroblasts |
| CD44 | Cluster of differentiation 44 |
| CD8 | Cluster of Differentiation 8 |
| CDH6 | cadherin6 |
| Cer18 | Ceramide 18 |
| cGAS | Cyclic GMP-AMP Synthase |
| CL | Cardiolipin |
| c-Myc | Proto-Oncogene C-Myc |
| CpG | regions of DNA where a cytosine nucleotide is followed by a guanine nucleotide |
| CSCs | Cancer stem cells |
| DAMPs | damage-associated molecular pattern |
| DNA | Deoxyribonucleic acid |
| DRP1 | Dynamin-related protein 1 |
| EMT | epithelial–mesenchymal transition |
| ER | endoplasmic reticulum |
| ESCRT | endosomal sorting complex |
| F-actin | Filamentous Actin |
| FAO | Fatty acid β-oxidation |
| FIS1 | Fission, Mitochondrial 1 |
| FKBP8/FKBP38 | FK506-binding protein 8 |
| FOXOa3 | Forkhead Box O3 isoform A |
| FUNDC1 | FUN14 Domain Containing 1 |
| GABARAP | Gamma-aminobutyric acid receptor-associated protein |
| GABARAPL1 | GABARAP-like 1 |
| GABARAPL2 | GABARAP-like 2 |
| HBx | Hepatitis B virus X protein |
| HeLa | human cervical cancer cell line |
| HIF1α | Hypoxia-Inducible Factor 1 alpha |
| HMGB1 | High Mobility Group Box 1 |
| HUWE1 | HECT, UBA And WWE Domain Containing E3 Ubiquitin Protein Ligase 1 |
| IECs | Intestinal epithelial cells |
| IGF-1 | insulin-like growth factor 1 |
| IKKα | Inhibitor of Nuclear Factor Kappa-B Kinase Subunit Alpha |
| IL-18 | Interleukin 18 |
| IL-1β | Interleukin 1 beta |
| IL-6 | Interleukin 6 |
| IMM | Inner mitochondrial membranes |
| IMS | Inter-membrane space |
| INF2 | Inverted formin 2 |
| K-RAS | Kirsten Rat Sarcoma Viral Oncogene |
| LC3A | Microtubule-associated proteins 1A/1B light chain 3A |
| LC3B | Microtubule-associated proteins 1A/1B light chain 3B |
| LC3B-II | lipidated from of LC3B |
| LC3C | Microtubule-associated proteins 1A/1B light chain 3C |
| LIR | LC3-interacting domain |
| LON | Lon Peptidase 1 |
| MAMs | ER-mitochondria contact sites |
| MAPK | Mitogen-Activated Protein Kinase |
| MARCH5 | Membrane Associated Ring-CH-Type Finger 5 |
| MAVS | mitochondria-associated adaptor protein |
| MCF-7 | human breast cancer cell line |
| MDA5 | Melanoma Differentiation-Associated Protein 5 |
| Mff | Mitochondrial Fission Factor |
| MFN1 | Mitofusin 1 |
| MFN2 | Mitofusin 2 |
| MiD49 | mitochondrial dynamics protein of 49 kDa |
| MiD51 | mitochondrial dynamics protein of 51 kDa |
| Miro1 | Mitochondrial Rho GTPase 1 |
| Miz1 | Zinc Finger and BTB Domain Containing 17 |
| MSCs | mesenchymal stem cells |
| mtDNA | mitochondrial DNA |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mTOR complex 1 |
| NANOG | Homeobox Transcription Factor Nanog |
| NDPK-D | Nucleoside Diphosphate Kinase isoform D |
| NF-kB | Nuclear Factor Kappa Beta |
| NK | Natural Killer |
| NLRP3 | NLR Family Pyrin Domain Containing 3 |
| OCT4 | Octamer-Binding Protein 4 |
| OIP5 | Opa-interacting protein 5 |
| OMA1 | OMA1 Zinc Metallopeptidase |
| OMM | Outer mitochondrial membrane |
| (S/L)-Opa1 | (Short/Long isoform of) Optic Atrophy 1 |
| OPTN | optineurin |
| OXPHOS | Oxidative phosphorylation |
| O2 | Oxygen |
| p53 | Tumor Protein P53 |
| p62/SQSTM1 | Sequestosome 1 |
| Park2 | gene encoding Parkin |
| PARL | Presenilin-associated rhomboid-like |
| PCYT1A | phosphate cytidylyltransferase 1 choline-α |
| PDK2 | Pyruvate Dehydrogenase Kinase 2 |
| PD-L1 | Programmed Cell Death 1 Ligand 1 |
| PGAM5 | phosphoglycerate mutase 5 |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator-1 alpha |
| PHB2 | Prohibitin 2 |
| Pink1 | gene encoding PINK1 |
| PINK1 | Serine/threonine PTEN-induced putative kinase 1 (PINK1) |
| PKM2 | Pyruvate kinase muscle isozyme 2 |
| PP2A | Protein phosphatase 2 A |
| PRRs | pattern recognition receptors |
| PTEN | Phosphatase and Tensin Homolog |
| RABGEF1 | RAB Guanine Nucleotide Exchange Factor 1 |
| RAS | Rat Sarcoma protein, family Small GTP Binding Protein |
| RAS-v12 | RAS mutated in the 12 aminoacid to be constitutively active |
| Rheb | Ras homolog enriched in brain protein |
| RIG-I | Retinoic Acid-Inducible Gene I Protein |
| ROS | Reactive oxygen species |
| SLC25A28/mitoferrin-2 | Solute Carrier Family 25 Member 28 |
| SLC25A37/mitoferrin-1 | Solute Carrier Family 25 Member 37 |
| SOX2 | SRY-Box 2 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| STING | Stimulator of Interferon Genes |
| TBC1D15 | TBC1 Domain Family Member 15 |
| TBC1D17 | TBC1 Domain Family Member 17 |
| TCA | Tricarboxylic acid (cycle) |
| TIM | Translocase of the Inner Membrane |
| TME | Tumor microenvironment |
| TNFα | Tumor Necrosis Factor alpha |
| TOM | Translocase of the Outer Membrane |
| UCP3 | Uncoupling Protein 3 |
| Ulk1 | Unc-51 Like Autophagy Activating Kinase 1 |
| VDAC1 | Voltage Dependent Anion Channel 1 |
| YME1L | YME1-Like Protein 1 |
| ρ0 cells | Cells depleted from mitochondrial content |
References
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Reviews. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef]
- Wild, P.; McEwan, D.G.; Dikic, I. The LC3 interactome at a glance. J. Cell Sci. 2014, 127, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef]
- Shirihai, O.S.; Song, M.; Dorn, G.W., 2nd. How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 2015, 116, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2018, 28, 67–76. [Google Scholar] [CrossRef]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; Kanazawa, T.; van der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.G.; et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J. Biol. Chem. 2006, 281, 37972–37979. [Google Scholar] [CrossRef]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Del Dotto, V.; Mishra, P.; Vidoni, S.; Fogazza, M.; Maresca, A.; Caporali, L.; McCaffery, J.M.; Cappelletti, M.; Baruffini, E.; Lenaers, G.; et al. OPA1 Isoforms in the Hierarchical Organization of Mitochondrial Functions. Cell Rep. 2017, 19, 2557–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Macdonald, P.J.; Francy, C.A.; Stepanyants, N.; Lehman, L.; Baglio, A.; Mears, J.A.; Qi, X.; Ramachandran, R. Distinct Splice Variants of Dynamin-related Protein 1 Differentially Utilize Mitochondrial Fission Factor as an Effector of Cooperative GTPase Activity. J. Biol. Chem. 2016, 291, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Francy, C.A.; Clinton, R.W.; Frohlich, C.; Murphy, C.; Mears, J.A. Cryo-EM Studies of Drp1 Reveal Cardiolipin Interactions that Activate the Helical Oligomer. Sci. Rep. 2017, 7, 10744. [Google Scholar] [CrossRef]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Jin, S.B.; Lendahl, U.; Nister, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Serasinghe, M.N.; Chipuk, J.E. Mitochondrial Fission in Human Diseases. Handb. Exp. Pharmacol. 2017, 240, 159–188. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Et Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef]
- Naon, D.; Scorrano, L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Et Biophys. Acta 2014, 1843, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, A.V.; Luchkina, E.A.; Gogvadze, V.; Zhivotovsky, B. Mitophagy: Link to cancer development and therapy. Biochem. Biophys. Res. Commun. 2017, 482, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; McQuibban, G.A. The Mitochondrial Rhomboid Protease PARL Is Regulated by PDK2 to Integrate Mitochondrial Quality Control and Metabolism. Cell Rep. 2017, 18, 1458–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Glauser, L.; Sonnay, S.; Stafa, K.; Moore, D.J. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J. Neurochem. 2011, 118, 636–645. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Lopez-Domenech, G.; Covill-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Schwarten, M.; Mohrluder, J.; Ma, P.; Stoldt, M.; Thielmann, Y.; Stangler, T.; Hersch, N.; Hoffmann, B.; Merkel, R.; Willbold, D. Nix directly binds to GABARAP: A possible crosstalk between apoptosis and autophagy. Autophagy 2009, 5, 690–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef]
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Et Biophys. Acta 2015, 1853, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiology. Heart Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef] [Green Version]
- Real, P.J.; Benito, A.; Cuevas, J.; Berciano, M.T.; de Juan, A.; Coffer, P.; Gomez-Roman, J.; Lafarga, M.; Lopez-Vega, J.M.; Fernandez-Luna, J.L. Blockade of epidermal growth factor receptors chemosensitizes breast cancer cells through up-regulation of Bnip3L. Cancer Res. 2005, 65, 8151–8157. [Google Scholar] [CrossRef]
- Dhingra, R.; Gang, H.; Wang, Y.; Biala, A.K.; Aviv, Y.; Margulets, V.; Tee, A.; Kirshenbaum, L.A. Bidirectional regulation of nuclear factor-kappaB and mammalian target of rapamycin signaling functionally links Bnip3 gene repression and cell survival of ventricular myocytes. Circ. Heart Fail. 2013, 6, 335–343. [Google Scholar] [CrossRef]
- Frazier, D.P.; Wilson, A.; Graham, R.M.; Thompson, J.W.; Bishopric, N.H.; Webster, K.A. Acidosis regulates the stability, hydrophobicity, and activity of the BH3-only protein Bnip3. Antioxid. Redox Signal. 2006, 8, 1625–1634. [Google Scholar] [CrossRef]
- Hendgen-Cotta, U.B.; Esfeld, S.; Rudi, K.; Miinalainen, I.; Klare, J.P.; Rassaf, T. Cytosolic BNIP3 Dimer Interacts with Mitochondrial BAX Forming Heterodimers in the Mitochondrial Outer Membrane under Basal Conditions. Int. J. Mol. Sci. 2017, 18, 687. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Kitamura, N.; Shinogi, D.; Yoshida, M.; Goda, O.; Murai, R.; Kamino, H.; Arakawa, H. BNIP3 and NIX mediate Mieap-induced accumulation of lysosomal proteins within mitochondria. PLoS ONE 2012, 7, e30767. [Google Scholar] [CrossRef]
- Gugnoni, M.; Sancisi, V.; Gandolfi, G.; Manzotti, G.; Ragazzi, M.; Giordano, D.; Tamagnini, I.; Tigano, M.; Frasoldati, A.; Piana, S.; et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene 2017, 36, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnaune-Pelloquin, L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef]
- Liu, K.E.; Frazier, W.A. Phosphorylation of the BNIP3 C-Terminus Inhibits Mitochondrial Damage and Cell Death without Blocking Autophagy. PLoS ONE 2015, 10, e0129667. [Google Scholar] [CrossRef]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef]
- Rogov, V.V.; Suzuki, H.; Marinkovic, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubasic, M.; Sprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Wang, C.; Li, F.; Peng, J.; Wen, B.; Gong, Q.; Shi, Y.; Tang, Y. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell 2017, 8, 25–38. [Google Scholar] [CrossRef]
- Wu, X.; Wu, F.H.; Wu, Q.; Zhang, S.; Chen, S.; Sima, M. Phylogenetic and Molecular Evolutionary Analysis of Mitophagy Receptors under Hypoxic Conditions. Front. Physiol. 2017, 8, 539. [Google Scholar] [CrossRef]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef]
- Sekine, S.; Kanamaru, Y.; Koike, M.; Nishihara, A.; Okada, M.; Kinoshita, H.; Kamiyama, M.; Maruyama, J.; Uchiyama, Y.; Ishihara, N.; et al. Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J. Biol. Chem. 2012, 287, 34635–34645. [Google Scholar] [CrossRef]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugo, M.; Kimura, H.; Arasaki, K.; Amemiya, T.; Hirota, N.; Dohmae, N.; Imai, Y.; Inoshita, T.; Shiba-Fukushima, K.; Hattori, N.; et al. Syntaxin 17 regulates the localization and function of PGAM5 in mitochondrial division and mitophagy. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Chen, Z.; Siraj, S.; Liu, L.; Chen, Q. MARCH5-FUNDC1 axis fine-tunes hypoxia-induced mitophagy. Autophagy 2017, 13, 1244–1245. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.T.; Bayir, H.; Kagan, V.E. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: Implications for Parkinson disease. Autophagy 2014, 10, 376–378. [Google Scholar] [CrossRef]
- Anton, Z.; Landajuela, A.; Hervas, J.H.; Montes, L.R.; Hernandez-Tiedra, S.; Velasco, G.; Goni, F.M.; Alonso, A. Human Atg8-cardiolipin interactions in mitophagy: Specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy 2016, 12, 2386–2403. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Jiang, J.; Huang, Z.; Tyurina, Y.Y.; Desbourdes, C.; Cottet-Rousselle, C.; Dar, H.H.; Verma, M.; Tyurin, V.A.; Kapralov, A.A.; et al. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ. 2016, 23, 1140–1151. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef]
- Di Rita, A.; D’Acunzo, P.; Simula, L.; Campello, S.; Strappazzon, F.; Cecconi, F. AMBRA1-Mediated Mitophagy Counteracts Oxidative Stress and Apoptosis Induced by Neurotoxicity in Human Neuroblastoma SH-SY5Y Cells. Front. Cell. Neurosci. 2018, 12, 92. [Google Scholar] [CrossRef]
- Di Rita, A.; Peschiaroli, A.; Pasquale, D.; Strobbe, D.; Hu, Z.; Gruber, J.; Nygaard, M.; Lambrughi, M.; Melino, G.; Papaleo, E.; et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKalpha. Nat. Commun. 2018, 9, 3755. [Google Scholar] [CrossRef]
- Murakawa, T.; Yamaguchi, O.; Hashimoto, A.; Hikoso, S.; Takeda, T.; Oka, T.; Yasui, H.; Ueda, H.; Akazawa, Y.; Nakayama, H.; et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015, 6, 7527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampert, M.A.; Orogo, A.M.; Najor, R.H.; Hammerling, B.C.; Leon, L.J.; Wang, B.J.; Kim, T.; Sussman, M.A.; Gustafsson, A.B. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 2019, 1–17. [Google Scholar] [CrossRef]
- Ju, L.; Chen, S.; Alimujiang, M.; Bai, N.; Yan, H.; Fang, Q.; Han, J.; Ma, X.; Yang, Y.; Jia, W. A novel role for Bcl2l13 in promoting beige adipocyte biogenesis. Biochem. Biophys. Res. Commun. 2018, 506, 485–491. [Google Scholar] [CrossRef]
- Bhujabal, Z.; Birgisdottir, A.B.; Sjottem, E.; Brenne, H.B.; Overvatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef]
- Saita, S.; Shirane, M.; Nakayama, K.I. Selective escape of proteins from the mitochondria during mitophagy. Nat. Commun. 2013, 4, 1410. [Google Scholar] [CrossRef] [Green Version]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Yamano, K.; Wang, C.; Sarraf, S.A.; Munch, C.; Kikuchi, R.; Noda, N.N.; Hizukuri, Y.; Kanemaki, M.T.; Harper, W.; Tanaka, K.; et al. Endosomal Rab cycles regulate Parkin-mediated mitophagy. eLife 2018, 7. [Google Scholar] [CrossRef]
- Hammerling, B.C.; Najor, R.H.; Cortez, M.Q.; Shires, S.E.; Leon, L.J.; Gonzalez, E.R.; Boassa, D.; Phan, S.; Thor, A.; Jimenez, R.E.; et al. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat. Commun. 2017, 8, 14050. [Google Scholar] [CrossRef]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, J.; Lyu, Z.; Chen, Y.; Ji, X.; Cao, H.; Jin, M.; Zhu, J.; Yang, J.; Ling, R.; et al. Positive feedback loop between mitochondrial fission and Notch signaling promotes survivin-mediated survival of TNBC cells. Cell Death Dis. 2018, 9, 1050. [Google Scholar] [CrossRef]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef]
- Philley, J.V.; Kannan, A.; Qin, W.; Sauter, E.R.; Ikebe, M.; Hertweck, K.L.; Troyer, D.A.; Semmes, O.J.; Dasgupta, S. Complex-I Alteration and Enhanced Mitochondrial Fusion Are Associated With Prostate Cancer Progression. J. Cell Physiol. 2016, 231, 1364–1374. [Google Scholar] [CrossRef]
- Chakraborty, P.K.; Murphy, B.; Mustafi, S.B.; Dey, A.; Xiong, X.; Rao, G.; Naz, S.; Zhang, M.; Yang, D.; Dhanasekaran, D.N.; et al. Cystathionine beta-synthase regulates mitochondrial morphogenesis in ovarian cancer. FASEB J. 2018, 32, 4145–4157. [Google Scholar] [CrossRef]
- Soares, C.D.; Morais, T.M.L.; Carlos, R.; de Almeida, O.P.; Mariano, F.V.; Altemani, A.; de Carvalho, M.G.F.; Correa, M.B.; Dos Reis, R.R.D.; Amorim, L.S.; et al. Prognostic importance of mitochondrial markers in mucosal and cutaneous head and neck melanomas. Hum. Pathol. 2019, 85, 279–289. [Google Scholar] [CrossRef]
- Fang, C.L.; Sun, D.P.; Chen, H.K.; Lin, C.C.; Hung, S.T.; Uen, Y.H.; Lin, K.Y. Overexpression of Mitochondrial GTPase MFN2 Represents a Negative Prognostic Marker in Human Gastric Cancer and Its Inhibition Exerts Anti-Cancer Effects. J. Cancer 2017, 8, 1153–1161. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, Y.; Sun, J.; Gong, W.; Sun, P.; Kong, X.; Yang, M.; Zhang, W. Mitofusin-2 acts as biomarker for predicting poor prognosis in hepatitis B virus related hepatocellular carcinoma. Infect. Agent Cancer 2018, 13, 36. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Chen, G.; Li, X.; Wu, X.; Chang, Z.; Xu, J.; Zhu, Y.; Yin, P.; Liang, X.; Dong, L. MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling. Sci. Rep. 2017, 7, 41718. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, D.; Xu, X.; Zhao, X.; Huang, P.; Zhou, X.; Song, W.; Guo, H.; Wang, W.; Zheng, S. Clinical significance of mitofusin-2 and its signaling pathways in hepatocellular carcinoma. World J. Surg. Oncol. 2016, 14, 179. [Google Scholar] [CrossRef]
- Cheng, C.T.; Kuo, C.Y.; Ouyang, C.; Li, C.F.; Chung, Y.; Chan, D.C.; Kung, H.J.; Ann, D.K. Metabolic Stress-Induced Phosphorylation of KAP1 Ser473 Blocks Mitochondrial Fusion in Breast Cancer Cells. Cancer Res. 2016, 76, 5006–5018. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xie, Q.; Zhou, X.; Yao, J.; Zhu, X.; Huang, P.; Zhang, L.; Wei, J.; Xie, H.; Zhou, L.; et al. Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 2015, 358, 47–58. [Google Scholar] [CrossRef]
- Kannan, A.; Wells, R.B.; Sivakumar, S.; Komatsu, S.; Singh, K.P.; Samten, B.; Philley, J.V.; Sauter, E.R.; Ikebe, M.; Idell, S.; et al. Mitochondrial Reprogramming Regulates Breast Cancer Progression. Clin. Cancer Res. 2016, 22, 3348–3360. [Google Scholar] [CrossRef] [Green Version]
- Cruz, M.D.; Ledbetter, S.; Chowdhury, S.; Tiwari, A.K.; Momi, N.; Wali, R.K.; Bliss, C.; Huang, C.; Lichtenstein, D.; Bhattacharya, S.; et al. Metabolic reprogramming of the premalignant colonic mucosa is an early event in carcinogenesis. Oncotarget 2017, 8, 20543–20557. [Google Scholar] [CrossRef]
- Fang, H.Y.; Chen, C.Y.; Chiou, S.H.; Wang, Y.T.; Lin, T.Y.; Chang, H.W.; Chiang, I.P.; Lan, K.J.; Chow, K.C. Overexpression of optic atrophy 1 protein increases cisplatin resistance via inactivation of caspase-dependent apoptosis in lung adenocarcinoma cells. Hum. Pathol. 2012, 43, 105–114. [Google Scholar] [CrossRef]
- Kang, J.U.; Koo, S.H.; Kwon, K.C.; Park, J.W.; Kim, J.M. Identification of novel candidate target genes, including EPHB3, MASP1 and SST at 3q26.2-q29 in squamous cell carcinoma of the lung. BMC Cancer 2009, 9, 237. [Google Scholar] [CrossRef]
- Zhao, X.; Tian, C.; Puszyk, W.M.; Ogunwobi, O.O.; Cao, M.; Wang, T.; Cabrera, R.; Nelson, D.R.; Liu, C. OPA1 downregulation is involved in sorafenib-induced apoptosis in hepatocellular carcinoma. Lab. Investig. 2013, 93, 8–19. [Google Scholar] [CrossRef]
- Tanwar, D.K.; Parker, D.J.; Gupta, P.; Spurlock, B.; Alvarez, R.D.; Basu, M.K.; Mitra, K. Crosstalk between the mitochondrial fission protein, Drp1, and the cell cycle is identified across various cancer types and can impact survival of epithelial ovarian cancer patients. Oncotarget 2016, 7, 60021–60037. [Google Scholar] [CrossRef]
- Kim, Y.Y.; Yun, S.H.; Yun, J. Downregulation of Drp1, a fission regulator, is associated with human lung and colon cancers. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Chiang, S.F.; Chen, W.T.; Ke, T.W.; Chen, T.W.; You, Y.S.; Lin, C.Y.; Chao, K.S.C.; Huang, C.Y. HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Dis. 2018, 9, 1004. [Google Scholar] [CrossRef]
- Wieder, S.Y.; Serasinghe, M.N.; Sung, J.C.; Choi, D.C.; Birge, M.B.; Yao, J.L.; Bernstein, E.; Celebi, J.T.; Chipuk, J.E. Activation of the Mitochondrial Fragmentation Protein DRP1 Correlates with BRAF(V600E) Melanoma. J. Investig. Dermatol. 2015, 135, 2544–2547. [Google Scholar] [CrossRef]
- Li, J.; Huang, Q.; Long, X.; Guo, X.; Sun, X.; Jin, X.; Li, Z.; Ren, T.; Yuan, P.; Huang, X.; et al. Mitochondrial elongation-mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene 2017, 36, 4901–4912. [Google Scholar] [CrossRef]
- Tak, H.; Kang, H.; Ji, E.; Hong, Y.; Kim, W.; Lee, E.K. Potential use of TIA-1, MFF, microRNA-200a-3p, and microRNA-27 as a novel marker for hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 497, 1117–1122. [Google Scholar] [CrossRef]
- Fan, S.; Liu, B.; Sun, L.; Lv, X.B.; Lin, Z.; Chen, W.; Chen, W.; Tang, Q.; Wang, Y.; Su, Y.; et al. Mitochondrial fission determines cisplatin sensitivity in tongue squamous cell carcinoma through the BRCA1-miR-593-5p-MFF axis. Oncotarget 2015, 6, 14885–14904. [Google Scholar] [CrossRef]
- Abo Elwafa, R.; Gamaleldin, M.; Ghallab, O. The clinical and prognostic significance of FIS1, SPI1, PDCD7 and Ang2 expression levels in acute myeloid leukemia. Cancer Genet. 2018. [Google Scholar] [CrossRef]
- Tian, Y.; Huang, Z.; Wang, Z.; Yin, C.; Zhou, L.; Zhang, L.; Huang, K.; Zhou, H.; Jiang, X.; Li, J.; et al. Identification of novel molecular markers for prognosis estimation of acute myeloid leukemia: Over-expression of PDCD7, FIS1 and Ang2 may indicate poor prognosis in pretreatment patients with acute myeloid leukemia. PLoS ONE 2014, 9, e84150. [Google Scholar] [CrossRef]
- Hsiao, C.P.; Wang, D.; Kaushal, A.; Saligan, L. Mitochondria-related gene expression changes are associated with fatigue in patients with nonmetastatic prostate cancer receiving external beam radiation therapy. Cancer Nurs. 2013, 36, 189–197. [Google Scholar] [CrossRef]
- Fan, S.; Chen, W.X.; Lv, X.B.; Tang, Q.L.; Sun, L.J.; Liu, B.D.; Zhong, J.L.; Lin, Z.Y.; Wang, Y.Y.; Li, Q.X.; et al. miR-483-5p determines mitochondrial fission and cisplatin sensitivity in tongue squamous cell carcinoma by targeting FIS1. Cancer Lett. 2015, 362, 183–191. [Google Scholar] [CrossRef]
- Chang, G.; Zhang, W.; Ma, Y.; Wen, Q. PINK1 Expression Is Associated with Poor Prognosis in Lung Adenocarcinoma. Tohoku J. Exp Med. 2018, 245, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, K.; Miyata, H.; Makino, T.; Masuike, Y.; Furukawa, H.; Tanaka, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. High Expression of the Mitophagy-Related Protein Pink1 is Associated with a Poor Response to Chemotherapy and a Poor Prognosis for Patients Treated with Neoadjuvant Chemotherapy for Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2017, 24, 4025–4032. [Google Scholar] [CrossRef]
- Unoki, M.; Nakamura, Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001, 20, 4457–4465. [Google Scholar] [CrossRef] [Green Version]
- Poulogiannis, G.; McIntyre, R.E.; Dimitriadi, M.; Apps, J.R.; Wilson, C.H.; Ichimura, K.; Luo, F.; Cantley, L.C.; Wyllie, A.H.; Adams, D.J.; et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 15145–15150. [Google Scholar] [CrossRef]
- Bhat, Z.I.; Kumar, B.; Bansal, S.; Naseem, A.; Tiwari, R.R.; Wahabi, K.; Sharma, G.D.; Alam Rizvi, M.M. Association of PARK2 promoter polymorphisms and methylation with colorectal cancer in North Indian population. Gene 2019, 682, 25–32. [Google Scholar] [CrossRef]
- da Silva-Camargo, C.C.V.; Svoboda Baldin, R.K.; Costacurta Polli, N.L.; Agostinho, A.P.; Olandosk, M.; de Noronha, L.; Sotomaior, V.S. Parkin protein expression and its impact on survival of patients with advanced colorectal cancer. Cancer Biol. Med. 2018, 15, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Kannengiesser, C.; Lesage, S.; Andre, J.; Mourah, S.; Michel, L.; Descamps, V.; Basset-Seguin, N.; Bagot, M.; Bensussan, A.; et al. PARKIN Inactivation Links Parkinson’s Disease to Melanoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Gao, G.; Kasperbauer, J.L.; Tombers, N.M.; Wang, V.; Mayer, K.; Smith, D.I. A selected group of large common fragile site genes have decreased expression in oropharyngeal squamous cell carcinomas. Genes Chromosom. Cancer 2014, 53, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Toma, M.I.; Wuttig, D.; Kaiser, S.; Herr, A.; Weber, T.; Zastrow, S.; Koch, R.; Meinhardt, M.; Baretton, G.B.; Wirth, M.P.; et al. PARK2 and PACRG are commonly downregulated in clear-cell renal cell carcinoma and are associated with aggressive disease and poor clinical outcome. Genes Chromosom. Cancer 2013, 52, 265–273. [Google Scholar] [CrossRef]
- Mehdi, S.J.; Ali, A.; Rizvi, M.M. Parkin gene alterations in ovarian carcinoma from northern Indian population. Pathol. Oncol. Res. 2011, 17, 579–586. [Google Scholar] [CrossRef]
- Agirre, X.; Roman-Gomez, J.; Vazquez, I.; Jimenez-Velasco, A.; Garate, L.; Montiel-Duarte, C.; Artieda, P.; Cordeu, L.; Lahortiga, I.; Calasanz, M.J.; et al. Abnormal methylation of the common PARK2 and PACRG promoter is associated with downregulation of gene expression in acute lymphoblastic leukemia and chronic myeloid leukemia. Int. J. Cancer 2006, 118, 1945–1953. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Wen, Q.; Xie, Y.; Liu, J.; Kroemer, G.; et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 2018, 46, 441–455. [Google Scholar] [CrossRef]
- Jiang, Z.; Yu, F.; Li, M. Upregulation of BCL2 19 kD Protein-Interacting Protein 3 (BNIP3) is Predictive of Unfavorable Prognosis in Uveal Melanoma. Med. Sci. Monit. 2018, 24, 4711–4717. [Google Scholar] [CrossRef]
- Schulten, H.J.; Bangash, M.; Karim, S.; Dallol, A.; Hussein, D.; Merdad, A.; Al-Thoubaity, F.K.; Al-Maghrabi, J.; Jamal, A.; Al-Ghamdi, F.; et al. Comprehensive molecular biomarker identification in breast cancer brain metastases. J. Transl. Med. 2017, 15, 269. [Google Scholar] [CrossRef]
- Xu, Q.; Junttila, S.; Scherer, A.; Giri, K.R.; Kivela, O.; Skovorodkin, I.; Roning, J.; Quaggin, S.E.; Marti, H.P.; Shan, J.; et al. Renal carcinoma/kidney progenitor cell chimera organoid as a novel tumorigenesis gene discovery model. Dis. Model. Mech. 2017, 10, 1503–1515. [Google Scholar] [CrossRef]
- Chen, J.L.; David, J.; Cook-Spaeth, D.; Casey, S.; Cohen, D.; Selvendiran, K.; Bekaii-Saab, T.; Hays, J.L. Autophagy Induction Results in Enhanced Anoikis Resistance in Models of Peritoneal Disease. Mol. Cancer Res. 2017, 15, 26–34. [Google Scholar] [CrossRef]
- Fujimoto, T.; Ohtsuka, T.; Date, K.; Kimura, H.; Matsunaga, T.; Mori, Y.; Miyasaka, Y.; Mochidome, N.; Oda, Y.; Nakamura, M. Expression of Bcl-2 19-kDa interacting protein 3 predicts prognosis after ampullary carcinoma resection. J. Hepatobiliary Pancreat Sci. 2016, 23, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.S.; Kim, J.W.; Yoon, J.S. Expression of Autophagy and Reactive Oxygen Species-Related Proteins in Lacrimal Gland Adenoid Cystic Carcinoma. Yonsei Med. J. 2016, 57, 482–489. [Google Scholar] [CrossRef] [Green Version]
- Petrova, V.; Mancini, M.; Agostini, M.; Knight, R.A.; Annicchiarico-Petruzzelli, M.; Barlev, N.A.; Melino, G.; Amelio, I. TAp73 transcriptionally represses BNIP3 expression. Cell Cycle 2015, 14, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Thongchot, S.; Yongvanit, P.; Loilome, W.; Seubwai, W.; Phunicom, K.; Tassaneeyakul, W.; Pairojkul, C.; Promkotra, W.; Techasen, A.; Namwat, N. High expression of HIF-1alpha, BNIP3 and PI3KC3: Hypoxia-induced autophagy predicts cholangiocarcinoma survival and metastasis. Asian Pac. J. Cancer Prev. 2014, 15, 5873–5878. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, C.; Dachs, G.U.; Munn, D.; Currie, M.J.; Robinson, B.A.; Pearson, J.F.; Vissers, M.C. Increased Tumor Ascorbate is Associated with Extended Disease-Free Survival and Decreased Hypoxia-Inducible Factor-1 Activation in Human Colorectal Cancer. Front. Oncol. 2014, 4, 10. [Google Scholar] [CrossRef]
- Karpathiou, G.; Sivridis, E.; Koukourakis, M.; Mikroulis, D.; Bouros, D.; Froudarakis, M.; Bougioukas, G.; Maltezos, E.; Giatromanolaki, A. Autophagy and Bcl-2/BNIP3 death regulatory pathway in non-small cell lung carcinomas. APMIS 2013, 121, 592–604. [Google Scholar] [CrossRef]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef]
- Petry, I.B.; Fieber, E.; Schmidt, M.; Gehrmann, M.; Gebhard, S.; Hermes, M.; Schormann, W.; Selinski, S.; Freis, E.; Schwender, H.; et al. ERBB2 induces an antiapoptotic expression pattern of Bcl-2 family members in node-negative breast cancer. Clin. Cancer Res. 2010, 16, 451–460. [Google Scholar] [CrossRef]
- Lukashova-v Zangen, I.; Kneitz, S.; Monoranu, C.M.; Rutkowski, S.; Hinkes, B.; Vince, G.H.; Huang, B.; Roggendorf, W. Ependymoma gene expression profiles associated with histological subtype, proliferation, and patient survival. Acta Neuropathol. 2007, 113, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.; Horn, L.C.; Hockel, M. Hypoxia and expression of the proapoptotic regulator BNIP3 in cervical cancer. Int. J. Gynecol. Cancer 2006, 16, 1314–1320. [Google Scholar] [CrossRef]
- An, J.S.; Huang, M.N.; Song, Y.M.; Li, N.; Wu, L.Y.; Zhan, Q.M. A preliminary study of genes related to concomitant chemoradiotherapy resistance in advanced uterine cervical squamous cell carcinoma. Chin. Med. J. (Engl.) 2013, 126, 4109–4115. [Google Scholar] [PubMed]
- Macher-Goeppinger, S.; Keith, M.; Hatiboglu, G.; Hohenfellner, M.; Schirmacher, P.; Roth, W.; Tagscherer, K.E. Expression and Functional Characterization of the BNIP3 Protein in Renal Cell Carcinomas. Transl. Oncol. 2017, 10, 869–875. [Google Scholar] [CrossRef]
- Niu, Y.; Lin, Z.; Wan, A.; Chen, H.; Liang, H.; Sun, L.; Wang, Y.; Li, X.; Xiong, X.F.; Wei, B.; et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol. Cancer 2019, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Pei, L.; Jiang, H.; Yang, W.; Chen, J.; Liang, H. Chemoresistance of colorectal cancer to 5-fluorouracil is associated with silencing of the BNIP3 gene through aberrant methylation. J. Cancer 2017, 8, 1187–1196. [Google Scholar] [CrossRef] [Green Version]
- Sambuudash, O.; Kim, H.S.; Cho, M.Y. Lack of Aberrant Methylation in an Adjacent Area of Left-Sided Colorectal Cancer. Yonsei Med. J. 2017, 58, 749–755. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Xue, H.; Lu, L.; Shi, P.; Wang, J.; Wang, J. Accumulated promoter methylation as a potential biomarker for esophageal cancer. Oncotarget 2017, 8, 679–691. [Google Scholar] [CrossRef]
- Kamino, H.; Nakamura, Y.; Tsuneki, M.; Sano, H.; Miyamoto, Y.; Kitamura, N.; Futamura, M.; Kanai, Y.; Taniguchi, H.; Shida, D.; et al. Mieap-regulated mitochondrial quality control is frequently inactivated in human colorectal cancer. Oncogenesis 2016, 4, e181. [Google Scholar] [CrossRef] [PubMed]
- Simsek, B.C.; Turk, B.A.; Ozen, F.; Tuzcu, M.; Kanter, M. Investigation of telomerase activity and apoptosis on invasive ductal carcinoma of the breast using immunohistochemical and Western blot methods. Eur. Rev. Med. Pharm. Sci. 2015, 19, 3089–3099. [Google Scholar]
- Liu, Z.; Zhang, J.; Gao, Y.; Pei, L.; Zhou, J.; Gu, L.; Zhang, L.; Zhu, B.; Hattori, N.; Ji, J.; et al. Large-scale characterization of DNA methylation changes in human gastric carcinomas with and without metastasis. Clin. Cancer Res. 2014, 20, 4598–4612. [Google Scholar] [CrossRef]
- Garcia-Baquero, R.; Puerta, P.; Beltran, M.; Alvarez-Mujica, M.; Alvarez-Ossorio, J.L.; Sanchez-Carbayo, M. Methylation of tumor suppressor genes in a novel panel predicts clinical outcome in paraffin-embedded bladder tumors. Tumour Biol.: J. Int. Soc. Oncodevelopmental Biol. Med. 2014, 35, 5777–5786. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Baquero, R.; Puerta, P.; Beltran, M.; Alvarez, M.; Sacristan, R.; Alvarez-Ossorio, J.L.; Sanchez-Carbayo, M. Methylation of a novel panel of tumor suppressor genes in urine moves forward noninvasive diagnosis and prognosis of bladder cancer: A 2-center prospective study. J. Urol. 2013, 190, 723–730. [Google Scholar] [CrossRef]
- Deng, Q.; Huang, C.M.; Chen, N.; Li, L.; Wang, X.D.; Zhang, W.; Bi, F.; Tang, Q.L.; Li, Z.P.; Wang, W. Chemotherapy and radiotherapy downregulate the activity and expression of DNA methyltransferase and enhance Bcl-2/E1B-19-kDa interacting protein-3-induced apoptosis in human colorectal cancer cells. Chemotherapy 2012, 58, 445–453. [Google Scholar] [CrossRef]
- Jin, T.; Lin, H.X.; Lin, H.; Guo, L.B.; Ge, N.; Cai, X.Y.; Sun, R.; Chen, W.K.; Li, Q.L.; Hu, W.H. Expression TGM2 and BNIP3 have prognostic significance in laryngeal cancer patients receiving surgery and postoperative radiotherapy: A retrospective study. J. Transl. Med. 2012, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Sugita, H.; Iida, S.; Inokuchi, M.; Kato, K.; Ishiguro, M.; Ishikawa, T.; Takagi, Y.; Enjoji, M.; Yamada, H.; Uetake, H.; et al. Methylation of BNIP3 and DAPK indicates lower response to chemotherapy and poor prognosis in gastric cancer. Oncol. Rep. 2011, 25, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Kitajima, Y.; Koga, Y.; Tanaka, T.; Nakamura, J.; Hashiguchi, K.; Noshiro, H.; Miyazaki, K. Aberrant gene methylation is a biomarker for the detection of cancer cells in peritoneal wash samples from advanced gastric cancer patients. Ann. Surg. Oncol. 2011, 18, 3013–3019. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Iida, S.; Ishiguro, M.; Uetake, H.; Ishikawa, T.; Takagi, Y.; Kobayashi, H.; Higuchi, T.; Enomoto, M.; Mogushi, K.; et al. Methylated BNIP3 gene in colorectal cancer prognosis. Oncol. Lett. 2010, 1, 865–872. [Google Scholar] [CrossRef]
- Hiraki, M.; Kitajima, Y.; Nakafusa, Y.; Nakamura, J.; Hashiguchi, K.; Sumi, K.; Noshiro, H.; Miyazaki, K. CpG island methylation of BNIP3 predicts resistance against S-1/CPT-11 combined therapy in colorectal cancer patients. Oncol. Rep. 2010, 23, 191–197. [Google Scholar]
- Jourdan, M.; Reme, T.; Goldschmidt, H.; Fiol, G.; Pantesco, V.; De Vos, J.; Rossi, J.F.; Hose, D.; Klein, B. Gene expression of anti- and pro-apoptotic proteins in malignant and normal plasma cells. Br. J. Haematol. 2009, 145, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.; Schmidt, W.M.; Ziegler, B.; Holzer, S.; Mullauer, L.; Bilban, M.; Zielinski, C.C.; Drach, J.; Zochbauer-Muller, S. Genome-wide transcriptional response to 5-aza-2’-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. 2008, 68, 44–54. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Esposito, I.; Giese, T.; Ketterer, K.; Buchler, M.W.; Giese, N.A.; Friess, H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 2005, 24, 4421–4432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akada, M.; Crnogorac-Jurcevic, T.; Lattimore, S.; Mahon, P.; Lopes, R.; Sunamura, M.; Matsuno, S.; Lemoine, N.R. Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clin. Cancer Res. 2005, 11, 3094–3101. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.; Grau, L.; Puerta, P.; Gimenez, L.; Venditti, J.; Quadrelli, S.; Sanchez-Carbayo, M. Multiplexed methylation profiles of tumor suppressor genes and clinical outcome in lung cancer. J. Transl. Med. 2010, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, J.W.; Wang, Y.; Chen, T.; Ren, N.; Yang, L.; Xu, W.; He, H.; Jiang, Y.; Chen, X.; et al. Abnormal miRNA-30e Expression is Associated with Breast Cancer Progression. Clin. Lab. 2016, 62, 121–128. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, L.; He, M.; Huang, W.; Li, H.; Wang, Y.; Kong, J.; Qi, S.; Ouyang, J.; Qiu, X. Nix protein positively regulates NF-kappaB activation in gliomas. PLoS ONE 2012, 7, e44559. [Google Scholar] [CrossRef]
- Liu, W.; Xie, C.C.; Zhu, Y.; Li, T.; Sun, J.; Cheng, Y.; Ewing, C.M.; Dalrymple, S.; Turner, A.R.; Sun, J.; et al. Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia 2008, 10, 897–907. [Google Scholar] [CrossRef]
- Eisele, L.; Klein-Hitpass, L.; Chatzimanolis, N.; Opalka, B.; Boes, T.; Seeber, S.; Moritz, T.; Flasshove, M. Differential expression of drug-resistance-related genes between sensitive and resistant blasts in acute myeloid leukemia. Acta Haematol. 2007, 117, 8–15. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, D.; Zhou, L.; Pei, Y.; Zhuang, Y.; Cui, W.; Chen, J. FUN14 domain-containing 1 promotes breast cancer proliferation and migration by activating calcium-NFATC1-BMI1 axis. EBioMedicine 2019, 41, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Wu, H.; Wang, T.W.; Yang, N.; Guo, X.; Jang, X.J. Hydrogen peroxide-induced mitophagy contributes to laryngeal cancer cells survival via the upregulation of FUNDC1. Clin. Transl. Oncol. 2018. [Google Scholar] [CrossRef]
- Hou, H.; Er, P.; Cheng, J.; Chen, X.; Ding, X.; Wang, Y.; Chen, X.; Yuan, Z.; Pang, Q.; Wang, P.; et al. High expression of FUNDC1 predicts poor prognostic outcomes and is a promising target to improve chemoradiotherapy effects in patients with cervical cancer. Cancer Med. 2017, 6, 1871–1881. [Google Scholar] [CrossRef]
- Cheng, J.; Qian, D.; Ding, X.; Song, T.; Cai, M.; Dan, X.; Wang, Y.; Zhao, J.; Liu, Z.; Wu, Z.; et al. High PGAM5 expression induces chemoresistance by enhancing Bcl-xL-mediated anti-apoptotic signaling and predicts poor prognosis in hepatocellular carcinoma patients. Cell Death Dis. 2018, 9, 991. [Google Scholar] [CrossRef]
- Ng Kee Kwong, F.; Nicholson, A.G.; Pavlidis, S.; Adcock, I.M.; Chung, K.F. PGAM5 expression and macrophage signatures in non-small cell lung cancer associated with chronic obstructive pulmonary disease (COPD). BMC Cancer 2018, 18, 1238. [Google Scholar] [CrossRef]
- Randall, E.C.; Zadra, G.; Chetta, P.; Lopez, B.G.C.; Syamala, S.; Basu, S.S.; Agar, J.N.; Loda, M.; Tempany, C.M.; Fennessy, F.M.; et al. Molecular Characterization of Prostate Cancer with Associated Gleason Score Using Mass Spectrometry Imaging. Mol. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Sapandowski, A.; Stope, M.; Evert, K.; Evert, M.; Zimmermann, U.; Peter, D.; Page, I.; Burchardt, M.; Schild, L. Cardiolipin composition correlates with prostate cancer cell proliferation. Mol. Cell Biochem. 2015, 410, 175–185. [Google Scholar] [CrossRef]
- Zhong, H.; Xiao, M.; Zarkovic, K.; Zhu, M.; Sa, R.; Lu, J.; Tao, Y.; Chen, Q.; Xia, L.; Cheng, S.; et al. Mitochondrial control of apoptosis through modulation of cardiolipin oxidation in hepatocellular carcinoma: A novel link between oxidative stress and cancer. Free Radic. Biol. Med. 2017, 102, 67–76. [Google Scholar] [CrossRef]
- Cai, X.W.; Yu, W.W.; Yu, W.; Zhang, Q.; Feng, W.; Liu, M.N.; Sun, M.H.; Xiang, J.Q.; Zhang, Y.W.; Fu, X.L. Tissue-based quantitative proteomics to screen and identify the potential biomarkers for early recurrence/metastasis of esophageal squamous cell carcinoma. Cancer Med. 2018, 7, 2504–2517. [Google Scholar] [CrossRef]
- Ross, J.A.; Robles-Escajeda, E.; Oaxaca, D.M.; Padilla, D.L.; Kirken, R.A. The prohibitin protein complex promotes mitochondrial stabilization and cell survival in hematologic malignancies. Oncotarget 2017, 8, 65445–65456. [Google Scholar] [CrossRef] [Green Version]
- Yoshimaru, T.; Ono, M.; Bando, Y.; Chen, Y.A.; Mizuguchi, K.; Shima, H.; Komatsu, M.; Imoto, I.; Izumi, K.; Honda, J.; et al. A-kinase anchoring protein BIG3 coordinates oestrogen signalling in breast cancer cells. Nat. Commun. 2017, 8, 15427. [Google Scholar] [CrossRef] [Green Version]
- Zuo, X.; Chen, L.; Liu, L.; Zhang, Z.; Zhang, X.; Yu, Q.; Feng, L.; Zhao, X.; Qin, T. Identification of a panel of complex autoantigens (LGALS3, PHB2, MUC1, and GK2) in combination with CA15-3 for the diagnosis of early-stage breast cancer. Tumour Biol.: J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 1309–1317. [Google Scholar] [CrossRef]
- Mengwasser, J.; Piau, A.; Schlag, P.; Sleeman, J.P. Differential immunization identifies PHB1/PHB2 as blood-borne tumor antigens. Oncogene 2004, 23, 7430–7435. [Google Scholar] [CrossRef] [Green Version]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef]
- Koybasi, S.; Senkal, C.E.; Sundararaj, K.; Spassieva, S.; Bielawski, J.; Osta, W.; Day, T.A.; Jiang, J.C.; Jazwinski, S.M.; Hannun, Y.A.; et al. Defects in cell growth regulation by C18:0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J. Biol. Chem. 2004, 279, 44311–44319. [Google Scholar] [CrossRef]
- Wang, Z.; Wen, L.; Zhu, F.; Wang, Y.; Xie, Q.; Chen, Z.; Li, Y. Overexpression of ceramide synthase 1 increases C18-ceramide and leads to lethal autophagy in human glioma. Oncotarget 2017, 8, 104022–104036. [Google Scholar] [CrossRef]
- Qu, B.; Yao, L.; Ma, H.L.; Chen, H.L.; Zhang, Z.; Xie, J. Prognostic significance of autophagy-related proteins expression in resected human gastric adenocarcinoma. J. Huazhong Univ. Sci. Technol. Med. Sci. 2017, 37, 37–43. [Google Scholar] [CrossRef]
- Nitta, T.; Sato, Y.; Ren, X.S.; Harada, K.; Sasaki, M.; Hirano, S.; Nakanuma, Y. Autophagy may promote carcinoma cell invasion and correlate with poor prognosis in cholangiocarcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 4913–4921. [Google Scholar]
- Ko, Y.H.; Cho, Y.S.; Won, H.S.; Jeon, E.K.; An, H.J.; Hong, S.U.; Park, J.H.; Lee, M.A. Prognostic significance of autophagy-related protein expression in resected pancreatic ductal adenocarcinoma. Pancreas 2013, 42, 829–835. [Google Scholar] [CrossRef]
- Falasca, L.; Torino, F.; Marconi, M.; Costantini, M.; Pompeo, V.; Sentinelli, S.; De Salvo, L.; Patrizio, M.; Padula, C.; Gallucci, M.; et al. AMBRA1 and SQSTM1 expression pattern in prostate cancer. Apoptosis 2015, 20, 1577–1586. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Yang, D.; Cheng, D.; Tu, Q.; Yang, H.; Sun, B.; Yan, L.; Dai, H.; Luo, J.; Mao, B.; Cao, Y.; et al. HUWE1 controls the development of non-small cell lung cancer through down-regulation of p53. Theranostics 2018, 8, 3517–3529. [Google Scholar] [CrossRef]
- Li, Z.; Peng, Z.; Gu, S.; Zheng, J.; Feng, D.; Qin, Q.; He, J. Global Analysis of miRNA-mRNA Interaction Network in Breast Cancer with Brain Metastasis. Anticancer Res. 2017, 37, 4455–4468. [Google Scholar] [CrossRef]
- Kodama, T.; Newberg, J.Y.; Kodama, M.; Rangel, R.; Yoshihara, K.; Tien, J.C.; Parsons, P.H.; Wu, H.; Finegold, M.J.; Copeland, N.G.; et al. Transposon mutagenesis identifies genes and cellular processes driving epithelial-mesenchymal transition in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, E3384–E3393. [Google Scholar] [CrossRef]
- Cheng, D.D.; Yu, T.; Hu, T.; Yao, M.; Fan, C.Y.; Yang, Q.C. MiR-542-5p is a negative prognostic factor and promotes osteosarcoma tumorigenesis by targeting HUWE1. Oncotarget 2015, 6, 42761–42772. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.L.; Lin, S.R.; Chen, J.S.; Lin, S.W.; Yu, S.L.; Chen, H.Y.; Yen, C.T.; Lin, C.Y.; Lin, J.F.; Lin, K.H.; et al. Expression and prognostic significance of the apoptotic genes BCL2L13, Livin, and CASP8AP2 in childhood acute lymphoblastic leukemia. Leuk Res. 2010, 34, 18–23. [Google Scholar] [CrossRef]
- Tahir, S.K.; Wass, J.; Joseph, M.K.; Devanarayan, V.; Hessler, P.; Zhang, H.; Elmore, S.W.; Kroeger, P.E.; Tse, C.; Rosenberg, S.H.; et al. Identification of expression signatures predictive of sensitivity to the Bcl-2 family member inhibitor ABT-263 in small cell lung carcinoma and leukemia/lymphoma cell lines. Mol. Cancer Ther. 2010, 9, 545–557. [Google Scholar] [CrossRef]
- Holleman, A.; den Boer, M.L.; de Menezes, R.X.; Cheok, M.H.; Cheng, C.; Kazemier, K.M.; Janka-Schaub, G.E.; Gobel, U.; Graubner, U.B.; Evans, W.E.; et al. The expression of 70 apoptosis genes in relation to lineage, genetic subtype, cellular drug resistance, and outcome in childhood acute lymphoblastic leukemia. Blood 2006, 107, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Millino, C.; Maretto, I.; Pacchioni, B.; Digito, M.; De Paoli, A.; Canzonieri, V.; D’Angelo, E.; Agostini, M.; Rizzolio, F.; Giordano, A.; et al. Gene and MicroRNA Expression Are Predictive of Tumor Response in Rectal Adenocarcinoma Patients Treated With Preoperative Chemoradiotherapy. J. Cell Physiol. 2017, 232, 426–435. [Google Scholar] [CrossRef]
- Leon-Mateos, L.; Casas, H.; Abalo, A.; Vieito, M.; Abreu, M.; Anido, U.; Gomez-Tato, A.; Lopez, R.; Abal, M.; Muinelo-Romay, L. Improving circulating tumor cells enumeration and characterization to predict outcome in first line chemotherapy mCRPC patients. Oncotarget 2017, 8, 54708–54721. [Google Scholar] [CrossRef]
- da Silva, S.D.; Marchi, F.A.; Xu, B.; Bijian, K.; Alobaid, F.; Mlynarek, A.; Rogatto, S.R.; Hier, M.; Kowalski, L.P.; Alaoui-Jamali, M.A. Predominant Rab-GTPase amplicons contributing to oral squamous cell carcinoma progression to metastasis. Oncotarget 2015, 6, 21950–21963. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Et Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.E.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef] [Green Version]
- Lyons, A.; Coleman, M.; Riis, S.; Favre, C.; O’Flanagan, C.H.; Zhdanov, A.V.; Papkovsky, D.B.; Hursting, S.D.; O’Connor, R. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J. Biol. Chem. 2017, 292, 16983–16998. [Google Scholar] [CrossRef] [Green Version]
- Agnihotri, S.; Golbourn, B.; Huang, X.; Remke, M.; Younger, S.; Cairns, R.A.; Chalil, A.; Smith, C.A.; Krumholtz, S.L.; Mackenzie, D.; et al. PINK1 Is a Negative Regulator of Growth and the Warburg Effect in Glioblastoma. Cancer Res. 2016, 76, 4708–4719. [Google Scholar] [CrossRef]
- Chang, H.W.; Kim, M.R.; Lee, H.J.; Lee, H.M.; Kim, G.C.; Lee, Y.S.; Nam, H.Y.; Lee, M.; Jang, H.J.; Lee, K.E.; et al. p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene 2019. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef] [PubMed]
- Maes, H.; Van Eygen, S.; Krysko, D.V.; Vandenabeele, P.; Nys, K.; Rillaerts, K.; Garg, A.D.; Verfaillie, T.; Agostinis, P. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. 2014, 5, e1127. [Google Scholar] [CrossRef]
- Kim, J.W.; Gao, P.; Liu, Y.C.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell. Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef]
- Dang, C.V.; Kim, J.W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, L.; Fei, X.C.; Jiang, X.F.; Zheng, Z.; Zhao, Y.; Wang, C.F.; Li, B.; Chen, S.J.; Janin, A.; et al. MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J. 2017, 7, e582. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The Role of PGC1alpha in Cancer Metabolism and its Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef]
- Park, J.H.; Ko, J.; Park, Y.S.; Park, J.; Hwang, J.; Koh, H.C. Clearance of Damaged Mitochondria Through PINK1 Stabilization by JNK and ERK MAPK Signaling in Chlorpyrifos-Treated Neuroblastoma Cells. Mol. Neurobiol. 2017, 54, 1844–1857. [Google Scholar] [CrossRef]
- Bazil, J.N.; Beard, D.A.; Vinnakota, K.C. Catalytic Coupling of Oxidative Phosphorylation, ATP Demand, and Reactive Oxygen Species Generation. Biophys. J. 2016, 110, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef]
- Melser, S.; Chatelain, E.H.; Lavie, J.; Mahfouf, W.; Jose, C.; Obre, E.; Goorden, S.; Priault, M.; Elgersma, Y.; Rezvani, H.R.; et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab. 2013, 17, 719–730. [Google Scholar] [CrossRef]
- Vara-Perez, M.; Maes, H.; Van Dingenen, S.; Agostinis, P. BNIP3 contributes to the glutamine-driven aggressive behavior of melanoma cells. Biol. Chem. 2019, 400, 187–193. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- McDonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Tan, Z.; Shi, F.; Tang, M.; Xie, L.; Zhao, L.; Li, Y.; Hu, J.; Zhou, M.; Bode, A.; et al. PGC1alpha/CEBPB/CPT1A axis promotes radiation resistance of nasopharyngeal carcinoma through activating fatty acid oxidation. Cancer Sci. 2019. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. PGC-1alpha buffers ROS-mediated removal of mitochondria during myogenesis. Cell Death Dis. 2014, 5, e1515. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef] [Green Version]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 706. [Google Scholar] [CrossRef]
- Inoue, S.; Hao, Z.; Elia, A.J.; Cescon, D.; Zhou, L.; Silvester, J.; Snow, B.; Harris, I.S.; Sasaki, M.; Li, W.Y.; et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 2013, 27, 1101–1114. [Google Scholar] [CrossRef] [Green Version]
- Strappazzon, F.; Cecconi, F. AMBRA1-induced mitophagy: A new mechanism to cope with cancer? Mol. Cell Oncol. 2015, 2, e975647. [Google Scholar] [CrossRef] [Green Version]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef]
- Gargini, R.; Garcia-Escudero, V.; Izquierdo, M.; Wandosell, F. Oncogene-mediated tumor transformation sensitizes cells to autophagy induction. Oncol. Rep. 2016, 35, 3689–3695. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.Y.; Li, D.; Chen, Z.X.; Huang, Y.H.; Gao, W.Y.; Zheng, B.Y.; Wang, X.Z. Hepatitis B Virus X protein elevates Parkin-mediated mitophagy through Lon Peptidase in starvation. Exp. Cell Res. 2018, 368, 75–83. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Xie, X.; Zhan, L.; Yang, Y.; Sharp, D.W.; Hu, Z.S.; Su, X.; Maganti, A.; Jiang, C.; Lu, W.; et al. Autophagy maintains tumour growth through circulating arginine. Nature 2018, 563, 569–573. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.F. The Regulation of Iron Absorption and Homeostasis. Clin. Biochem. Rev. 2016, 37, 51–62. [Google Scholar]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef]
- Kang, R.; Xie, Y.; Zeh, H.J.; Klionsky, D.J.; Tang, D. Mitochondrial quality control mediated by PINK1 and PRKN: Links to iron metabolism and tumor immunity. Autophagy 2019, 15, 172–173. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, P.K.; Bollrath, J.; Pallangyo, C.K.; Matsutani, T.; Canli, O.; De Oliveira, T.; Diamanti, M.A.; Muller, N.; Gamrekelashvili, J.; Putoczki, T.; et al. Mitophagy in Intestinal Epithelial Cells Triggers Adaptive Immunity during Tumorigenesis. Cell 2018, 174, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904. [Google Scholar] [CrossRef]
- Fessler, E.; Dijkgraaf, F.E.; De Sousa, E.M.F.; Medema, J.P. Cancer stem cell dynamics in tumor progression and metastasis: Is the microenvironment to blame? Cancer Lett. 2013, 341, 97–104. [Google Scholar] [CrossRef]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292. [Google Scholar] [CrossRef]
- Lathia, J.D.; Liu, H. Overview of Cancer Stem Cells and Stemness for Community Oncologists. Target Oncol. 2017, 12, 387–399. [Google Scholar] [CrossRef] [Green Version]
- Whelan, K.A.; Chandramouleeswaran, P.M.; Tanaka, K.; Natsuizaka, M.; Guha, M.; Srinivasan, S.; Darling, D.S.; Kita, Y.; Natsugoe, S.; Winkler, J.D.; et al. Autophagy supports generation of cells with high CD44 expression via modulation of oxidative stress and Parkin-mediated mitochondrial clearance. Oncogene 2017, 36, 4843–4858. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.A.; Wang, C.Y.; Hsieh, Y.T.; Chen, Y.J.; Wei, Y.H. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle 2015, 14, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 2017, 6, e22187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhu, R.; Shen, J.; Wu, J.; Lu, W.; Zhang, J.; Zhang, J.; Liu, K. Human Bone Marrow Mesenchymal Stem Cells Rescue Endothelial Cells Experiencing Chemotherapy Stress by Mitochondrial Transfer Via Tunneling Nanotubes. Stem Cells Dev. 2019. [Google Scholar] [CrossRef]
- Wang, X.; Gerdes, H.H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, A.; Fritz, V.; Brondello, J.M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; de Fraipont, F.; Moisan, A.; Prouteau, C.A.; Boukhaddaoui, H.; et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 2015, 5, 9073. [Google Scholar] [CrossRef] [PubMed]
- Nzigou Mombo, B.; Gerbal-Chaloin, S.; Bokus, A.; Daujat-Chavanieu, M.; Jorgensen, C.; Hugnot, J.P.; Vignais, M.L. MitoCeption: Transferring Isolated Human MSC Mitochondria to Glioblastoma Stem Cells. J. Vis. Exp.: JoVE 2017. [Google Scholar] [CrossRef]
- Lu, J.; Zheng, X.; Li, F.; Yu, Y.; Chen, Z.; Liu, Z.; Wang, Z.; Xu, H.; Yang, W. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 2017, 8, 15539–15552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hough, K.P.; Trevor, J.L.; Strenkowski, J.G.; Wang, Y.; Chacko, B.K.; Tousif, S.; Chanda, D.; Steele, C.; Antony, V.B.; Dokland, T.; et al. Exosomal transfer of mitochondria from airway myeloid-derived regulatory cells to T cells. Redox Biol. 2018, 18, 54–64. [Google Scholar] [CrossRef]
- Torralba, D.; Baixauli, F.; Villarroya-Beltri, C.; Fernandez-Delgado, I.; Latorre-Pellicer, A.; Acin-Perez, R.; Martin-Cofreces, N.B.; Jaso-Tamame, A.L.; Iborra, S.; Jorge, I.; et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun. 2018, 9, 2658. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kim, J.H.; Kim, M.; Park, S.J.; Koh, S.H.; Ahn, H.S.; Kang, G.H.; Lee, J.B.; Park, K.S.; Lee, H.K. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS ONE 2012, 7, e32778. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef]
- Lou, E.; Zhai, E.; Sarkari, A.; Desir, S.; Wong, P.; Iizuka, Y.; Yang, J.; Subramanian, S.; McCarthy, J.; Bazzaro, M.; et al. Cellular and Molecular Networking Within the Ecosystem of Cancer Cell Communication via Tunneling Nanotubes. Front. Cell Dev. Biol. 2018, 6, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patheja, P.; Sahu, K. Macrophage conditioned medium induced cellular network formation in MCF-7 cells through enhanced tunneling nanotube formation and tunneling nanotube mediated release of viable cytoplasmic fragments. Exp. Cell Res. 2017, 355, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef] [PubMed]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Miro1 Enhances Mitochondria Transfer from Multipotent Mesenchymal Stem Cells (MMSC) to Neural Cells and Improves the Efficacy of Cell Recovery. Molecules 2018, 23, 687. [Google Scholar] [CrossRef]
- Roger, A.J.; Munoz-Gomez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Munoz-Planillo, R.; Nunez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Bhat, N.; Fitzgerald, K.A. Recognition of cytosolic DNA by cGAS and other STING-dependent sensors. Eur. J. Immunol. 2014, 44, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Quicke, K.M.; Diamond, M.S.; Suthar, M.S. Negative regulators of the RIG-I-like receptor signaling pathway. Eur. J. Immunol. 2017, 47, 615–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, D.; Park, E.; Kang, S.J. Stimulator of IFN genes-mediated DNA-sensing pathway is suppressed by NLRP3 agonists and regulated by mitofusin 1 and TBC1D15, mitochondrial dynamics mediators. FASEB J. 2017, 31, 4866–4878. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassano, M.L.; van Vliet, A.R.; Agostinis, P. Mitochondria-Associated Membranes As Networking Platforms and Regulators of Cancer Cell Fate. Front. Oncol. 2017, 7, 174. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [Green Version]
- McLelland, G.L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 2018, 7, e32866. [Google Scholar] [CrossRef]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Zhang, N.P.; Liu, X.J.; Xie, L.; Shen, X.Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Investig. 2019. [Google Scholar] [CrossRef]
- Kim, M.J.; Bae, S.H.; Ryu, J.C.; Kwon, Y.; Oh, J.H.; Kwon, J.; Moon, J.S.; Kim, K.; Miyawaki, A.; Lee, M.G.; et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 2016, 12, 1272–1291. [Google Scholar] [CrossRef] [Green Version]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef]
- Gomez-Cabanas, L.; Lopez-Cotarelo, P.; Criado-Garcia, O.; Murphy, M.P.; Boya, P.; Rodriguez-Fernandez, J.L. Immunological Synapse Formation Induces Mitochondrial Clustering and Mitophagy in Dendritic Cells. J. Immunol. 2019, 202, 1715–1723. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, T.E.; Johnson, L.R.; Kang, H.H.; Sun, J.C. BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 2015, 43, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Abdrakhmanov, A.; Kulikov, A.V.; Luchkina, E.A.; Zhivotovsky, B.; Gogvadze, V. Involvement of mitophagy in cisplatin-induced cell death regulation. Biol. Chem. 2019, 400, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Shao, L.J.; Jiang, Z.F.; Liu, R.Y. Gemcitabine-Induced Autophagy Protects Human Lung Cancer Cells from Apoptotic Death. Lung 2016, 194, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Mackeh, R.; Perdiz, D.; Lorin, S.; Codogno, P.; Pous, C. Autophagy and microtubules - new story, old players. J. Cell Sci. 2013, 126, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Cao, J.; Yao, S. Matrine promotes liver cancer cell apoptosis by inhibiting mitophagy and PINK1/Parkin pathways. Cell Stress Chaperones 2018, 23, 1295–1309. [Google Scholar] [CrossRef]
- Zheng, R.; Yao, Q.; Xie, G.; Du, S.; Ren, C.; Wang, Y.; Yuan, Y. TAT-ODD-p53 enhances the radiosensitivity of hypoxic breast cancer cells by inhibiting Parkin-mediated mitophagy. Oncotarget 2015, 6, 17417–17429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Fang, Y.; Yan, L.; Yuan, N.; Zhang, S.; Xu, L.; Nie, M.; Zhang, X.; Wang, J. Erythroleukemia cells acquire an alternative mitophagy capability. Sci. Rep. 2016, 6, 24641. [Google Scholar] [CrossRef] [Green Version]
- MacKeigan, J.P.; Murphy, L.O.; Blenis, J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 2005, 7, 591–600. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Z.; Guo, J.; Wang, L.; Liu, X. Ambra1 induces autophagy and desensitizes human prostate cancer cells to cisplatin. Biosci. Rep. 2017. [Google Scholar] [CrossRef]
- Villa, E.; Proics, E.; Rubio-Patino, C.; Obba, S.; Zunino, B.; Bossowski, J.P.; Rozier, R.M.; Chiche, J.; Mondragon, L.; Riley, J.S.; et al. Parkin-Independent Mitophagy Controls Chemotherapeutic Response in Cancer Cells. Cell Rep. 2017, 20, 2846–2859. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Wang, J.; Roginskaya, V.; McDermott, L.A.; Edwards, R.P.; Stolz, D.B.; Llambi, F.; Green, D.R.; Van Houten, B. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget 2014, 5, 4180–4194. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Wang, C.; Hu, N.; Li, Y.; Liu, M.; Lei, Y.; Chen, M.; Chen, L.; Chen, C.; Lan, P.; et al. Inhibition of PINK1/Parkin-dependent mitophagy sensitizes multidrug-resistant cancer cells to B5G1, a new betulinic acid analog. Cell Death Dis. 2019, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Cristofani, R.; Montagnani Marelli, M.; Cicardi, M.E.; Fontana, F.; Marzagalli, M.; Limonta, P.; Poletti, A.; Moretti, R.M. Dual role of autophagy on docetaxel-sensitivity in prostate cancer cells. Cell Death Dis. 2018, 9, 889. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, S.J.; Park, Y.J.; Park, S.Y.; Oh, B.M.; Park, Y.S.; Kim, B.Y.; Lee, Y.H.; Cho, H.J.; Yoon, S.R.; et al. Opa-interacting protein 5 modulates docetaxel-induced cell death via regulation of mitophagy in gastric cancer. Tumour Biol.: J. Int. Soc. Oncodevelopmental Biol. Med. 2017, 39, 1010428317733985. [Google Scholar] [CrossRef]
- Yan, C.; Luo, L.; Guo, C.Y.; Goto, S.; Urata, Y.; Shao, J.H.; Li, T.S. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017, 388, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.A.; Van Wickle, J.; Hill, R.B.; Marchese, A.; Kalyanaraman, B.; Dwinell, M.B. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J. Biol. Chem. 2018, 293, 14891–14904. [Google Scholar] [CrossRef] [Green Version]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Naik, P.P.; Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, C.K.; Mishra, R.; Patil, S.; Bhutia, S.K. Autophagy regulates cisplatin-induced stemness and chemoresistance via the upregulation of CD44, ABCB1 and ADAM17 in oral squamous cell carcinoma. Cell Prolif. 2018, 51. [Google Scholar] [CrossRef]
- Takeda, M.; Koseki, J.; Takahashi, H.; Miyoshi, N.; Nishida, N.; Nishimura, J.; Hata, T.; Matsuda, C.; Mizushima, T.; Yamamoto, H.; et al. Disruption of Endolysosomal RAB5/7 Efficiently Eliminates Colorectal Cancer Stem Cells. Cancer Res. 2019, 79, 1426–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.M.; Lan, K.L.; Huang, W.S.; Lee, Y.J.; Lee, T.W.; Chang, C.H.; Chuang, C.M. (188)Re-Liposome Can Induce Mitochondrial Autophagy and Reverse Drug Resistance for Ovarian Cancer: From Bench Evidence to Preliminary Clinical Proof-of-Concept. Int. J. Mol. Sci. 2017, 18, 903. [Google Scholar] [CrossRef]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef]
- Held, N.M.; Houtkooper, R.H. Mitochondrial quality control pathways as determinants of metabolic health. Bioessays: News Rev. Mol. Cell. Dev. Biol. 2015, 37, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Guo, J.; Zhang, Q.; Cui, L.; Zhang, L.; Zhang, T.; Zhao, J.; Li, J.; Middleton, A.; Carmichael, P.L.; et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol. Vitr.: Int. J. Publ. Assoc. BIBRA 2018, 51, 1–10. [Google Scholar] [CrossRef]
- Gharanei, M.; Hussain, A.; Janneh, O.; Maddock, H. Attenuation of doxorubicin-induced cardiotoxicity by mdivi-1: A mitochondrial division/mitophagy inhibitor. PLoS ONE 2013, 8, e77713. [Google Scholar] [CrossRef]
- Du, Q.; Zhu, B.; Zhai, Q.; Yu, B. Sirt3 attenuates doxorubicin-induced cardiac hypertrophy and mitochondrial dysfunction via suppression of Bnip3. Am. J. Transl. Res. 2017, 9, 3360–3373. [Google Scholar]
- Warren, S. The immediate causes of death in cancer. Am. J. Med. Sci. 1932, 184, 610–615. [Google Scholar] [CrossRef]
- Martin, L.; Birdsell, L.; Macdonald, N.; Reiman, T.; Clandinin, M.T.; McCargar, L.J.; Murphy, R.; Ghosh, S.; Sawyer, M.B.; Baracos, V.E. Cancer cachexia in the age of obesity: Skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 2013, 31, 1539–1547. [Google Scholar] [CrossRef]
- Skipworth, R.J.; Stewart, G.D.; Dejong, C.H.; Preston, T.; Fearon, K.C. Pathophysiology of cancer cachexia: Much more than host-tumour interaction? Clin. Nutr. 2007, 26, 667–676. [Google Scholar] [CrossRef]
- von Haehling, S.; Anker, S.D. Prevalence, incidence and clinical impact of cachexia: Facts and numbers-update 2014. J. Cachexiasarcopenia Muscle 2014, 5, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Caillet, P.; Liuu, E.; Raynaud Simon, A.; Bonnefoy, M.; Guerin, O.; Berrut, G.; Lesourd, B.; Jeandel, C.; Ferry, M.; Rolland, Y.; et al. Association between cachexia, chemotherapy and outcomes in older cancer patients: A systematic review. Clin. Nutr. 2017, 36, 1473–1482. [Google Scholar] [CrossRef]
- Op den Kamp, C.M.; Langen, R.C.; Snepvangers, F.J.; de Theije, C.C.; Schellekens, J.M.; Laugs, F.; Dingemans, A.M.; Schols, A.M. Nuclear transcription factor kappa B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am. J. Clin. Nutr. 2013, 98, 738–748. [Google Scholar] [CrossRef]
- Johns, N.; Hatakeyama, S.; Stephens, N.A.; Degen, M.; Degen, S.; Frieauff, W.; Lambert, C.; Ross, J.A.; Roubenoff, R.; Glass, D.J.; et al. Clinical classification of cancer cachexia: Phenotypic correlates in human skeletal muscle. PLoS ONE 2014, 9, e83618. [Google Scholar] [CrossRef]
- Aversa, Z.; Pin, F.; Lucia, S.; Penna, F.; Verzaro, R.; Fazi, M.; Colasante, G.; Tirone, A.; Rossi Fanelli, F.; Ramaccini, C.; et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 2016, 6, 30340. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Manne, N.D.; Lima, M.; Enos, R.T.; Wehner, P.; Carson, J.A.; Blough, E. Altered cardiac muscle mTOR regulation during the progression of cancer cachexia in the ApcMin/+ mouse. Int. J. Oncol. 2013, 42, 2134–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, N.A.; Gallagher, I.J.; Rooyackers, O.; Skipworth, R.J.; Tan, B.H.; Marstrand, T.; Ross, J.A.; Guttridge, D.C.; Lundell, L.; Fearon, K.C.; et al. Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome Med. 2010, 2, 1. [Google Scholar] [CrossRef]
- Asp, M.L.; Tian, M.; Wendel, A.A.; Belury, M.A. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int. J. Cancer 2010, 126, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Feather, C.E.; Lees, J.G.; Makker, P.G.S.; Goldstein, D.; Kwok, J.B.; Moalem-Taylor, G.; Polly, P. Oxaliplatin induces muscle loss and muscle-specific molecular changes in Mice. Muscle Nerve 2018, 57, 650–658. [Google Scholar] [CrossRef]
- Barreto, R.; Mandili, G.; Witzmann, F.A.; Novelli, F.; Zimmers, T.A.; Bonetto, A. Cancer and Chemotherapy Contribute to Muscle Loss by Activating Common Signaling Pathways. Front. Physiol. 2016, 7, 472. [Google Scholar] [CrossRef] [Green Version]
- Julienne, C.M.; Tardieu, M.; Chevalier, S.; Pinault, M.; Bougnoux, P.; Labarthe, F.; Couet, C.; Servais, S.; Dumas, J.F. Cardiolipin content is involved in liver mitochondrial energy wasting associated with cancer-induced cachexia without the involvement of adenine nucleotide translocase. Biochim. Et Biophys. Acta 2014, 1842, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Dumas, J.F.; Goupille, C.; Julienne, C.M.; Pinault, M.; Chevalier, S.; Bougnoux, P.; Servais, S.; Couet, C. Efficiency of oxidative phosphorylation in liver mitochondria is decreased in a rat model of peritoneal carcinosis. J. Hepatol. 2011, 54, 320–327. [Google Scholar] [CrossRef]
- Tzika, A.A.; Fontes-Oliveira, C.C.; Shestov, A.A.; Constantinou, C.; Psychogios, N.; Righi, V.; Mintzopoulos, D.; Busquets, S.; Lopez-Soriano, F.J.; Milot, S.; et al. Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int. J. Oncol. 2013, 43, 886–894. [Google Scholar] [CrossRef] [Green Version]
- Bing, C.; Brown, M.; King, P.; Collins, P.; Tisdale, M.J.; Williams, G. Increased gene expression of brown fat uncoupling protein (UCP)1 and skeletal muscle UCP2 and UCP3 in MAC16-induced cancer cachexia. Cancer Res. 2000, 60, 2405–2410. [Google Scholar]
- Pettersen, K.; Andersen, S.; Degen, S.; Tadini, V.; Grosjean, J.; Hatakeyama, S.; Tesfahun, A.N.; Moestue, S.; Kim, J.; Nonstad, U.; et al. Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci. Rep. 2017, 7, 2046. [Google Scholar] [CrossRef] [PubMed]
- Lijie, G.; Yueyue, Z.; Nan, Z.; Ling, W.; Xuan, W.; Weijie, Y. Mitsugumin 53 promotes mitochondrial autophagy through regulating Ambra1 expression in C2C12 myoblast cells. Cell Biol. Int. 2019, 43, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Jang, G.M.; Huttenhain, R.; Gordon, D.E.; Du, D.; Newton, B.W.; Johnson, J.R.; Hiatt, J.; Hultquist, J.F.; Johnson, T.L.; et al. CRL4(AMBRA1) targets Elongin C for ubiquitination and degradation to modulate CRL5 signaling. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, Z.; Qi, J.; Duan, S.; Huang, Z.; Zhang, C.; Wu, L.; Zeng, M.; Zhang, B.; Wang, N.; et al. Drp1-dependent mitophagy protects against cisplatin-induced apoptosis of renal tubular epithelial cells by improving mitochondrial function. Oncotarget 2017, 8, 20988–21000. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018, 9, 1113. [Google Scholar] [CrossRef]
Figure 1.
The figure schematically summarizes the main mechanisms and players of canonical mitophagy pathways upon autophagosome recognition of the different receptors in a damaged mitochondrion (see main text for further details). First, PINK1/Parkin mediated ubiquitination of mitochondrial proteins enables the autophagy cargo receptors p62 and OPTN to bridge the mitochondria/autophagosome interaction. Alternatively, BNIP3, NIX and FUNDC1 can directly bind the LC3 molecules decorating the autophagosome, through a mechanism modulated by their phosphorylation status.
Figure 1.
The figure schematically summarizes the main mechanisms and players of canonical mitophagy pathways upon autophagosome recognition of the different receptors in a damaged mitochondrion (see main text for further details). First, PINK1/Parkin mediated ubiquitination of mitochondrial proteins enables the autophagy cargo receptors p62 and OPTN to bridge the mitochondria/autophagosome interaction. Alternatively, BNIP3, NIX and FUNDC1 can directly bind the LC3 molecules decorating the autophagosome, through a mechanism modulated by their phosphorylation status.

Figure 2.
Emerging links connecting oncogene-driven metabolic pathways and key modulators of the mitophagic machinery. (a) Upregulation of BNIP3 and NIX expression induced by HIF-1α diminishes mitochondrial mass and O2 consumption. (b) Loss of BNIP3 or PINK1/Parkin expression reduces mitophagy, leading to the accumulation of damaged mitochondria and ROS, which promotes HIF-1α stabilization and subsequently glycolysis. (c) p53 and BNIP3-dependent mitophagy removes abnormal mitochondria, reducing glycolysis and promoting O2 consumption. (d) RAS driven tumors require elevated autophagy/mitophagy levels to maintain mitochondrial functions and carry out proficient FAO. PGC1α and c-Myc are key regulators of mitochondrial functions to provide cancer cells with metabolic flexibility.
Figure 2.
Emerging links connecting oncogene-driven metabolic pathways and key modulators of the mitophagic machinery. (a) Upregulation of BNIP3 and NIX expression induced by HIF-1α diminishes mitochondrial mass and O2 consumption. (b) Loss of BNIP3 or PINK1/Parkin expression reduces mitophagy, leading to the accumulation of damaged mitochondria and ROS, which promotes HIF-1α stabilization and subsequently glycolysis. (c) p53 and BNIP3-dependent mitophagy removes abnormal mitochondria, reducing glycolysis and promoting O2 consumption. (d) RAS driven tumors require elevated autophagy/mitophagy levels to maintain mitochondrial functions and carry out proficient FAO. PGC1α and c-Myc are key regulators of mitochondrial functions to provide cancer cells with metabolic flexibility.

Figure 3.
The figure schematically summarizes how mitophagy would modulate the response and resistance of cancer cells to anticancer therapies impinging on mitochondrial functions. Upon anti-cancer treatment, cancer cell mitochondria depolarize and start producing ROS, which if accumulating to lethal levels, trigger apoptosis as major cell death pathway. High mitophagy levels within the treated cancer cell contribute to withstand the damage caused by the treatment and counteract the onset of cell death programs, by maintaining a healthier mitochondrial network (limiting ROS production and accumulation of depolarized mitochondria, preventing cytochrome C release).
Figure 3.
The figure schematically summarizes how mitophagy would modulate the response and resistance of cancer cells to anticancer therapies impinging on mitochondrial functions. Upon anti-cancer treatment, cancer cell mitochondria depolarize and start producing ROS, which if accumulating to lethal levels, trigger apoptosis as major cell death pathway. High mitophagy levels within the treated cancer cell contribute to withstand the damage caused by the treatment and counteract the onset of cell death programs, by maintaining a healthier mitochondrial network (limiting ROS production and accumulation of depolarized mitochondria, preventing cytochrome C release).


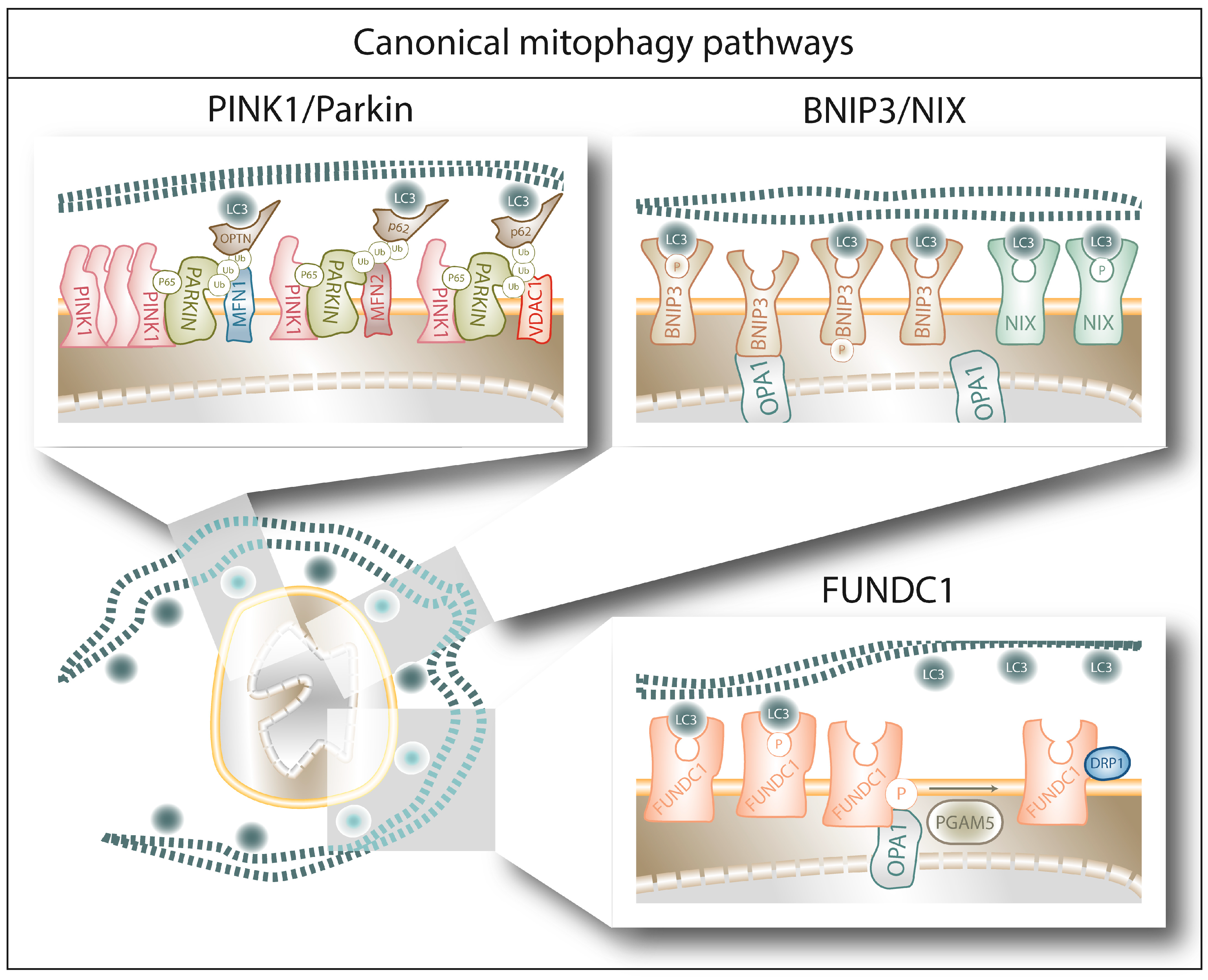{kind=link}
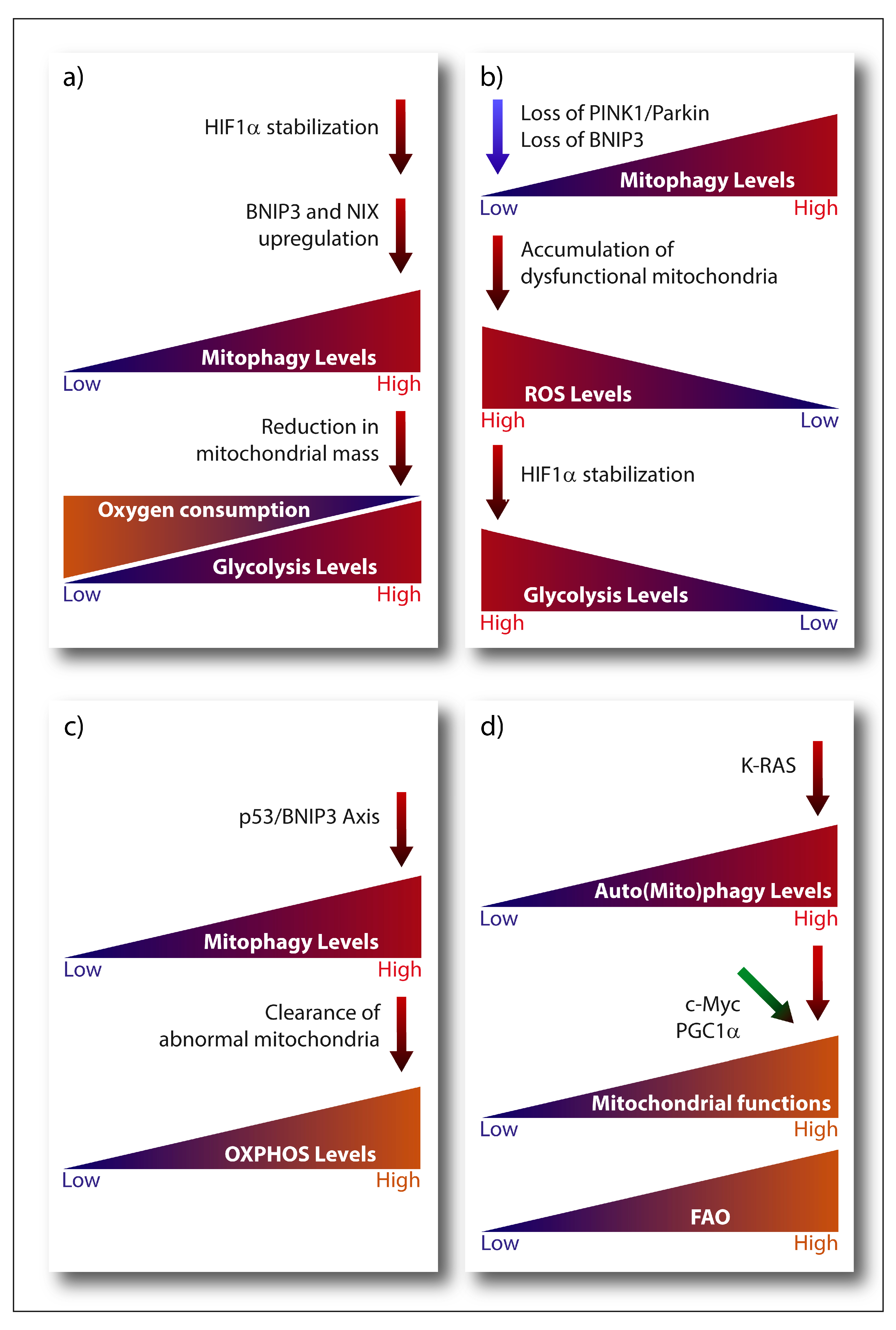{kind=link}
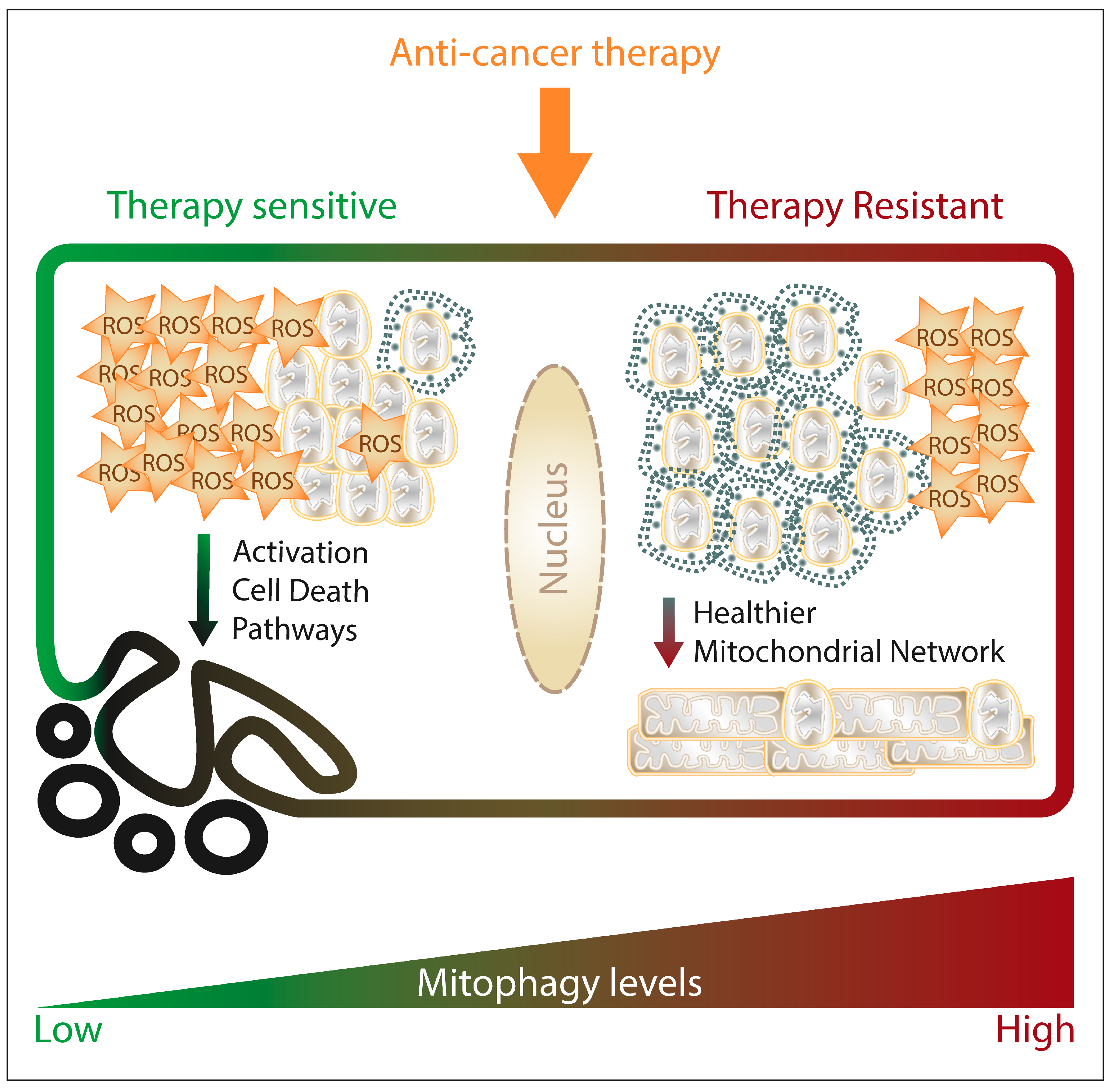{kind=link}
Table 1.
Expression levels of mitophagy regulators in samples from cancer patients. The levels of regulation are defined by the correlation of the expression levels with cancer progression and/or poor prognosis.
Table 1.
Expression levels of mitophagy regulators in samples from cancer patients. The levels of regulation are defined by the correlation of the expression levels with cancer progression and/or poor prognosis.
| Protein | Expression Levels in Patients | Cancer Type | Reference(s) |
|---|---|---|---|
| Mitochondrial Dynamics | |||
| MFN 1 | Downregulation | Triple Negative Breast Cancer 1,2, Hepatocellular Carcinoma 1,2 | [89,90] |
| MFN 2 | Upregulation | Cutaneous Melanoma 2, Gastric Cancer 2, Ovarian Cancer 1, Prostate Cancer 2 | [91,92,93,94] |
| MFN 2 | Downregulation | Breast Cancer 1,2, Hepatocellular Carcinoma 1,2, Non-Small Cell Lung Cancer 1 | [95,96,97,98,99,100] |
| Opa1 | Upregulation | Lung Cancer 2,3, Colorectal Cancer 1 | [101,102,103] |
| Opa1 | Downregulation | Hepatocellular Carcinoma 2 | [104] |
| DRP1 | Upregulation | Triple Negative Breast Cancer 1,2, Colorectal Cancer1, Hepatocellular Carcinoma 1,2, Ovarian Cancer 1,3 | [89,90,101,105] |
| DRP1 | Downregulation | Colorectal Cancer 2, Lung Cancer 2 | [106] |
| pDRP1 (Ser616) | Upregulation | Colorectal Cancer 2, Melanoma 2 | [107,108] |
| pDRP1 (Ser637) | Upregulation | Hepatocellular Carcinoma 2 | [109] |
| Mff | Upregulation | Hepatocellular Carcinoma 1,2 | [110] |
| Mff | Downregulation | Tongue Squamous Cell Carcinoma 2 | [111] |
| FIS1 | Upregulation | Acute Myeloid Leukemia 1,2, Oral Melanoma 2, Prostate Cancer 1 | [93,112,113,114] |
| FIS1 | Downregulation | Tongue Squamous Cell Carcinoma 2 | [115] |
| Canonical Mitophagy Pathways | |||
| PINK1 | Upregulation | Lung Cancer 2, Esophageal Squamous Cell Carcinoma 2 | [116,117] |
| PINK1 | Downregulation | Ovarian cancer 1 | [118] |
| Parkin | Downregulation | Acute Lymphoblastic Leukemia 4, Colorectal Cancer 1,2,4, Clear Cell Renal Cell Carcinoma 1,2, Melanoma 1,3, Oropharyngeal Squamous Cell Carcinoma 1, Ovarian Cancer 1,3, Pancreatic Cancer 1,2,3 | [119,120,121,122,123,124,125,126,127] |
| BNIP3 | Upregulation | Adenoid Cystic Carcinoma 2, Ampullary Carcinoma 2, Breast Cancer 1, Cervical Cancer 1,2, Cholangiocarcinoma 2, Colorectal Cancer 2, Ependydoma 1, Glioblastoma 2, Lung Cancer 1,2, Melanoma 2, Ovarian Cancer 1,2, Renal Carcinoma 1,2, Uterine-Cervical Squamous Cell Carcinoma 1 | [128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143] |
| BNIP3 | Downregulation | Bladder Cancer 4, Breast Cancer 1,2, Colorectal Cancer 2,4, Esophageal Cancer 4, Gastric Carcinoma 4, Laryngeal Squamous Cell Carcinoma 2, Lung Cancer 4, Multiple Myeloma 1,4, Pancreatic Cancer 1,2, | [142,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163] |
| NIX | Upregulation | Breast Cancer 1, Glioma 1,2 | [164,165] |
| NIX | Downregulation | Acute Myeloid Leukemia 1, Prostate Cancer 3 | [166,167] |
| FUNDC1 | Upregulation | Breast Cancer 1,2, Cervical Cancer 2, Laryngeal Cancer 2 | [168,169,170] |
| PGAM5 | Upregulation | Hepatocellular Carcinoma 2, Non-Small Cell Lung Cancer 2 | [171,172] |
| Non-Canonical Mitophagy Pathways | |||
| CL | Upregulation | Prostate Cancer | [173,174] |
| CL | Downregulation | Hepatocellular Carcinoma | [175] |
| PHB2 | Upregulation | Breast Cancer 1,2, Colorectal Cancer 2, Esophageal Squamous Cell carcinoma 1,2, Leukemia 2, Lymphoma 2 | [167,176,177,178,179,180] |
| C18-Ceramide | Downregulation | Glioblastoma, Glioma, Head and Neck Squamous Cell Carcinoma | [181,182,183] |
| AMBRA1 | Upregulation | Cholangiocarcinoma 2, Gastric Adenocarcinoma 2, Pancreatic Ductal Adenocarcinoma 2, Prostate Cancer 1,2 | [184,185,186,187] |
| HUWE1 | Upregulation | Lung Cancer 1,2, Multiple Myeloma 1 | [188,189] |
| HUWE1 | Downregulation | Breast Cancer 1, Hepatocellular Carcinoma 1, Osteosarcoma 1 | [190,191,192] |
| BCL2L13 (BCL-RAMBO) | Upregulation | Leukemia 1 | [193,194,195] |
| BCL2L13 (BCL-RAMBO) | Downregulation | Breast Cancer 1, Locally Advanced Rectal Cancer 1 | [139,196] |
| RAB7 | Upregulation | Oral Squamous Cell Carcinoma 2, Prostate Cancer 1, | [197,198] |
1 mRNA expression levels. 2 Protein expression levels. 3 Copy Number Variation levels. 4 Promoter methylation levels.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050493
AMA Style
Vara-Perez M, Felipe-Abrio B, Agostinis P. Mitophagy in Cancer: A Tale of Adaptation. Cells. 2019; 8(5):493. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050493
Chicago/Turabian StyleVara-Perez, Monica, Blanca Felipe-Abrio, and Patrizia Agostinis. 2019. "Mitophagy in Cancer: A Tale of Adaptation" Cells 8, no. 5: 493. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050493
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.