Hematological Malignancy-Derived Small Extracellular Vesicles and Tumor Microenvironment: The Art of Turning Foes into Friends



Abstract
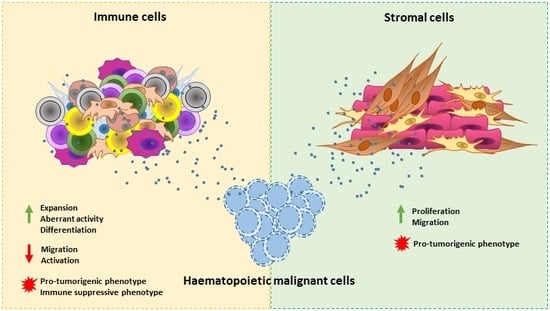
:
1. Introduction
2. Generality on Small EVs and the Tumor Microenvironment
2.1. Architecture and Content
2.2. The Complex Interactions between TEVs and Cells in the Microenvironment
2.3. TEVs as Potential Biomarkers
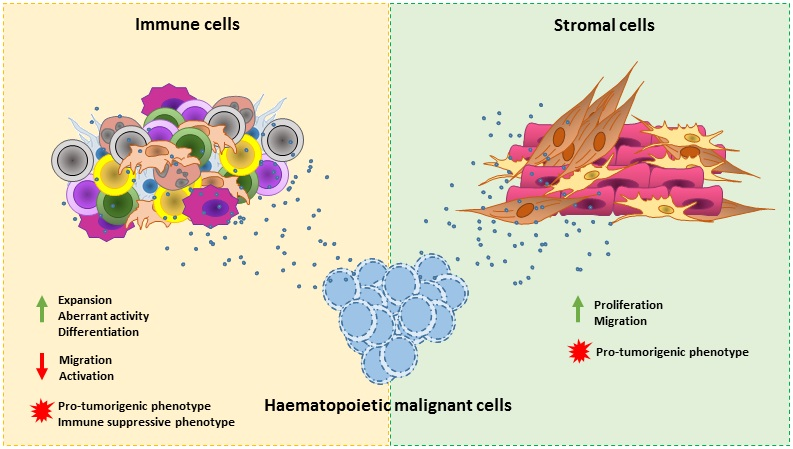
3. Hematological Malignancy-Derived Small EVs and Immune Cells
3.1. B Cells
3.2. T Cells
3.3. Dendritic Cells
3.4. Natural Killer Cells
3.5. Monocytes
3.6. Macrophages
3.7. Granulocytes
3.8. Myeloid-Derived Suppressor Cells
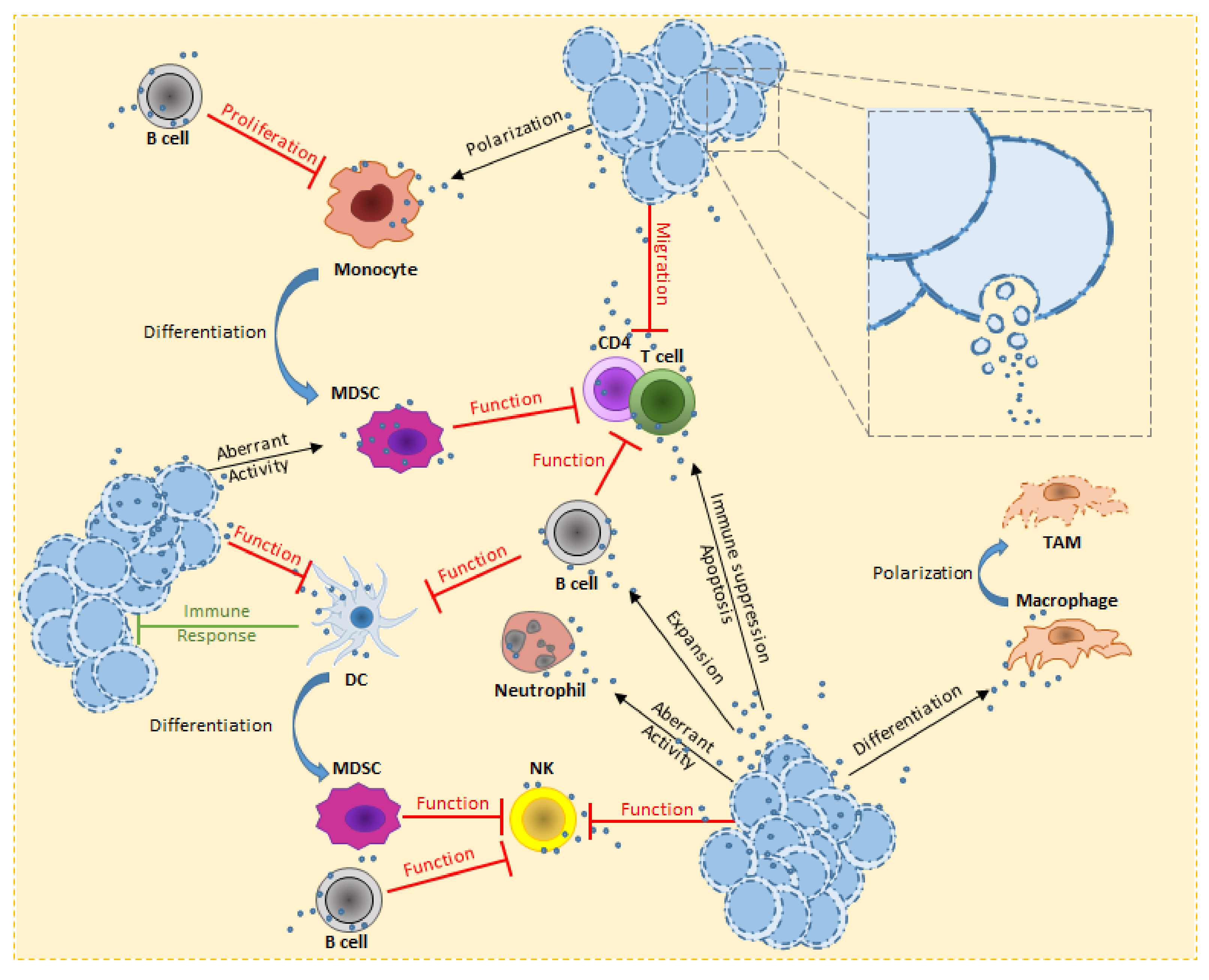
4. Hematological Malignancies-Derived Small EVs and Stromal Cells
4.1. Fibroblasts
4.2. Mesenchymal Stem Cells
4.3. Endothelial Cells
4.4. Osteoblasts and Osteoclasts
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Keller, S.; Ridinger, J.; Rupp, A.K.; Janssen, J.W.; Altevogt, P. Body fluid derived exosomes as a novel template for clinical diagnostics. J. Transl. Med. 2011, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Poliakov, A.; Spilman, M.; Dokland, T.; Amling, C.L.; Mobley, J.A. Structural heterogeneity and protein composition of exosome-like vesicles (prostasomes) in human semen. Prostate 2009, 69, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Rasool, H.I.; Palanisamy, V.; Mathisen, C.; Schmidt, M.; Wong, D.T.; Gimzewski, J.K. Structural-mechanical characterization of nanoparticle exosomes in human saliva, using correlative AFM, FESEM, and force spectroscopy. ACS Nano 2010, 4, 1921–1926. [Google Scholar] [CrossRef]
- Skriner, K.; Adolph, K.; Jungblut, P.R.; Burmester, G.R. Association of citrullinated proteins with synovial exosomes. Arthritis Rheum. 2006, 54, 3809–3814. [Google Scholar] [CrossRef]
- Zhang, H.G.; Grizzle, W.E. Exosomes: A novel pathway of local and distant intercellular communication that facilitates the growth and metastasis of neoplastic lesions. Am. J. Pathol. 2014, 184, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [PubMed]
- Wieckowski, E.; Whiteside, T.L. Human tumor-derived vs dendritic cell-derived exosomes have distinct biologic roles and molecular profiles. Immunol. Res. 2006, 36, 247–254. [Google Scholar] [CrossRef]
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Ruger, R. The Multiple Roles of Exosomes in Metastasis. Cancer Genomics Proteomics 2017, 14, 1–15. [Google Scholar] [CrossRef]
- Ichim, T.E.; Zhong, Z.; Kaushal, S.; Zheng, X.; Ren, X.; Hao, X.; Joyce, J.A.; Hanley, H.H.; Riordan, N.H.; Koropatnick, J.; et al. Exosomes as a tumor immune escape mechanism: Possible therapeutic implications. J. Transl. Med. 2008, 6, 37. [Google Scholar] [CrossRef]
- Keerthikumar, S.; Chisanga, D.; Ariyaratne, D.; Al Saffar, H.; Anand, S.; Zhao, K.; Samuel, M.; Pathan, M.; Jois, M.; Chilamkurti, N.; et al. ExoCarta: A web-based compendium of exosomal cargo. J. Mol. Biol. 2016, 428, 688–692. [Google Scholar] [CrossRef]
- Mathivanan, S.; Fahner, C.J.; Reid, G.E.; Simpson, R.J. ExoCarta 2012: Database of exosomal proteins, RNA and lipids. Nucleic Acids Res. 2012, 40, D1241–D1244. [Google Scholar] [CrossRef] [PubMed]
- Mathivanan, S.; Simpson, R.J. ExoCarta: A compendium of exosomal proteins and RNA. Proteomics 2009, 9, 4997–5000. [Google Scholar] [CrossRef]
- Jan, A.T.; Rahman, S.; Khan, S.; Tasduq, S.A.; Choi, I. Biology, pathophysiological role, and clinical implications of exosomes: A critical appraisal. Cells 2019, 8, 99. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of exosome composition. Cells 2019, 177, 428–445. [Google Scholar] [CrossRef]
- Van Niel, G.; Porto-Carreiro, I.; Simoes, S.; Raposo, G. Exosomes: A common pathway for a specialized function. J. Biochem. 2006, 140, 13–21. [Google Scholar] [CrossRef]
- Hemler, M.E. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu. Rev. Cell Dev. Biol. 2003, 19, 397–422. [Google Scholar] [CrossRef] [PubMed]
- Zoller, M. Tetraspanins: Push and pull in suppressing and promoting metastasis. Nat. Rev. Cancer 2009, 9, 40–55. [Google Scholar] [CrossRef]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Nazarenko, I.; Rana, S.; Baumann, A.; McAlear, J.; Hellwig, A.; Trendelenburg, M.; Lochnit, G.; Preissner, K.T.; Zoller, M. Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res. 2010, 70, 1668–1678. [Google Scholar] [CrossRef] [PubMed]
- Hannafon, B.N.; Ding, W.Q. Intercellular communication by exosome-derived microRNAs in cancer. Int. J. Mol. Sci. 2013, 14, 14240–14269. [Google Scholar] [CrossRef]
- Banno, K.; Iida, M.; Yanokura, M.; Kisu, I.; Iwata, T.; Tominaga, E.; Tanaka, K.; Aoki, D. MicroRNA in cervical cancer: OncomiRs and tumor suppressor miRs in diagnosis and treatment. Sci. World J. 2014, 2014, 178075. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Raimondo, S.; Chiesi, A.; Ciccia, F.; De Leo, G.; Alessandro, R. Exosomes as intercellular signaling organelles involved in health and disease: Basic science and clinical applications. Int. J. Mol. Sci. 2013, 14, 5338–5366. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cells 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Gutierrez-Vazquez, C.; Sanchez-Cabo, F.; Perez-Hernandez, D.; Vazquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sanchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, D.J.; Franklin, J.L.; Dou, Y.; Liu, Q.; Higginbotham, J.N.; Demory Beckler, M.; Weaver, A.M.; Vickers, K.; Prasad, N.; Levy, S.; et al. KRAS-dependent sorting of miRNA to exosomes. Elife 2015, 4, e07197. [Google Scholar] [CrossRef]
- Vidal, M.; Sainte-Marie, J.; Philippot, J.R.; Bienvenue, A. Asymmetric distribution of phospholipids in the membrane of vesicles released during in vitro maturation of guinea pig reticulocytes: Evidence precluding a role for “aminophospholipid translocase”. J. Cell Physiol. 1989, 140, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Pienimaeki-Roemer, A.; Kuhlmann, K.; Bottcher, A.; Konovalova, T.; Black, A.; Orso, E.; Liebisch, G.; Ahrens, M.; Eisenacher, M.; Meyer, H.E.; et al. Lipidomic and proteomic characterization of platelet extracellular vesicle subfractions from senescent platelets. Transfusion 2015, 55, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Durcin, M.; Fleury, A.; Taillebois, E.; Hilairet, G.; Krupova, Z.; Henry, C.; Truchet, S.; Trotzmuller, M.; Kofeler, H.; Mabilleau, G.; et al. Characterisation of adipocyte-derived extracellular vesicle subtypes identifies distinct protein and lipid signatures for large and small extracellular vesicles. J. Extracell Vesicles 2017, 6, 1305677. [Google Scholar] [CrossRef] [PubMed]
- Osteikoetxea, X.; Balogh, A.; Szabo-Taylor, K.; Nemeth, A.; Szabo, T.G.; Paloczi, K.; Sodar, B.; Kittel, A.; Gyorgy, B.; Pallinger, E.; et al. Improved characterization of EV preparations based on protein to lipid ratio and lipid properties. PLoS ONE 2015, 10, e0121184. [Google Scholar] [CrossRef] [PubMed]
- Lydic, T.A.; Townsend, S.; Adda, C.G.; Collins, C.; Mathivanan, S.; Reid, G.E. Rapid and comprehensive ‘shotgun’ lipidome profiling of colorectal cancer cell derived exosomes. Methods 2015, 87, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Laulagnier, K.; Motta, C.; Hamdi, S.; Roy, S.; Fauvelle, F.; Pageaux, J.F.; Kobayashi, T.; Salles, J.P.; Perret, B.; Bonnerot, C.; et al. Mast cell- and dendritic cell-derived exosomes display a specific lipid composition and an unusual membrane organization. Biochem. J. 2004, 380, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Haraszti, R.A.; Didiot, M.C.; Sapp, E.; Leszyk, J.; Shaffer, S.A.; Rockwell, H.E.; Gao, F.; Narain, N.R.; DiFiglia, M.; Kiebish, M.A.; et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell Vesicles 2016, 5, 32570. [Google Scholar] [CrossRef]
- Record, M.; Poirot, M.; Silvente-Poirot, S. Emerging concepts on the role of exosomes in lipid metabolic diseases. Biochimie 2014, 96, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Subra, C.; Grand, D.; Laulagnier, K.; Stella, A.; Lambeau, G.; Paillasse, M.; De Medina, P.; Monsarrat, B.; Perret, B.; Silvente-Poirot, S.; et al. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J. Lipid Res. 2010, 51, 2105–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altadill, T.; Campoy, I.; Lanau, L.; Gill, K.; Rigau, M.; Gil-Moreno, A.; Reventos, J.; Byers, S.; Colas, E.; Cheema, A.K. Enabling metabolomics based biomarker discovery studies using molecular phenotyping of exosome-like vesicles. PLoS ONE 2016, 11, e0151339. [Google Scholar] [CrossRef]
- Zeczycki, T.N.; Whelan, J.; Hayden, W.T.; Brown, D.A.; Shaikh, S.R. Increasing levels of cardiolipin differentially influence packing of phospholipids found in the mitochondrial inner membrane. Biochem. Biophys. Res. Commun. 2014, 450, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Yeshurun, A.; Willner, D.; Trembovler, V.; Alexandrovich, A.; Mechoulam, R.; Shohami, E.; Leker, R.R. N-arachidonoyl-L-serine (AraS) possesses proneurogenic properties in vitro and in vivo after traumatic brain injury. J. Cereb. Blood Flow Metab. 2013, 33, 1242–1250. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Menck, K.; Klemm, F.; Gross, J.C.; Pukrop, T.; Wenzel, D.; Binder, C. Induction and transport of Wnt 5a during macrophage-induced malignant invasion is mediated by two types of extracellular vesicles. Oncotarget 2013, 4, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Ding, Y.; Liu, G.; Yang, X.; Zhao, R.; Zhang, Y.; Zhao, X.; Anderson, G.J.; Nie, G. Cancer Cell-derived Exosomes Induce Mitogen-activated Protein Kinase-dependent Monocyte Survival by Transport of Functional Receptor Tyrosine Kinases. J. Biol. Chem. 2016, 291, 8453–8464. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, T.K.; Dworacki, G.; Tsukihiro, T.; Meidenbauer, N.; Gooding, W.; Johnson, J.T.; Whiteside, T.L. Spontaneous apoptosis of circulating T lymphocytes in patients with head and neck cancer and its clinical importance. Clin. Cancer Res. 2002, 8, 2553–2562. [Google Scholar] [PubMed]
- Schuler, P.J.; Schilling, B.; Harasymczuk, M.; Hoffmann, T.K.; Johnson, J.; Lang, S.; Whiteside, T.L. Phenotypic and functional characteristics of CD4+ CD39+ FOXP3+ and CD4+ CD39+ FOXP3neg T-cell subsets in cancer patients. Eur. J. Immunol. 2012, 42, 1876–1885. [Google Scholar] [CrossRef]
- Wierz, M.; Pierson, S.; Guyonnet, L.; Viry, E.; Lequeux, A.; Oudin, A.; Niclou, S.P.; Ollert, M.; Berchem, G.; Janji, B.; et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood 2018, 131, 1617–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, T.; Ward, J.P.; Gubin, M.M.; Arthur, C.D.; Lee, S.H.; Hundal, J.; Selby, M.J.; Graziano, R.F.; Mardis, E.R.; Korman, A.J.; et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol. Res. 2017, 5, 106–117. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chen, S.H.; Wang, J.Y.; Yang, B.C. Fas ligand on tumor cells mediates inactivation of neutrophils. J. Immunol. 2003, 171, 1183–1191. [Google Scholar] [CrossRef]
- Lin, H.C.; Lai, P.Y.; Lin, Y.P.; Huang, J.Y.; Yang, B.C. Fas ligand enhances malignant behavior of tumor cells through interaction with Met, hepatocyte growth factor receptor, in lipid rafts. J. Biol. Chem. 2012, 287, 20664–20673. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.; Cheung, J.; Navarro, A.; Lianoglou, S.; Haley, B.; Totpal, K.; Sanders, L.; Koeppen, H.; Caplazi, P.; McBride, J.; et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat. Commun. 2017, 8, 14572. [Google Scholar] [CrossRef] [PubMed]
- Wierz, M.; Janji, B.; Berchem, G.; Moussay, E.; Paggetti, J. High-dimensional mass cytometry analysis revealed microenvironment complexity in chronic lymphocytic leukemia. Oncoimmunology 2018, 7, e1465167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Wieckowski, E.; Taylor, D.D.; Reichert, T.E.; Watkins, S.; Whiteside, T.L. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin. Cancer Res. 2005, 11, 1010–1020. [Google Scholar] [PubMed]
- Andreola, G.; Rivoltini, L.; Castelli, C.; Huber, V.; Perego, P.; Deho, P.; Squarcina, P.; Accornero, P.; Lozupone, F.; Lugini, L.; et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J. Exp. Med. 2002, 195, 1303–1316. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE 2010, 5, e11469. [Google Scholar] [CrossRef]
- Bobrie, A.; Krumeich, S.; Reyal, F.; Recchi, C.; Moita, L.F.; Seabra, M.C.; Ostrowski, M.; Thery, C. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012, 72, 4920–4930. [Google Scholar] [CrossRef]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef]
- Kenific, C.M.; Nogues, L.; Lyden, D. Pre-metastatic niche formation has taken its TOLL. Cancer Cell 2016, 30, 189–191. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.; Han, Y.; Zhang, Q.; Jiang, Z.; Zhang, X.; Huang, B.; Xu, X.; Zheng, J.; Cao, X. Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell 2016, 30, 243–256. [Google Scholar] [CrossRef]
- Hornick, N.I.; Doron, B.; Abdelhamed, S.; Huan, J.; Harrington, C.A.; Shen, R.; Cambronne, X.A.; Chakkaramakkil Verghese, S.; Kurre, P. AML suppresses hematopoiesis by releasing exosomes that contain microRNAs targeting c-MYB. Sci. Signal. 2016, 9, ra88. [Google Scholar] [CrossRef]
- Li, A.; Zhang, T.; Zheng, M.; Liu, Y.; Chen, Z. Exosomal proteins as potential markers of tumor diagnosis. J. Hematol. Oncol. 2017, 10, 175. [Google Scholar] [CrossRef]
- Soung, Y.H.; Ford, S.; Zhang, V.; Chung, J. Exosomes in Cancer Diagnostics. Cancers (Basel) 2017, 9, 8. [Google Scholar] [CrossRef]
- Hon, K.W.; Abu, N.; Ab Mutalib, N.S.; Jamal, R. Exosomes as potential biomarkers and targeted therapy in colorectal cancer: A mini-review. Front. Pharmacol. 2017, 8, 583. [Google Scholar] [CrossRef]
- De Luca, L.; D’Arena, G.; Simeon, V.; Trino, S.; Laurenzana, I.; Caivano, A.; La Rocca, F.; Villani, O.; Mansueto, G.; Deaglio, S.; et al. Characterization and prognostic relevance of circulating microvesicles in chronic lymphocytic leukemia. Leuk. Lymphoma 2017, 58, 1424–1432. [Google Scholar] [CrossRef]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.H.; Zhang, Z.J.; Shang, L.R.; Luo, Y.W.; Lin, Y.F.; Yuan, Y.; Zhuang, S.M. Hepatoma cell-secreted exosomal microRNA-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology 2018, 68, 1459–1475. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Manier, S.; Liu, C.J.; Avet-Loiseau, H.; Park, J.; Shi, J.; Campigotto, F.; Salem, K.Z.; Huynh, D.; Glavey, S.V.; Rivotto, B.; et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood 2017, 129, 2429–2436. [Google Scholar] [CrossRef]
- Manier, S.; Powers, J.T.; Sacco, A.; Glavey, S.V.; Huynh, D.; Reagan, M.R.; Salem, K.Z.; Moschetta, M.; Shi, J.; Mishima, Y.; et al. The LIN28B/let-7 axis is a novel therapeutic pathway in multiple myeloma. Leukemia 2017, 31, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Krutilina, R.; Sun, W.; Sethuraman, A.; Brown, M.; Seagroves, T.N.; Pfeffer, L.M.; Ignatova, T.; Fan, M. MicroRNA-18a inhibits hypoxia-inducible factor 1alpha activity and lung metastasis in basal breast cancers. Breast Cancer Res. 2014, 16, R78. [Google Scholar] [CrossRef]
- Hornick, N.I.; Huan, J.; Doron, B.; Goloviznina, N.A.; Lapidus, J.; Chang, B.H.; Kurre, P. Serum exosome MicroRNA as a minimally-invasive early biomarker of AML. Sci. Rep. 2015, 5, 11295. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- Hong, C.S.; Muller, L.; Whiteside, T.L.; Boyiadzis, M. Plasma exosomes as markers of therapeutic response in patients with acute myeloid leukemia. Front. Immunol. 2014, 5, 160. [Google Scholar] [CrossRef] [PubMed]
- An, M.; Lohse, I.; Tan, Z.; Zhu, J.; Wu, J.; Kurapati, H.; Morgan, M.A.; Lawrence, T.S.; Cuneo, K.C.; Lubman, D.M. Quantitative proteomic analysis of serum exosomes from patients with locally advanced pancreatic cancer undergoing chemoradiotherapy. J. Proteome Res. 2017, 16, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The potential of tumor-derived exosomes for noninvasive cancer monitoring. Expert Rev. Mol. Diagn. 2015, 15, 1293–1310. [Google Scholar] [CrossRef]
- Boyiadzis, M.; Whiteside, T.L. Plasma-derived exosomes in acute myeloid leukemia for detection of minimal residual disease: Are we ready? Expert Rev. Mol. Diagn. 2016, 16, 623–629. [Google Scholar] [CrossRef]
- Wierz, M.; Pierson, S.; Gargiulo, E.; Guerin, C.; Moussay, E.; Paggetti, J. Purification of leukemia-derived exosomes to study microenvironment modulation. Methods Mol. Biol. 2019, 1884, 231–245. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cells 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Czernek, L.; Duchler, M. Functions of cancer-derived extracellular vesicles in immunosuppression. Arch. Immunol. Ther. Exp. (Warsz) 2017, 65, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, X.; Shi, H.; Wu, L.; Qian, H.; Xu, W. Exosomes in cancer: Small particle, big player. J. Hematol. Oncol. 2015, 8, 83. [Google Scholar] [CrossRef]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Richter, G.; Schuler, T.; Ibe, S.; Cao, X.; Blankenstein, T. B cells inhibit induction of T cell-dependent tumor immunity. Nat. Med. 1998, 4, 627–630. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Korets, L.V.; Coussens, L.M. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 2005, 7, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiLillo, D.J.; Yanaba, K.; Tedder, T.F. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: Therapeutic B cell depletion enhances B16 melanoma growth in mice. J. Immunol. 2010, 184, 4006–4016. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Teitz-Tennenbaum, S.; Donald, E.J.; Li, M.; Chang, A.E. In vivo sensitized and in vitro activated B cells mediate tumor regression in cancer adoptive immunotherapy. J. Immunol. 2009, 183, 3195–3203. [Google Scholar] [CrossRef] [PubMed]
- Largeot, A.; Pagano, G.; Gonder, S.; Moussay, E.; Paggetti, J. The B-side of cancer immunity: The underrated tune. Cells 2019, 8, 449. [Google Scholar] [CrossRef]
- Yanaba, K.; Bouaziz, J.D.; Haas, K.M.; Poe, J.C.; Fujimoto, M.; Tedder, T.F. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 2008, 28, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.A.; Norena, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Matsushita, T.; Tedder, T.F. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann. NY Acad. Sci. 2010, 1183, 38–57. [Google Scholar] [CrossRef]
- Khoder, A.; Sarvaria, A.; Alsuliman, A.; Chew, C.; Sekine, T.; Cooper, N.; Mielke, S.; de Lavallade, H.; Muftuoglu, M.; Fernandez Curbelo, I.; et al. Regulatory B cells are enriched within the IgM memory and transitional subsets in healthy donors but are deficient in chronic GVHD. Blood 2014, 124, 2034–2045. [Google Scholar] [CrossRef]
- Wang, W.W.; Yuan, X.L.; Chen, H.; Xie, G.H.; Ma, Y.H.; Zheng, Y.X.; Zhou, Y.L.; Shen, L.S. CD19+CD24hiCD38hiBregs involved in downregulate helper T cells and upregulate regulatory T cells in gastric cancer. Oncotarget 2015, 6, 33486–33499. [Google Scholar] [CrossRef]
- Han, S.; Feng, S.; Ren, M.; Ma, E.; Wang, X.; Xu, L.; Xu, M. Glioma cell-derived placental growth factor induces regulatory B cells. Int. J. Biochem. Cell Biol. 2014, 57, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Bankoti, R.; Gupta, K.; Levchenko, A.; Stager, S. Marginal zone B cells regulate antigen-specific T cell responses during infection. J. Immunol. 2012, 188, 3961–3971. [Google Scholar] [CrossRef]
- Das, A.; Ellis, G.; Pallant, C.; Lopes, A.R.; Khanna, P.; Peppa, D.; Chen, A.; Blair, P.; Dusheiko, G.; Gill, U.; et al. IL-10-producing regulatory B cells in the pathogenesis of chronic hepatitis B virus infection. J. Immunol. 2012, 189, 3925–3935. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Leitner, W.W.; Golding, B.; Scott, D. Inhibitory effects of B cells on antitumor immunity. Cancer Res. 2006, 66, 7741–7747. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Matsushita, T.; Horikawa, M.; Dilillo, D.J.; Yanaba, K.; Venturi, G.M.; Szabolcs, P.M.; Bernstein, S.H.; Magro, C.M.; Williams, A.D.; et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 2011, 117, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Moulin, V.; Andris, F.; Thielemans, K.; Maliszewski, C.; Urbain, J.; Moser, M. B lymphocytes regulate dendritic cell (DC) function in vivo: Increased interleukin 12 production by DCs from B cell-deficient mice results in T helper cell type 1 deviation. J. Exp. Med. 2000, 192, 475–482. [Google Scholar] [CrossRef]
- Flores-Borja, F.; Bosma, A.; Ng, D.; Reddy, V.; Ehrenstein, M.R.; Isenberg, D.A.; Mauri, C. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci. Transl. Med. 2013, 5, 173ra123. [Google Scholar] [CrossRef]
- Carter, N.A.; Rosser, E.C.; Mauri, C. Interleukin-10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis Res. Ther. 2012, 14, R32. [Google Scholar] [CrossRef]
- Yang, C.; Chalasani, G.; Ng, Y.H.; Robbins, P.D. Exosomes released from Mycoplasma infected tumor cells activate inhibitory B cells. PLoS ONE 2012, 7, e36138. [Google Scholar] [CrossRef]
- Gutzeit, C.; Nagy, N.; Gentile, M.; Lyberg, K.; Gumz, J.; Vallhov, H.; Puga, I.; Klein, E.; Gabrielsson, S.; Cerutti, A.; et al. Exosomes derived from Burkitt’s lymphoma cell lines induce proliferation, differentiation, and class-switch recombination in B cells. J. Immunol. 2014, 192, 5852–5862. [Google Scholar] [CrossRef]
- Vaiselbuh, S.H.a.S.R. Leukemia-derived exosomes induce paracrine and autocrine cell proliferation in pediatric ALL. Blood 2016, 128, 4080. [Google Scholar]
- Patel, S.J.; Darie, C.C.; Clarkson, B.D. Exosome mediated growth effect on the non-growing pre-B acute lymphoblastic leukemia cells at low starting cell density. Am. J. Transl Res. 2016, 8, 3614–3629. [Google Scholar]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef]
- Matsuda, M.; Ulfgren, A.K.; Lenkei, R.; Petersson, M.; Ochoa, A.C.; Lindblad, S.; Andersson, P.; Klareskog, L.; Kiessling, R. Decreased expression of signal-transducing CD3 zeta chains in T cells from the joints and peripheral blood of rheumatoid arthritis patients. Scand. J. Immunol. 1998, 47, 254–262. [Google Scholar] [CrossRef]
- Berg, L.; Ronnelid, J.; Klareskog, L.; Bucht, A. Down-regulation of the T cell receptor CD3 zeta chain in rheumatoid arthritis (RA) and its influence on T cell responsiveness. Clin. Exp. Immunol. 2000, 120, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, K.; Setoyama, Y.; Tsuzaka, K.; Abe, T.; Takeuchi, T. Reduced expression of TCR zeta is involved in the abnormal production of cytokines by peripheral T cells of patients with systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef]
- Renner, C.; Ohnesorge, S.; Held, G.; Bauer, S.; Jung, W.; Pfitzenmeier, J.P.; Pfreundschuh, M. T cells from patients with Hodgkin’s disease have a defective T-cell receptor zeta chain expression that is reversible by T-cell stimulation with CD3 and CD28. Blood 1996, 88, 236–241. [Google Scholar]
- Scott, D.W.; Gascoyne, R.D. The tumour microenvironment in B cell lymphomas. Nat. Rev. Cancer 2014, 14, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Radulovic, K.; Rossini, V.; Manta, C.; Holzmann, K.; Kestler, H.A.; Niess, J.H. The early activation marker CD69 regulates the expression of chemokines and CD4 T cell accumulation in intestine. PLoS ONE 2013, 8, e65413. [Google Scholar] [CrossRef] [PubMed]
- Shiow, L.R.; Rosen, D.B.; Brdickova, N.; Xu, Y.; An, J.; Lanier, L.L.; Cyster, J.G.; Matloubian, M. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006, 440, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, D.T.; Apollonio, B.; Willimott, S.; Lezina, L.; Alharthi, A.; Ambrose, A.R.; De Rossi, G.; Ramsay, A.G.; Wagner, S.D. Extracellular vesicles released by CD40/IL-4-stimulated CLL cells confer altered functional properties to CD4+ T cells. Blood 2016, 128, 542–552. [Google Scholar] [CrossRef]
- Chen, Z.; You, L.; Wang, L.; Huang, X.; Liu, H.; Wei, J.Y.; Zhu, L.; Qian, W. Dual effect of DLBCL-derived EXOs in lymphoma to improve DC vaccine efficacy in vitro while favor tumorgenesis in vivo. J. Exp. Clin. Cancer Res. 2018, 37, 190. [Google Scholar] [CrossRef]
- Zhang, F.; Li, R.; Yang, Y.; Shi, C.; Shen, Y.; Lu, C.; Chen, Y.; Zhou, W.; Lin, A.; Yu, L.; et al. Specific decrease in B-cell-derived extracellular vesicles enhances post-chemotherapeutic CD8(+) T cell responses. Immunity 2019. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Bruchard, M.; Chalmin, F.; Rebe, C. Production of adenosine by ectonucleotidases: A key factor in tumor immunoescape. J. Biomed. Biotechnol. 2012, 2012, 473712. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.; Cao, X. The exosomes in tumor immunity. Oncoimmunology 2015, 4, e1027472. [Google Scholar] [CrossRef] [Green Version]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Kim, S.H.; Bianco, N.R.; Robbins, P.D. Tumor-derived exosomes confer antigen-specific immunosuppression in a murine delayed-type hypersensitivity model. PLoS ONE 2011, 6, e22517. [Google Scholar] [CrossRef]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Thery, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Leaf, R.K.; Stroopinsky, D.; Pyzer, A.R.; Kruisbeek, A.M.; van Wetering, S.; Washington, A.; Ephraim, A.; Cole, L.; Morin, A.; Jain, S.; et al. DCOne as an allogeneic cell-based vaccine for multiple myeloma. J. Immunother 2017, 40, 315–322. [Google Scholar] [CrossRef]
- Santegoets, S.J.; Schreurs, M.W.; Masterson, A.J.; Liu, Y.P.; Goletz, S.; Baumeister, H.; Kueter, E.W.; Lougheed, S.M.; van den Eertwegh, A.J.; Scheper, R.J.; et al. In vitro priming of tumor-specific cytotoxic T lymphocytes using allogeneic dendritic cells derived from the human MUTZ-3 cell line. Cancer Immunol. Immunother 2006, 55, 1480–1490. [Google Scholar] [CrossRef]
- Arjan, A.; van de Loosdrecht, S.v.W.; Saskia, S.; Satwinder, K.S.; Malika, K.; Hall, A.; Ada, M.; Kruisbeek, T.; de Gruijl, D.; Gert, J.O. Clinical and immunological results of a phase I/IIa study of allogeneic dendritic cell (DC) vaccination, An “off The shelf” treatment to prevent or delay relapse in elderly patients with acute myeloid leukemia. Blood 2013, 122, 2651. [Google Scholar]
- Mac Keon, S.; Ruiz, M.S.; Gazzaniga, S.; Wainstok, R. Dendritic cell-based vaccination in cancer: Therapeutic implications emerging from murine models. Front. Immunol. 2015, 6, 243. [Google Scholar] [CrossRef]
- Gu, X.; Erb, U.; Buchler, M.W.; Zoller, M. Improved vaccine efficacy of tumor exosome compared to tumor lysate loaded dendritic cells in mice. Int. J. Cancer 2015, 136, E74–E84. [Google Scholar] [CrossRef]
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012, 12, 239–252. [Google Scholar] [CrossRef]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef]
- Mincheva-Nilsson, L.; Baranov, V. Cancer exosomes and NKG2D receptor-ligand interactions: Impairing NKG2D-mediated cytotoxicity and anti-tumour immune surveillance. Semin. Cancer Biol. 2014, 28, 24–30. [Google Scholar] [CrossRef]
- Hedlund, M.; Nagaeva, O.; Kargl, D.; Baranov, V.; Mincheva-Nilsson, L. Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PLoS ONE 2011, 6, e16899. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, S.; Sun, K.; Chng, W.J. The emerging roles of exosomes in leukemogeneis. Oncotarget 2016, 7, 50698–50707. [Google Scholar] [CrossRef]
- Vulpis, E.; Cecere, F.; Molfetta, R.; Soriani, A.; Fionda, C.; Peruzzi, G.; Caracciolo, G.; Palchetti, S.; Masuelli, L.; Simonelli, L.; et al. Genotoxic stress modulates the release of exosomes from multiple myeloma cells capable of activating NK cell cytokine production: Role of HSP70/TLR2/NF-kB axis. Oncoimmunology 2017, 6, e1279372. [Google Scholar] [CrossRef]
- Garg, T.K.G.J.I.; Malaviarachchi, P.A.; Stone, K.; Macleod, V.; Greenway, A.D.; Morgan, G.J. Myeloma-derived exosomes and soluble factors suppress natural killer cell function. In Proceedings of the 58th ASH Annual Meeting and Exposition, San Diego, CA, USA, 3–6 December 2016. [Google Scholar]
- Chillemi, A.; Quarona, V.; Antonioli, L.; Ferrari, D.; Horenstein, A.L.; Malavasi, F. Roles and Modalities of Ectonucleotidases in Remodeling the Multiple Myeloma Niche. Front. Immunol. 2017, 8, 305. [Google Scholar] [CrossRef]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A.; et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Rogers, T.L.; Holen, I. Tumour macrophages as potential targets of bisphosphonates. J. Transl. Med. 2011, 9, 177. [Google Scholar] [CrossRef]
- Quatromoni, J.G.; Eruslanov, E. Tumor-associated macrophages: Function, phenotype, and link to prognosis in human lung cancer. Am. J. Transl. Res. 2012, 4, 376–389. [Google Scholar]
- Schiavoni, G.; Gabriele, L.; Mattei, F. The tumor microenvironment: A pitch for multiple players. Front. Oncol. 2013, 3, 90. [Google Scholar] [CrossRef] [PubMed]
- Manček-Keber, M.D.L.; Mojca, B.; Jiaji, G.; Chen, R.; Romih, Z.R.; Hunter, S.P.T.; Jerala, R. Extracellular vesicle–mediated transfer of constitutively active MyD88L265P engages MyD88wt and activates signaling. Blood 2018, 131, 1720–1729. [Google Scholar]
- Jafarzadeh, N.; Safari, Z.; Pornour, M.; Amirizadeh, N.; Forouzandeh Moghadam, M.; Sadeghizadeh, M. Alteration of cellular and immune-related properties of bone marrow mesenchymal stem cells and macrophages by K562 chronic myeloid leukemia cell derived exosomes. J. Cell Physiol. 2019, 234, 3697–3710. [Google Scholar] [CrossRef] [PubMed]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Tecchio, C.; Scapini, P.; Pizzolo, G.; Cassatella, M.A. On the cytokines produced by human neutrophils in tumors. Semin. Cancer Biol. 2013, 23, 159–170. [Google Scholar] [CrossRef]
- Gregory, A.D.; Houghton, A.M. Tumor-associated neutrophils: New targets for cancer therapy. Cancer Res. 2011, 71, 2411–2416. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Cai, J.; Han, Y.; Ren, H.; Chen, C.; He, D.; Zhou, L.; Eisner, G.M.; Asico, L.D.; Jose, P.A.; Zeng, C. Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J. Mol. Cell Biol. 2013, 5, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Hansen, H.P.; Engels, H.M.; Dams, M.; Paes Leme, A.F.; Pauletti, B.A.; Simhadri, V.L.; Durkop, H.; Reiners, K.S.; Barnert, S.; Engert, A.; et al. Protrusion-guided extracellular vesicles mediate CD30 trans-signalling in the microenvironment of Hodgkin’s lymphoma. J. Pathol. 2014, 232, 405–414. [Google Scholar] [CrossRef]
- Dorhoi, A.; Du Plessis, N. Monocytic myeloid-derived suppressor cells in chronic infections. Front. Immunol. 2017, 8, 1895. [Google Scholar] [CrossRef]
- Mareel, M.; Madani, I. Tumour-associated host cells participating at invasion and metastasis: Targets for therapy? Acta Chir. Belg. 2006, 106, 635–640. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gabrilovich, D.I. Myeloid-derived suppressor cells. Adv. Exp. Med. Biol. 2007, 601, 213–223. [Google Scholar]
- Serafini, P.; Borrello, I.; Bronte, V. Myeloid suppressor cells in cancer: Recruitment, phenotype, properties, and mechanisms of immune suppression. Semin. Cancer Biol. 2006, 16, 53–65. [Google Scholar] [CrossRef]
- Yu, S.; Liu, C.; Su, K.; Wang, J.; Liu, Y.; Zhang, L.; Li, C.; Cong, Y.; Kimberly, R.; Grizzle, W.E.; et al. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J. Immunol. 2007, 178, 6867–6875. [Google Scholar] [CrossRef]
- Bartz, H.; Avalos, N.M.; Baetz, A.; Heeg, K.; Dalpke, A.H. Involvement of suppressors of cytokine signaling in toll-like receptor-mediated block of dendritic cell differentiation. Blood 2006, 108, 4102–4108. [Google Scholar] [CrossRef]
- Isomoto, H.; Mott, J.L.; Kobayashi, S.; Werneburg, N.W.; Bronk, S.F.; Haan, S.; Gores, G.J. Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing. Gastroenterology 2007, 132, 384–396. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Nagaraj, S.; Gabrilovich, D.I. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J. Immunol. 2005, 175, 4583–4592. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; De Veirman, K.; De Beule, N.; Maes, K.; De Bruyne, E.; Van Valckenborgh, E.; Vanderkerken, K.; Menu, E. The bone marrow microenvironment enhances multiple myeloma progression by exosome-mediated activation of myeloid-derived suppressor cells. Oncotarget 2015, 6, 43992–44004. [Google Scholar] [CrossRef]
- Schor, S.L.; Ellis, I.R.; Jones, S.J.; Baillie, R.; Seneviratne, K.; Clausen, J.; Motegi, K.; Vojtesek, B.; Kankova, K.; Furrie, E.; et al. Migration-stimulating factor: A genetically truncated onco-fetal fibronectin isoform expressed by carcinoma and tumor-associated stromal cells. Cancer Res. 2003, 63, 8827–8836. [Google Scholar] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cells 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Jordana, M.; Sarnstrand, B.; Sime, P.J.; Ramis, I. Immune-inflammatory functions of fibroblasts. Eur. Respir. J. 1994, 7, 2212–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cells 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Dror, S.; Sander, L.; Schwartz, H.; Sheinboim, D.; Barzilai, A.; Dishon, Y.; Apcher, S.; Golan, T.; Greenberger, S.; Barshack, I.; et al. Melanoma miRNA trafficking controls tumour primary niche formation. Nat. Cell Biol. 2016, 18, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Raimondo, S.; Saieva, L.; Flugy, A.M.; De Leo, G.; Alessandro, R. Exosome-mediated crosstalk between chronic myelogenous leukemia cells and human bone marrow stromal cells triggers an interleukin 8-dependent survival of leukemia cells. Cancer Lett. 2014, 348, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.; Hornick, N.I.; Shurtleff, M.J.; Skinner, A.M.; Goloviznina, N.A.; Roberts, C.T., Jr.; Kurre, P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013, 73, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Frassanito, M.A.; Desantis, V.; Di Marzo, L.; Craparotta, I.; Beltrame, L.; Marchini, S.; Annese, T.; Visino, F.; Arciuli, M.; Saltarella, I.; et al. Bone marrow fibroblasts overexpress miR-27b and miR-214 in step with multiple myeloma progression, dependent on tumour cell-derived exosomes. J. Pathol. 2008. [Google Scholar] [CrossRef]
- Gutkin, A.; Uziel, O.; Beery, E.; Nordenberg, J.; Pinchasi, M.; Goldvaser, H.; Henick, S.; Goldberg, M.; Lahav, M. Tumor cells derived exosomes contain hTERT mRNA and transform nonmalignant fibroblasts into telomerase positive cells. Oncotarget 2016, 7, 59173–59188. [Google Scholar] [CrossRef]
- Jaiswal, R.K.; Kumar, P.; Yadava, P.K. Telomerase and its extracurricular activities. Cell Mol. Biol. Lett. 2013, 18, 538–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witas, R.; Chaput, D.; Khan, H.; Stevens, S.M., Jr.; Kang, D. Isolation and proteomic analysis of microvesicles and exosomes from HT22 cells and primary neurons. Methods Mol. Biol. 2017, 1598, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G.; Jorgensen, C. Concise review: Adult multipotent stromal cells and cancer: Risk or benefit? Stem Cells 2008, 26, 1387–1394. [Google Scholar] [CrossRef]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.W.; Chen, H.Y.; Wang, L.T.; Wang, F.H.; Fang, L.W.; Lai, H.Y.; Chen, H.H.; Lu, J.; Hung, M.S.; Cheng, Y.; et al. Mesenchymal stem cells tune the development of monocyte-derived dendritic cells toward a myeloid-derived suppressive phenotype through growth-regulated oncogene chemokines. J. Immunol. 2013, 190, 5065–5077. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Li, X.; Liu, J.; Ye, Q.; Chen, Y.; Tan, S.; Liu, J. Multiple myeloma-derived exosomes regulate the functions of mesenchymal stem cells partially via modulating mir-21 and mir-146a. Stem Cells Int. 2017, 2017, 9012152. [Google Scholar] [CrossRef]
- El-Saghir, J.; Nassar, F.; Tawil, N.; El-Sabban, M. ATL-derived exosomes modulate mesenchymal stem cells: Potential role in leukemia progression. Retrovirology 2016, 13, 73. [Google Scholar] [CrossRef]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiguchi, H.; Kobune, M.; Kikuchi, S.; Yoshida, M.; Murata, M.; Murase, K.; Iyama, S.; Takada, K.; Sato, T.; Ono, K.; et al. Extracellular vesicle miR-7977 is involved in hematopoietic dysfunction of mesenchymal stromal cells via poly(rC) binding protein 1 reduction in myeloid neoplasms. Haematologica 2016, 101, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Secreto, C.R.; Knox, T.R.; Ding, W.; Mukhopadhyay, D.; Kay, N.E. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: Implications for disease progression. Blood 2010, 115, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; Van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef]
- Fei, F.; Joo, E.J.; Tarighat, S.S.; Schiffer, I.; Paz, H.; Fabbri, M.; Abdel-Azim, H.; Groffen, J.; Heisterkamp, N. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3. Oncotarget 2015, 6, 11378–11394. [Google Scholar] [CrossRef] [Green Version]
- Corrado, C.; Saieva, L.; Raimondo, S.; Santoro, A.; De Leo, G.; Alessandro, R. Chronic myelogenous leukaemia exosomes modulate bone marrow microenvironment through activation of epidermal growth factor receptor. J. Cell Mol. Med. 2016, 20, 1829–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridge, S.M.; Sullivan, F.J.; Glynn, S.A. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 31. [Google Scholar] [CrossRef]
- Norozi, F.; Ahmadzadeh, A.; Shahrabi, S.; Vosoughi, T.; Saki, N. Mesenchymal stem cells as a double-edged sword in suppression or progression of solid tumor cells. Tumour Biol. 2016, 37, 11679–11689. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G.; Lam, P.Y. Recent discoveries concerning the tumor-mesenchymal stem cell interactions. Biochim. Biophys. Acta 2016, 1866, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yin, Y.; Lai, R.C.; Tan, S.S.; Choo, A.B.; Lim, S.K. Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev. 2014, 23, 1233–1244. [Google Scholar] [CrossRef]
- Burrello, J.; Monticone, S.; Gai, C.; Gomez, Y.; Kholia, S.; Camussi, G. Stem Cell-Derived Extracellular Vesicles and Immune-Modulation. Front. Cell Dev. Biol. 2016, 4, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marigo, I.; Dazzi, F. The immunomodulatory properties of mesenchymal stem cells. Semin. Immunopathol. 2011, 33, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Amarnath, S.; Foley, J.E.; Farthing, D.E.; Gress, R.E.; Laurence, A.; Eckhaus, M.A.; Metais, J.Y.; Rose, J.J.; Hakim, F.T.; Felizardo, T.C.; et al. Bone marrow-derived mesenchymal stromal cells harness purinergenic signaling to tolerize human Th1 cells in vivo. Stem Cells 2015, 33, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Yeo, R.W.; Lai, R.C.; Zhang, B.; Tan, S.S.; Yin, Y.; Teh, B.J.; Lim, S.K. Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 336–341. [Google Scholar] [CrossRef]
- Kilpinen, L.; Impola, U.; Sankkila, L.; Ritamo, I.; Aatonen, M.; Kilpinen, S.; Tuimala, J.; Valmu, L.; Levijoki, J.; Finckenberg, P.; et al. Extracellular membrane vesicles from umbilical cord blood-derived MSC protect against ischemic acute kidney injury, a feature that is lost after inflammatory conditioning. J. Extracell Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Mokarizadeh, A.; Delirezh, N.; Morshedi, A.; Mosayebi, G.; Farshid, A.A.; Mardani, K. Microvesicles derived from mesenchymal stem cells: Potent organelles for induction of tolerogenic signaling. Immunol. Lett. 2012, 147, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Huang, Y.; Han, J.; Yu, L.; Li, Y.; Lu, Z.; Li, H.; Liu, Z.; Shi, C.; Duan, F.; et al. Immunomodulatory effects of mesenchymal stromal cells-derived exosome. Immunol. Res. 2016, 64, 831–840. [Google Scholar] [CrossRef] [PubMed]
- English, K.; Ryan, J.M.; Tobin, L.; Murphy, M.J.; Barry, F.P.; Mahon, B.P. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4+CD25(High) forkhead box P3+ regulatory T cells. Clin. Exp. Immunol. 2009, 156, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Tannock, I.F. The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br. J. Cancer 1968, 22, 258–273. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wang, X.; Hou, D.; Huang, Q.; Zhan, W.; Chen, C.; Liu, J.; You, R.; Xie, J.; Chen, P.; et al. Exosomes derived from acute myeloid leukemia cells promote chemoresistance by enhancing glycolysis-mediated vascular remodeling. J. Cell Physiol. 2019, 234, 10602–10614. [Google Scholar] [CrossRef]
- Zarfati, M.; Avivi, I.; Brenner, B.; Katz, T.; Aharon, A. Extracellular vesicles of multiple myeloma cells utilize the proteasome inhibitor mechanism to moderate endothelial angiogenesis. Angiogenesis 2019, 22, 185–196. [Google Scholar] [CrossRef]
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito, M.A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J. Pathol. 2016, 239, 162–173. [Google Scholar] [CrossRef]
- Taverna, S.; Fontana, S.; Monteleone, F.; Pucci, M.; Saieva, L.; De Caro, V.; Cardinale, V.G.; Giallombardo, M.; Vicario, E.; Rolfo, C.; et al. Curcumin modulates chronic myelogenous leukemia exosomes composition and affects angiogenic phenotype via exosomal miR-21. Oncotarget 2016, 7, 30420–30439. [Google Scholar] [CrossRef]
- Taverna, S.; Flugy, A.; Saieva, L.; Kohn, E.C.; Santoro, A.; Meraviglia, S.; De Leo, G.; Alessandro, R. Role of exosomes released by chronic myelogenous leukemia cells in angiogenesis. Int. J. Cancer 2012, 130, 2033–2043. [Google Scholar] [CrossRef]
- Umezu, T.; Ohyashiki, K.; Kuroda, M.; Ohyashiki, J.H. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 2013, 32, 2747–2755. [Google Scholar] [CrossRef]
- Taverna, S.; Amodeo, V.; Saieva, L.; Russo, A.; Giallombardo, M.; De Leo, G.; Alessandro, R. Exosomal shuttling of miR-126 in endothelial cells modulates adhesive and migratory abilities of chronic myelogenous leukemia cells. Mol. Cancer 2014, 13, 169. [Google Scholar] [CrossRef]
- Li, B.; Hong, J.; Hong, M.; Wang, Y.; Yu, T.; Zang, S.; Wu, Q. piRNA-823 delivered by multiple myeloma-derived extracellular vesicles promoted tumorigenesis through re-educating endothelial cells in the tumor environment. Oncogene 2019. [Google Scholar] [CrossRef] [PubMed]
- Storti, P.; Bolzoni, M.; Donofrio, G.; Airoldi, I.; Guasco, D.; Toscani, D.; Martella, E.; Lazzaretti, M.; Mancini, C.; Agnelli, L.; et al. Hypoxia-inducible factor (HIF)-1alpha suppression in myeloma cells blocks tumoral growth in vivo inhibiting angiogenesis and bone destruction. Leukemia 2013, 27, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Tadokoro, H.; Azuma, K.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014, 124, 3748–3757. [Google Scholar] [CrossRef]
- Tadokoro, H.; Umezu, T.; Ohyashiki, K.; Hirano, T.; Ohyashiki, J.H. Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. J. Biol. Chem. 2013, 288, 34343–34351. [Google Scholar] [CrossRef]
- Fang, Y.; Garnier, D.; Lee, T.H.; D’Asti, E.; Montermini, L.; Meehan, B.; Rak, J. PML-RARa modulates the vascular signature of extracellular vesicles released by acute promyelocytic leukemia cells. Angiogenesis 2016, 19, 25–38. [Google Scholar] [CrossRef]
- Blair, H.C.; Zaidi, M.; Huang, C.L.; Sun, L. The developmental basis of skeletal cell differentiation and the molecular basis of major skeletal defects. Biol. Rev. Camb. Philos. Soc. 2008, 83, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.C.; Sun, L.; Kohanski, R.A. Balanced regulation of proliferation, growth, differentiation, and degradation in skeletal cells. Ann. NY Acad. Sci. 2007, 1116, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.J. Osteoblast dysfunctions in bone diseases: From cellular and molecular mechanisms to therapeutic strategies. Cell Mol. Life Sci. 2015, 72, 1347–1361. [Google Scholar] [CrossRef]
- Rizzoli, R.; Body, J.J.; Brandi, M.L.; Cannata-Andia, J.; Chappard, D.; El Maghraoui, A.; Gluer, C.C.; Kendler, D.; Napoli, N.; Papaioannou, A.; et al. Cancer-associated bone disease. Osteoporos Int. 2013, 24, 2929–2953. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Bar-Shavit, Z. The osteoclast: A multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J. Cell Biochem. 2007, 102, 1130–1139. [Google Scholar] [CrossRef]
- Frisch, B.J.; Ashton, J.M.; Xing, L.; Becker, M.W.; Jordan, C.T.; Calvi, L.M. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood 2012, 119, 540–550. [Google Scholar] [CrossRef] [Green Version]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef]
- Bowers, M.; Zhang, B.; Ho, Y.; Agarwal, P.; Chen, C.C.; Bhatia, R. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood 2015, 125, 2678–2688. [Google Scholar] [CrossRef] [Green Version]
- Krevvata, M.; Silva, B.C.; Manavalan, J.S.; Galan-Diez, M.; Kode, A.; Matthews, B.G.; Park, D.; Zhang, C.A.; Galili, N.; Nickolas, T.L.; et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood 2014, 124, 2834–2846. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef]
- Hameed, A.; Brady, J.J.; Dowling, P.; Clynes, M.; O’Gorman, P. Bone disease in multiple myeloma: Pathophysiology and management. Cancer Growth Metastasis 2014, 7, 33–42. [Google Scholar] [CrossRef]
- Abe, M.; Hiura, K.; Wilde, J.; Shioyasono, A.; Moriyama, K.; Hashimoto, T.; Kido, S.; Oshima, T.; Shibata, H.; Ozaki, S.; et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: A vicious cycle between bone destruction and myeloma expansion. Blood 2004, 104, 2484–2491. [Google Scholar] [CrossRef]
- Yaccoby, S.; Wezeman, M.J.; Henderson, A.; Cottler-Fox, M.; Yi, Q.; Barlogie, B.; Epstein, J. Cancer and the microenvironment: Myeloma-osteoclast interactions as a model. Cancer Res. 2004, 64, 2016–2023. [Google Scholar] [CrossRef]
- Abe, M.; Kido, S.; Hiasa, M.; Nakano, A.; Oda, A.; Amou, H.; Matsumoto, T. BAFF and APRIL as osteoclast-derived survival factors for myeloma cells: A rationale for TACI-Fc treatment in patients with multiple myeloma. Leukemia 2006, 20, 1313–1315. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, S.; Saieva, L.; Vicario, E.; Pucci, M.; Toscani, D.; Manno, M.; Raccosta, S.; Giuliani, N.; Alessandro, R. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J. Hematol. Oncol. 2019, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; De Luca, A.; Amodio, N.; Manno, M.; Raccosta, S.; Taverna, S.; Bellavia, D.; Naselli, F.; Fontana, S.; Schillaci, O.; et al. Involvement of multiple myeloma cell-derived exosomes in osteoclast differentiation. Oncotarget 2015, 6, 13772–13789. [Google Scholar] [CrossRef]
- Faict, S.; Muller, J.; De Veirman, K.; De Bruyne, E.; Maes, K.; Vrancken, L.; Heusschen, R.; De Raeve, H.; Schots, R.; Vanderkerken, K.; et al. Exosomes play a role in multiple myeloma bone disease and tumor development by targeting osteoclasts and osteoblasts. Blood Cancer J. 2018, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Galan-Diez, M.; Kousteni, S. The osteoblastic niche in hematopoiesis and hematological myeloid malignancies. Curr. Mol. Biol. Rep. 2017, 3, 53–62. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Hematopoietic Malignancies | Small EVs Content | Target Cells | Effects | References |
|---|---|---|---|---|
| Lymphoma | LMP1 | B cells | Increased proliferation | [103] |
| CLL | miR-363 | T cells | Migration inhibition | [114] |
| DLBCL | Fas, FasL, and TRAIL | T cells | Increased apoptosis | [55,115] |
| Lymphoma | CD39 and CD73 | T cells Macrophages | Accumulation of Adenosine | [116,117] |
| T Lymphoma | TGF-β, IL-6 and PGE2 | DCs | Block of maturation | [121] |
| T- and B-leukemia/lymphoma | NKG2D ligands and TGF-β | NK cells | Reduced cytotoxicity | [130,131] |
| CML | TGF-β | NK cells | Reduced cytotoxicity | [133] |
| AML | CD33, CD34, CD117, MICA/MICB, and TGF-β | NK cells | Reduced cytotoxicity | [74,106] |
| MM | HSP70 | NK cells | Activation of the NF-κB pathway and IFN-γ released | [134] |
| CLL | Y RNA hY4 | Monocytes | Polarization induction | [138] |
| DLBCL | MyD88 | Macrophages | Pro-inflammatory signals | [144] |
| CML | BCR/ABL | Neutrophils | Aberrant BCR/ABL expression and CML-like symptoms | [152] |
| Lymphoma | HSP72 | MDSCs | Suppressive functions | [137] |
| MM | HSP72 | MDSCs | Proliferation increased and activity enhanced | [164] |
| Hematopoietic Malignancies | Small EVs Content | Target Cells | Effects | References |
|---|---|---|---|---|
| AML | CXCR4 and IGF-IR | Fibroblasts | Proliferation and VEGF expression | [177] |
| MM | miR-27b-3p and miR-214-3p | Fibroblasts | Myeloma-fibroblasts proliferation and survival boost | [178] |
| T cells Leukemia and CML | hTERT | Fibroblasts | CAF phenotype acquisition | [179,180,181] |
| CLL | microRNA and Y RNA | MSCs | CAF phenotype acquisition | [187] |
| MM | miR-21 and miR-146a | MSCs | Proliferation and CAF induction | [185] |
| ATLL | miR-21 and miR-155 | MSCs | MSCs proliferation | [186] |
| AML | miR-7977 | MSCs | Reduced ability to support CD34+ cells in the bone marrow | [177] |
| CML | AREG | MSCs | Aberrant activating of EGFR signalling | [192] |
| MM | AREG | MSCs | Block differentiation towards osteoblasts | [235] |
| AML | VEGF and VEGFR mRNA | ECs | Pro-angiogenesis mediated by increased glycolysis | [208] |
| MM | STAT3, JNK1/2/3, ERK1/2 and P53 | ECs | Enhanced migration and tube formation | [209,210] |
| MM | miR-135b | ECs | Aberrant HIF-FIH signalling | [217] |
| CML | miR-210 | ECs | Enhanced tube formation | [218] |
| CML | miR-17 -92 cluster | ECs | Enhanced migration and tube formation | [213] |
| MM | piRNA-823 | ECs | Enhanced proliferation, tube formation and invasion | [215] |
| CML (Blast crisis) | mir-126 | ECs | Increased tumor cells migration | [214] |
| APML; AML-M3 | PML-RARα mRNA | ECs | Acquisition of pro-coagulant and tissue factor properties | [219] |
| MM | AREG | Pre-osteoclasts | Increased differentiation towards osteoclasts | [235] |
| MM | DKK-1 | Osteoblasts | Block of function and differentiation | [237] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gargiulo, E.; Paggetti, J.; Moussay, E. Hematological Malignancy-Derived Small Extracellular Vesicles and Tumor Microenvironment: The Art of Turning Foes into Friends. Cells 2019, 8, 511. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050511
Gargiulo E, Paggetti J, Moussay E. Hematological Malignancy-Derived Small Extracellular Vesicles and Tumor Microenvironment: The Art of Turning Foes into Friends. Cells. 2019; 8(5):511. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050511
Chicago/Turabian StyleGargiulo, Ernesto, Jerome Paggetti, and Etienne Moussay. 2019. "Hematological Malignancy-Derived Small Extracellular Vesicles and Tumor Microenvironment: The Art of Turning Foes into Friends" Cells 8, no. 5: 511. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8050511