Iron Oxide Nanoparticle-Induced Autophagic Flux Is Regulated by Interplay between p53-mTOR Axis and Bcl-2 Signaling in Hepatic Cells

 , , and
, , and
Abstract
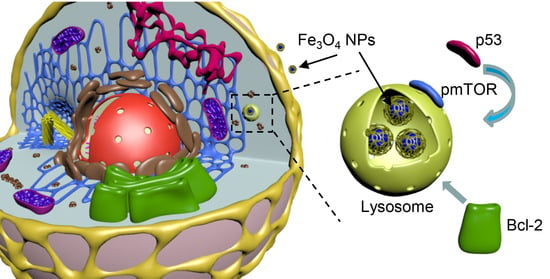
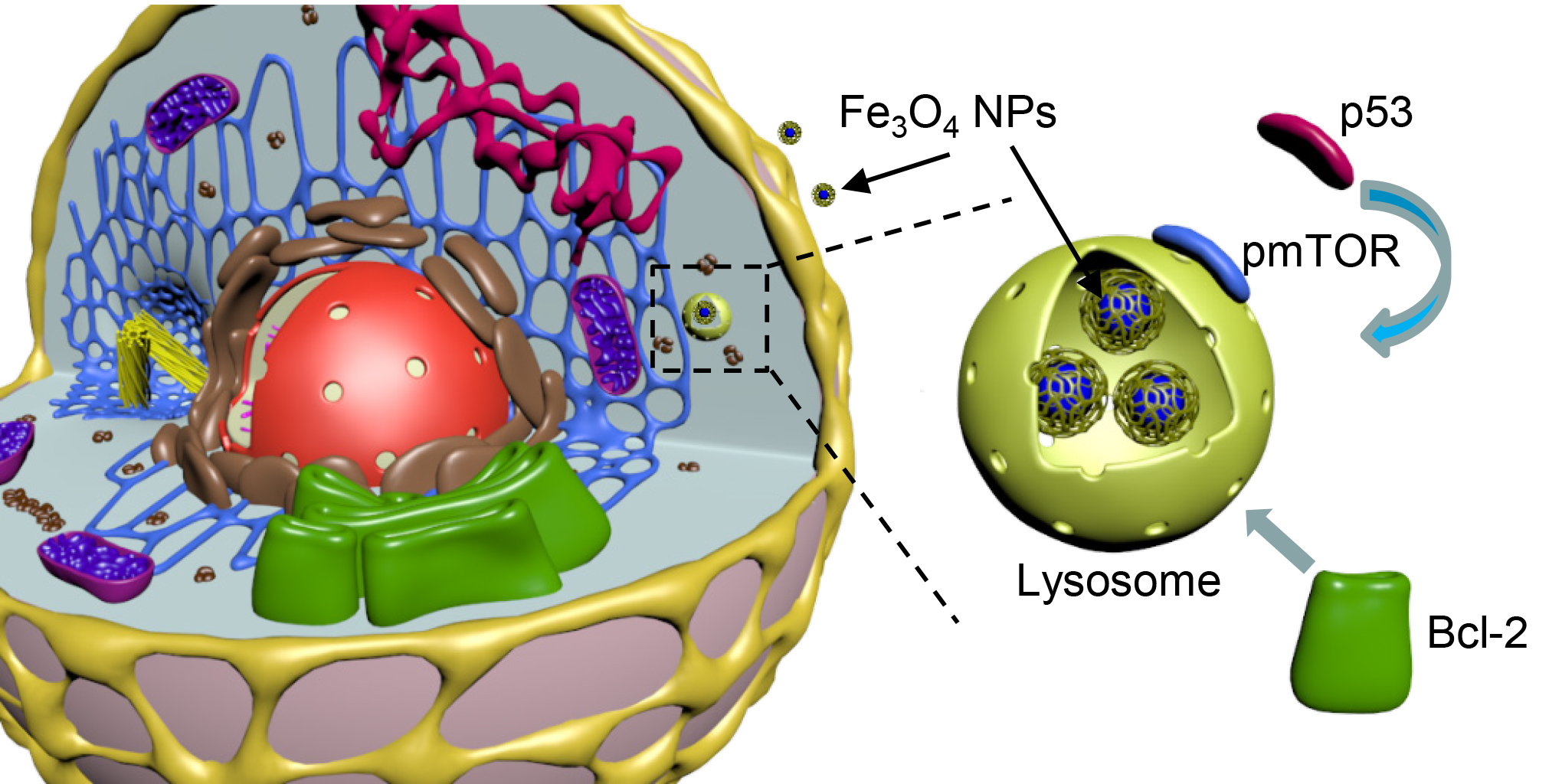
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Cell Culture
2.3. Nanoparticles
2.4. Nanoparticle-Protein Interaction
2.5. Cell Viability Assay
2.6. Lipid Peroxidation Assay
2.7. Analysis of Nanoparticles Uptake Kinetics
2.8. Immunofluorescence
2.9. Lysosomal Integrity Assay
2.10. Assessment of Mitochondrial Membrane Potential
2.11. Cell Extracts and Western Blot Analysis
2.12. Detection of Intracellular ROS Levels
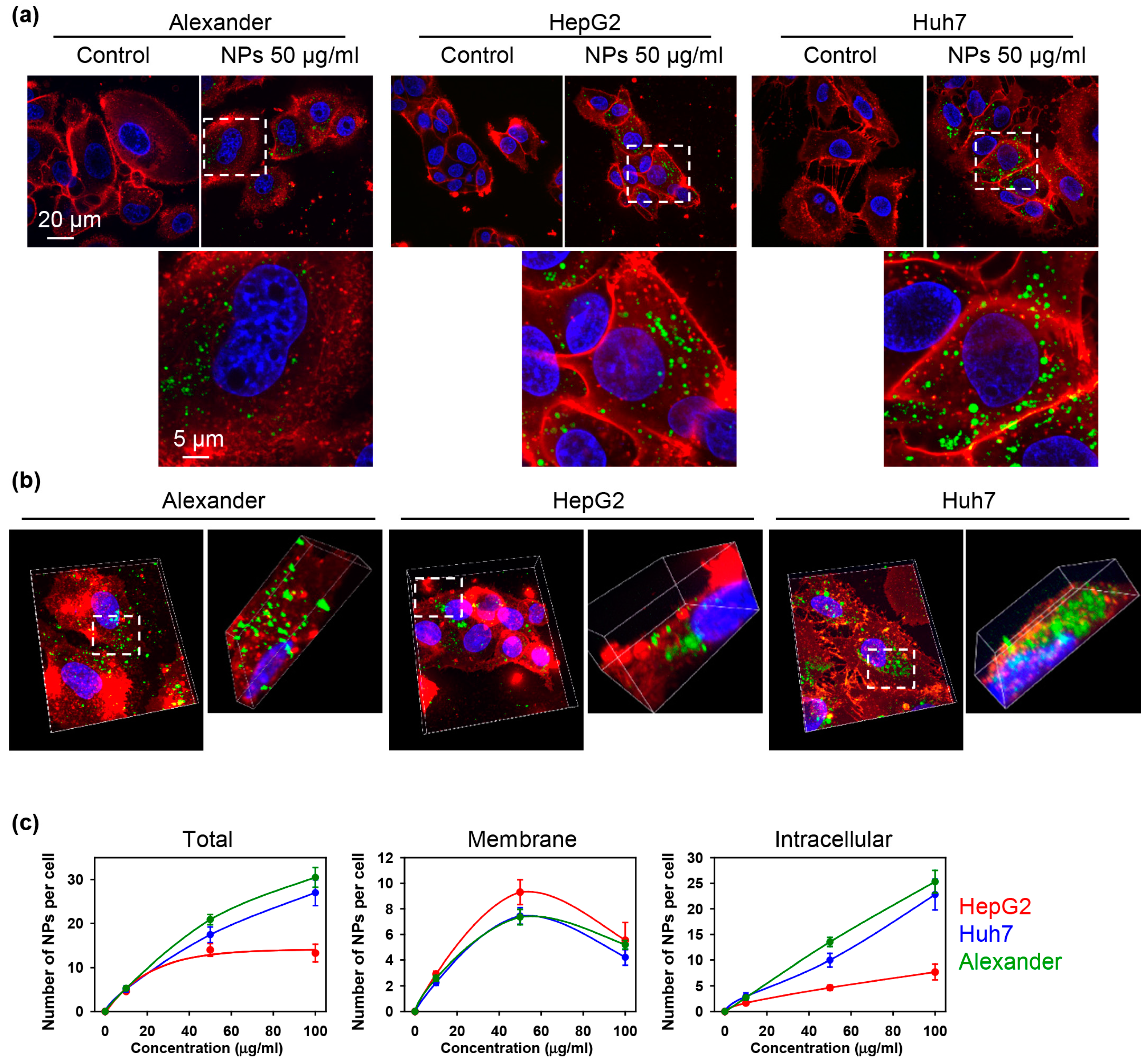
2.13. Nanoparticle Quantifications from Confocal Microscopy Images
2.14. Super-Resolution Spinning Disk Confocal Microscopy
2.15. Statistical Analysis
3. Results
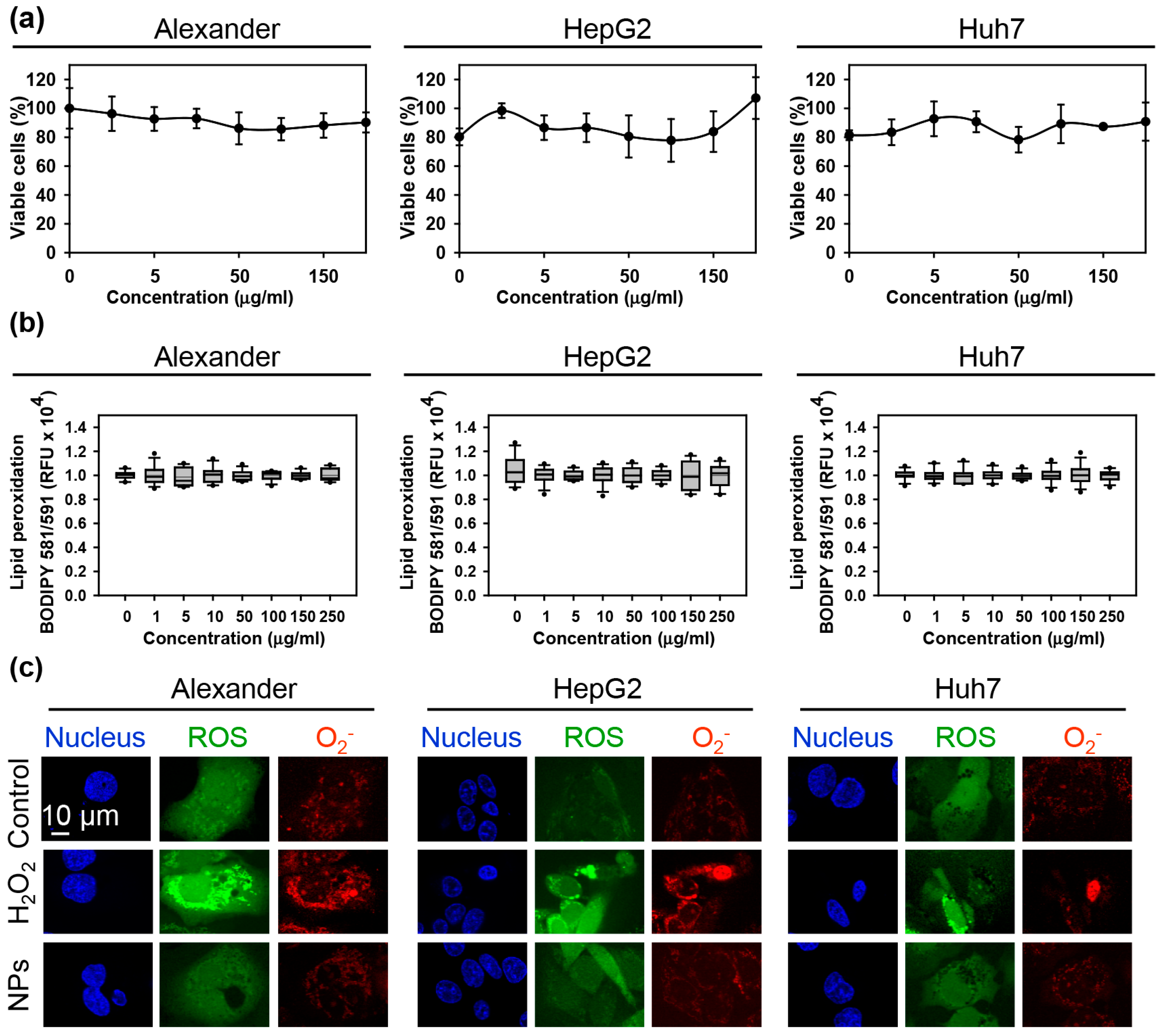
3.1. Effect of IRON Oxide Nanoparticles on Cell Viability and Oxidative Stress
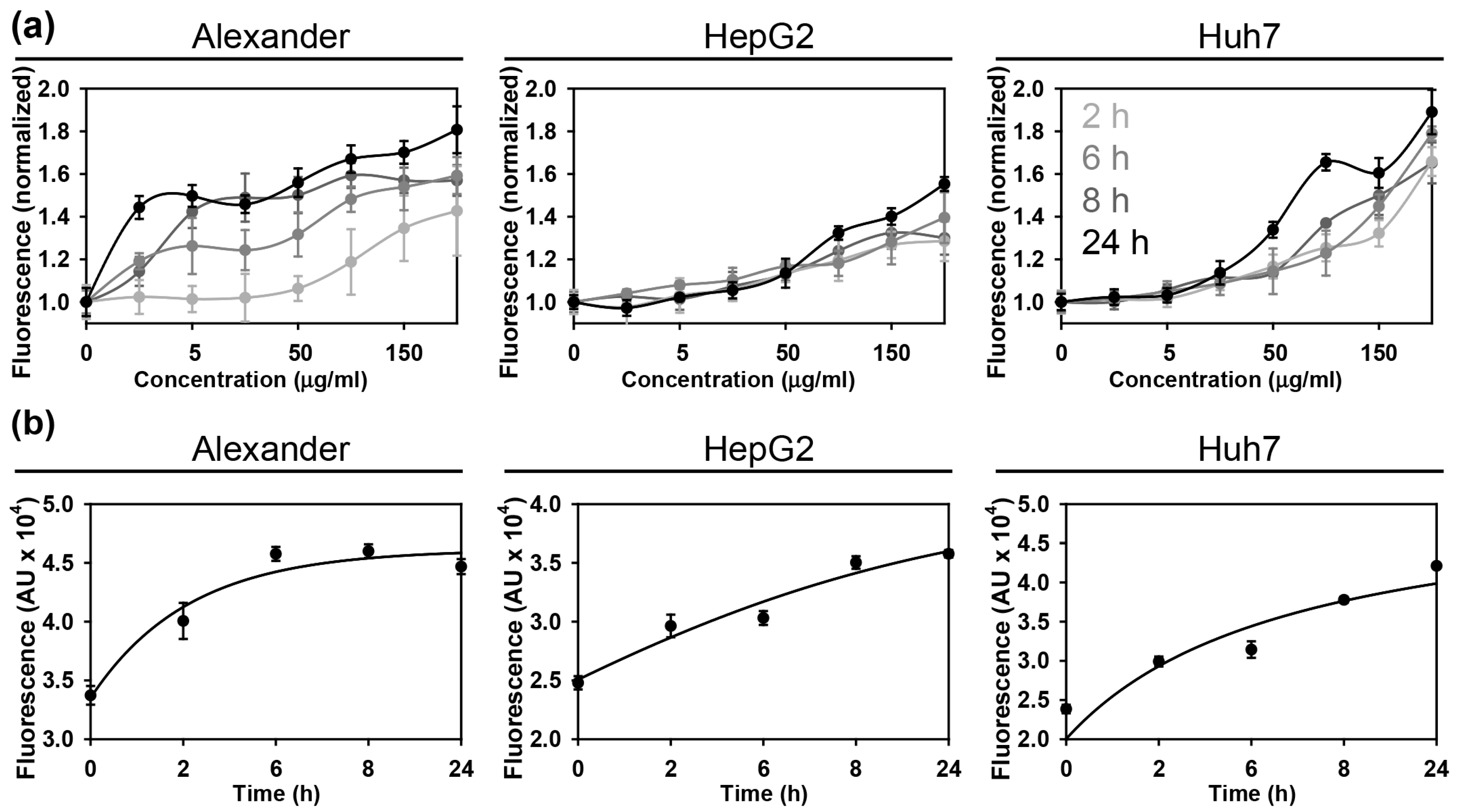
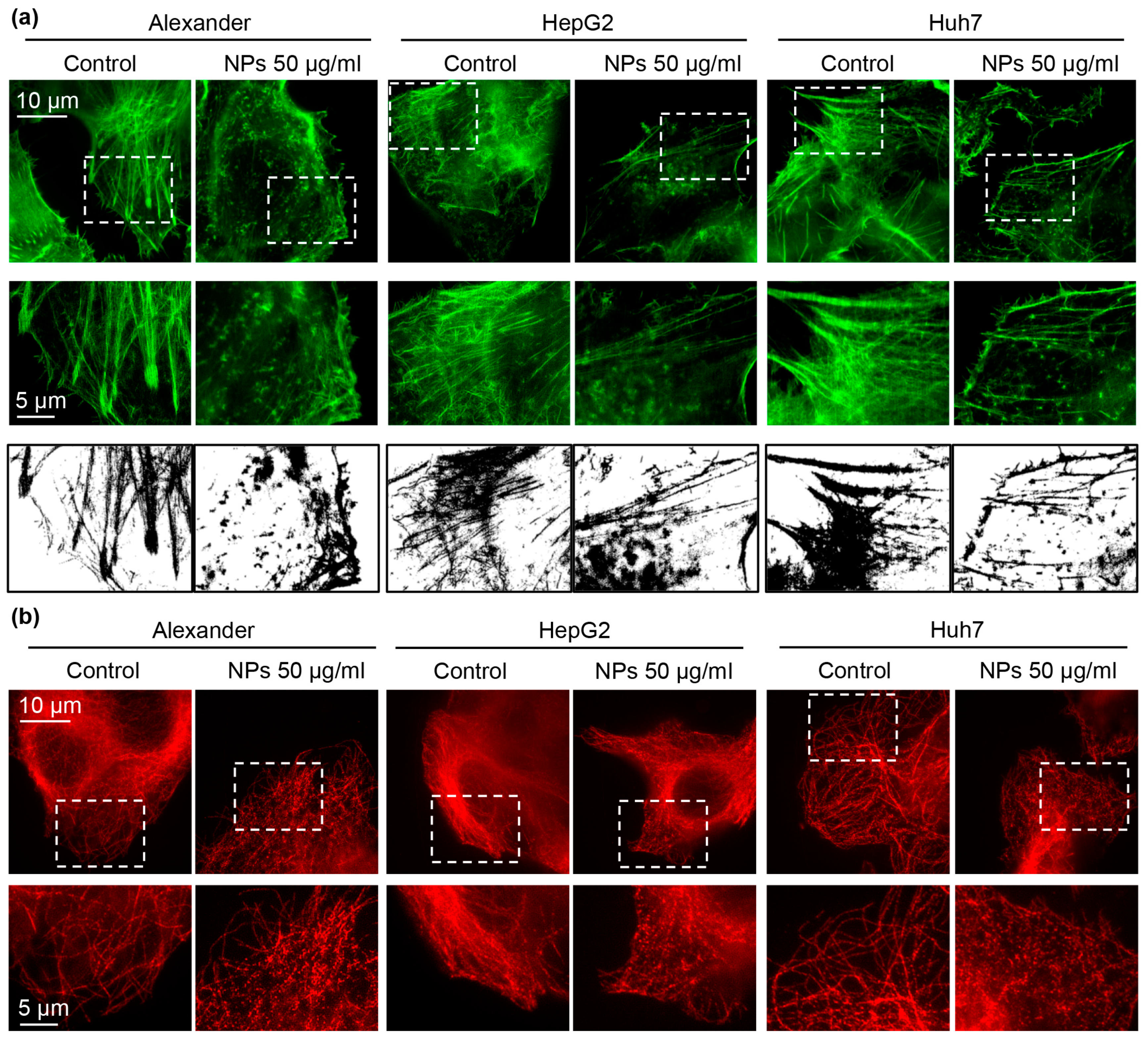
3.2. Nanoparticle Uptake and Cytoskeleton Remodeling
3.3. Nanoparticles Alter Lysosomal Function
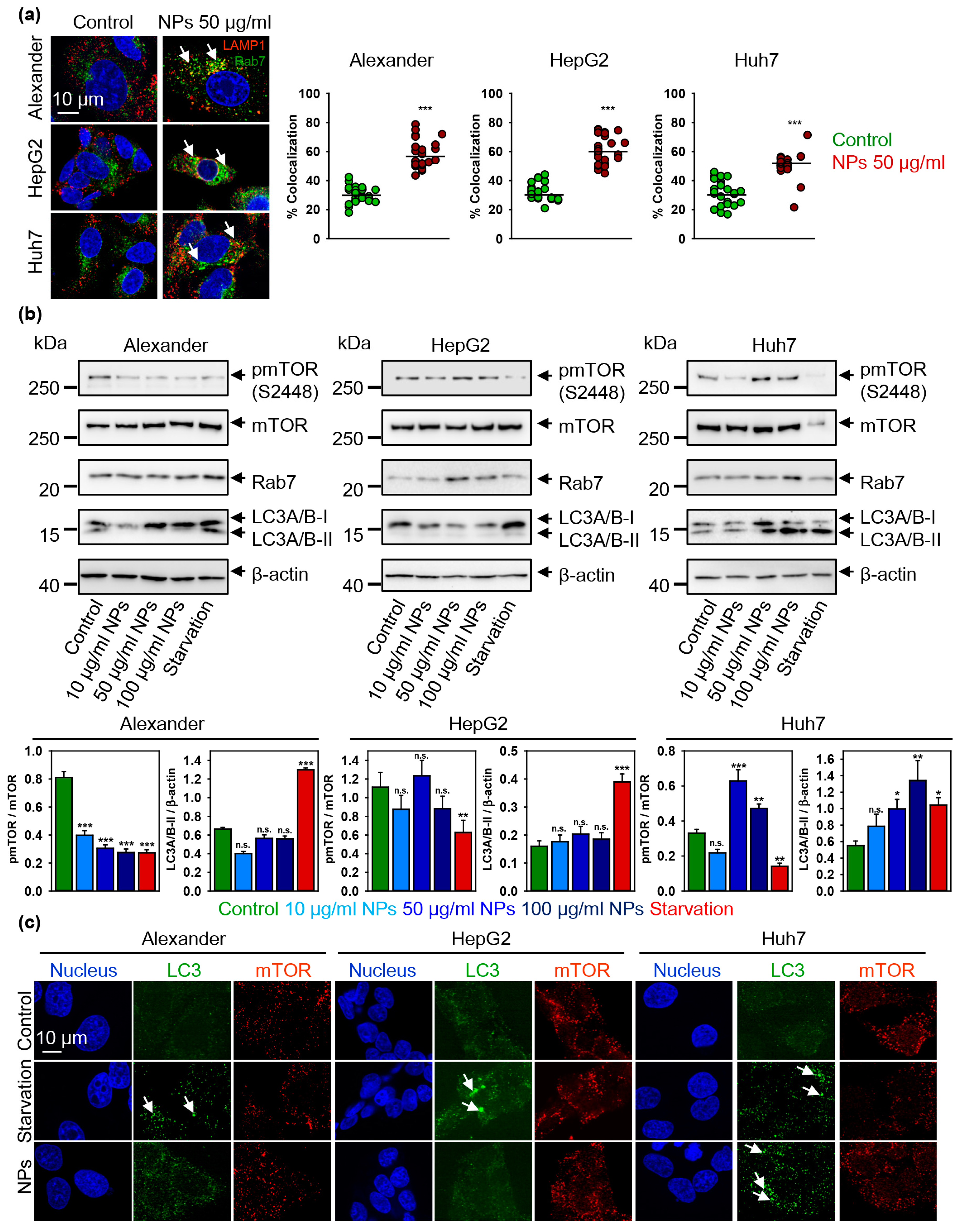
3.4. Iron Oxide Nanoparticles Modulate Autophagic Flux and Mtor Activity in Hepatic Cells
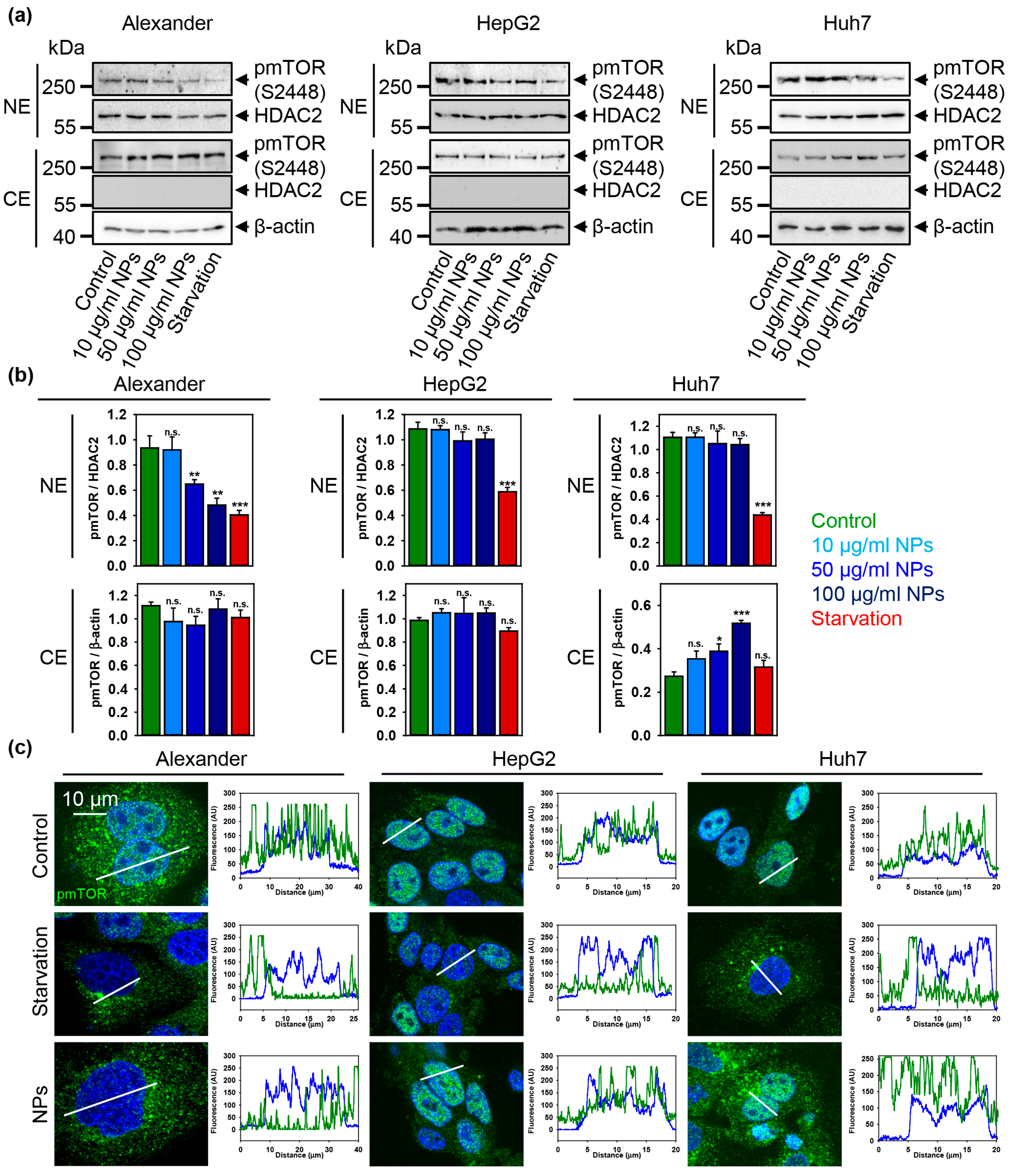
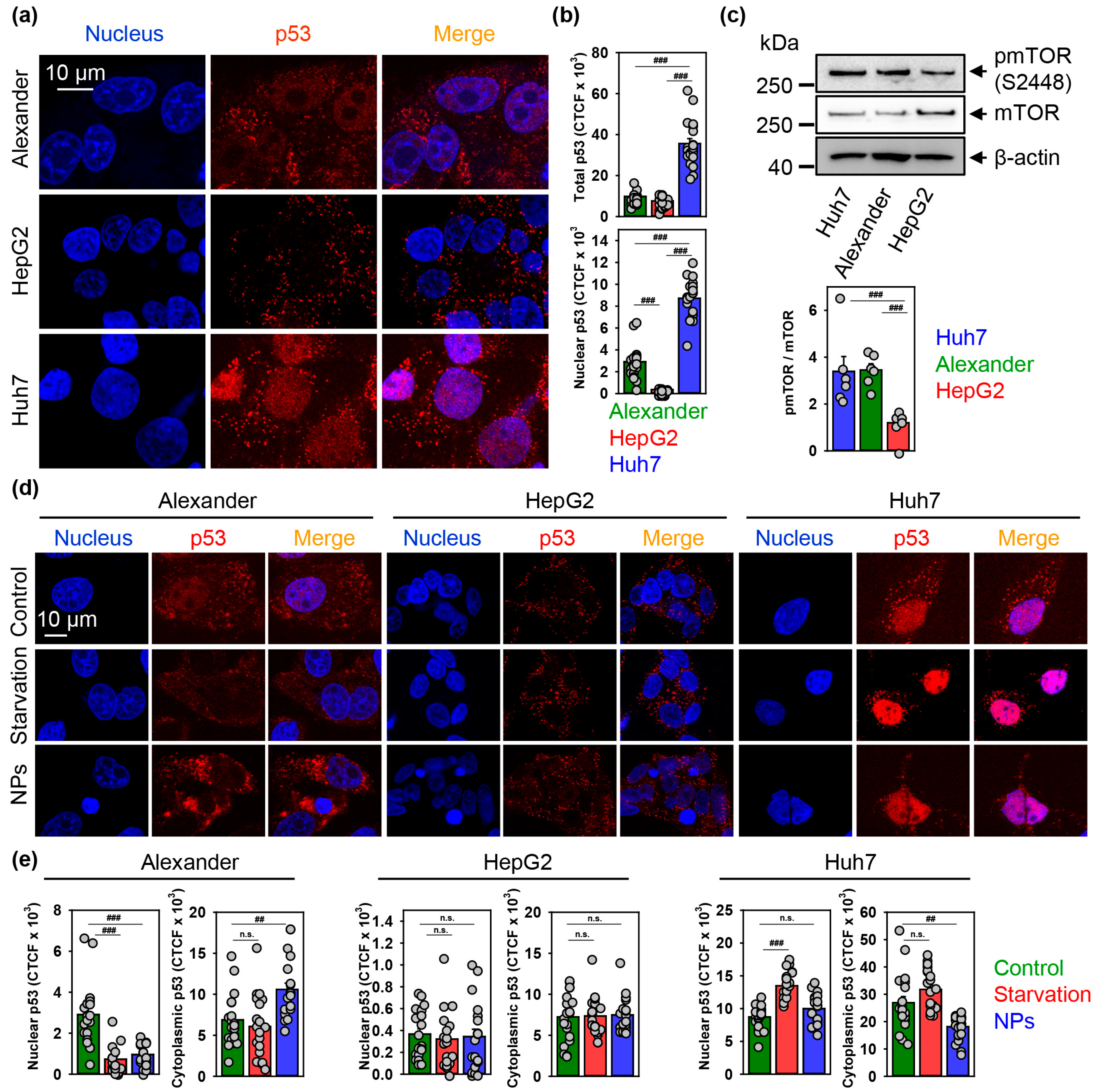
3.5. p53 Sub-Cellular Localization Mediates Hepatic Cell Response to IRON Oxide Nanoparticles
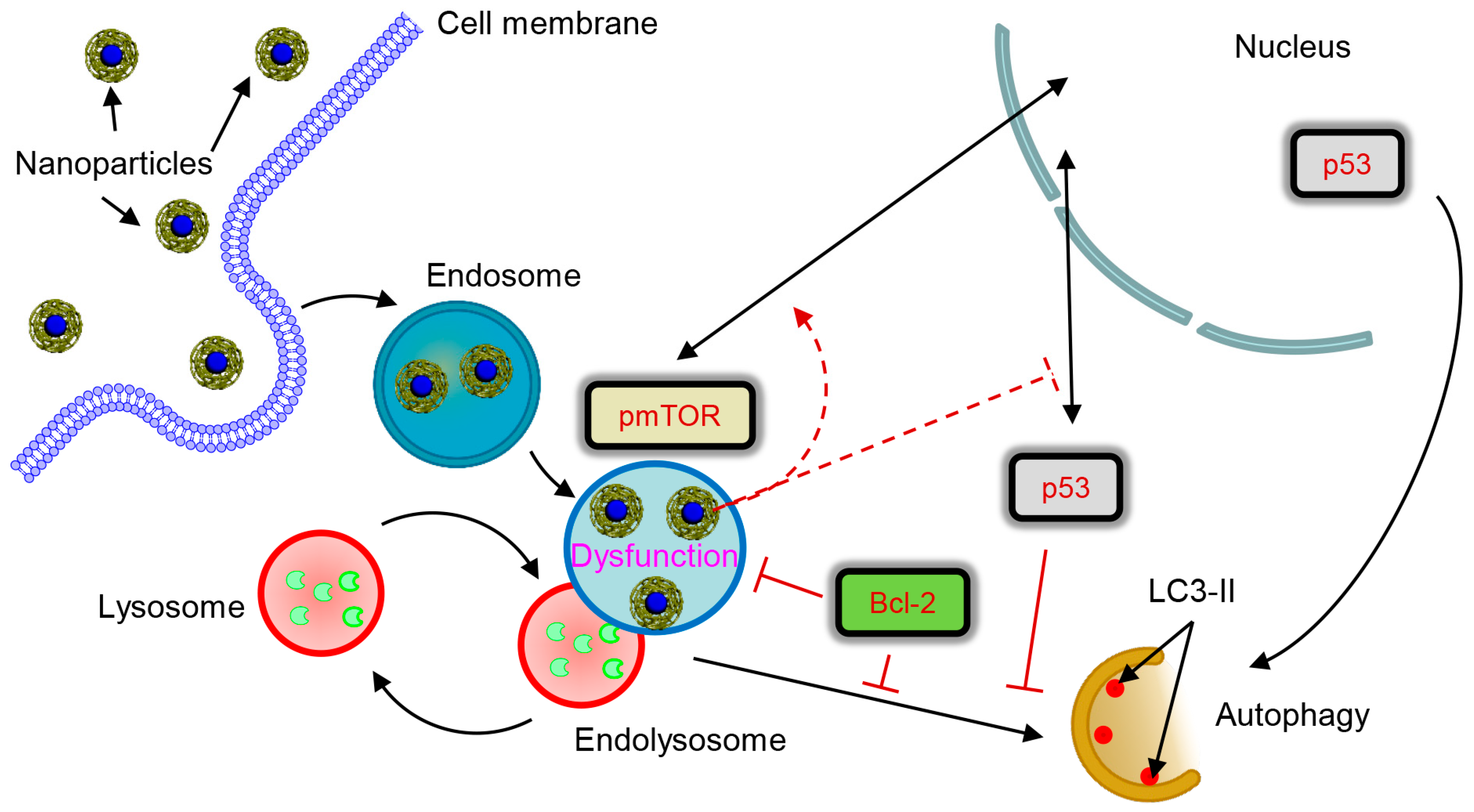
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, W.; Jiang, C.Z.; Roy, V.A. Designed synthesis and surface engineering strategies of magnetic iron oxide nanoparticles for biomedical applications. Nanoscale 2016, 8, 19421–19474. [Google Scholar] [CrossRef]
- Shi, J.J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Guo, Z.; Gao, F.; Gao, Q.; Wang, D.; Liaw, B.S.; Cai, Q.; Sun, X.; Wang, X.; Zhao, L. Shape-, size- and structure-controlled synthesis and biocompatibility of iron oxide nanoparticles for magnetic theranostics. Theranostics 2018, 8, 3284–3307. [Google Scholar] [CrossRef]
- Martinez-Banderas, A.I.; Aires, A.; Quintanilla, M.; Holguin-Lerma, J.A.; Lozano-Pedraza, C.; Teran, F.J.; Moreno, J.A.; Perez, J.E.; Ooi, B.S.; Ravasi, T.; et al. Iron-based core-shell nanowires for combinatorial drug delivery and photothermal and magnetic therapy. ACS Appl. Mater. Interfaces 2019, 11, 43976–43988. [Google Scholar] [CrossRef] [PubMed]
- Lunov, O.; Uzhytchak, M.; Smolkova, B.; Lunova, M.; Jirsa, M.; Dempsey, N.M.; Dias, A.L.; Bonfim, M.; Hof, M.; Jurkiewicz, P.; et al. Remote actuation of apoptosis in liver cancer cells via magneto-mechanical modulation of iron oxide nanoparticles. Cancers 2019, 11, 1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzhytchak, M.; Lynnyk, A.; Zablotskii, V.; Dempsey, N.M.; Dias, A.L.; Bonfim, M.; Lunova, M.; Jirsa, M.; Kubinova, S.; Lunov, O.; et al. The use of pulsed magnetic fields to increase the uptake of iron oxide nanoparticles by living cells. Appl. Phys. Lett. 2017, 111, 243703. [Google Scholar] [CrossRef]
- Wang, Y.-X.J. Superparamagnetic iron oxide based MRI contrast agents: Current status of clinical application. Quant. Imaging Med. Surg. 2011, 1, 35–40. [Google Scholar]
- Wang, Y.X.J.; Idee, J.M. A comprehensive literatures update of clinical researches of superparamagnetic resonance iron oxide nanoparticles for magnetic resonance imaging. Quant. Imaging Med. Surg. 2017, 7, 88–122. [Google Scholar] [CrossRef] [Green Version]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-based medicines: A review of FDA-approved materials and clinical trials to date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Kendall, M.; Lynch, I. Long-term monitoring for nanomedicine implants and drugs. Nat. Nanotechnol. 2016, 11, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.K.; Chu, H.H.; Wang, Y.H.; Lai, C.W.; Chou, P.T.; Hsieh, S.T.; Wang, J.L.; Liu, H.M. Macrophage physiological function after superparamagnetic iron oxide labeling. NMR Biomed. 2008, 21, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Lunov, O.; Syrovets, T.; Buchele, B.; Jiang, X.; Rocker, C.; Tron, K.; Nienhaus, G.U.; Walther, P.; Mailander, V.; Landfester, K.; et al. The effect of carboxydextran-coated superparamagnetic iron oxide nanoparticles on c-Jun N-terminal kinase-mediated apoptosis in human macrophages. Biomaterials 2010, 31, 5063–5071. [Google Scholar] [CrossRef]
- Lunov, O.; Syrovets, T.; Rocker, C.; Tron, K.; Nienhaus, G.U.; Rasche, V.; Mailander, V.; Landfester, K.; Simmet, T. Lysosomal degradation of the carboxydextran shell of coated superparamagnetic iron oxide nanoparticles and the fate of professional phagocytes. Biomaterials 2010, 31, 9015–9022. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.E.; Huh, M.I.; Ryu, B.K.; Do, J.Y.; Jin, S.U.; Moon, M.J.; Jung, J.C.; Chang, Y.; Kim, E.; Chi, S.G.; et al. The effect of static magnetic fields on the aggregation and cytotoxicity of magnetic nanoparticles. Biomaterials 2011, 32, 9401–9414. [Google Scholar] [CrossRef]
- Mirshafiee, V.; Sun, B.; Chang, C.H.; Liao, Y.P.; Jiang, W.; Jiang, J.; Liu, X.; Wang, X.; Xia, T.; Nel, A.E. Toxicological profiling of metal oxide nanoparticles in liver context reveals pyroptosis in kupffer cells and macrophages versus apoptosis in hepatocytes. ACS Nano 2018, 12, 3836–3852. [Google Scholar] [CrossRef]
- Ma, X.; Hartmann, R.; Jimenez de Aberasturi, D.; Yang, F.; Soenen, S.J.H.; Manshian, B.B.; Franz, J.; Valdeperez, D.; Pelaz, B.; Feliu, N.; et al. Colloidal gold nanoparticles induce changes in cellular and subcellular morphology. ACS Nano 2017, 11, 7807–7820. [Google Scholar] [CrossRef]
- Lunova, M.; Smolkova, B.; Lynnyk, A.; Uzhytchak, M.; Jirsa, M.; Kubinova, S.; Dejneka, A.; Lunov, O. Targeting the mTOR signaling pathway utilizing nanoparticles: A critical overview. Cancers 2019, 11, 82. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C.W. Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination. J. Control. Release 2016, 240, 332–348. [Google Scholar] [CrossRef]
- Tsoi, K.M.; MacParland, S.A.; Ma, X.Z.; Spetzler, V.N.; Echeverri, J.; Ouyang, B.; Fadel, S.M.; Sykes, E.A.; Goldaracena, N.; Kaths, J.M.; et al. Mechanism of hard-nanomaterial clearance by the liver. Nat. Mater. 2016, 15, 1212–1221. [Google Scholar] [CrossRef]
- Peng, M.L.; Li, H.L.; Luo, Z.Y.; Kong, J.; Wan, Y.S.; Zheng, L.M.; Zhang, Q.L.; Niu, H.X.; Vermorken, A.; Van de Ven, W.; et al. Dextran-coated superparamagnetic nanoparticles as potential cancer drug carriers in vivo. Nanoscale 2015, 7, 11155–11162. [Google Scholar] [CrossRef]
- Tassa, C.; Shaw, S.Y.; Weissleder, R. Dextran-coated iron oxide nanoparticles: A versatile platform for targeted molecular imaging, molecular diagnostics, and therapy. Acc. Chem. Res. 2011, 44, 842–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunov, O.; Zablotskii, V.; Syrovets, T.; Rocker, C.; Tron, K.; Nienhaus, G.U.; Simmet, T. Modeling receptor-mediated endocytosis of polymer-functionalized iron oxide nanoparticles by human macrophages. Biomaterials 2011, 32, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.Y.; Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef]
- Jin, R.R.; Liu, L.; Zhu, W.C.; Li, D.Y.; Yang, L.; Duan, J.M.; Cai, Z.Y.; Nie, Y.; Zhang, Y.J.; Gong, Q.Y.; et al. Iron oxide nanoparticles promote macrophage autophagy and inflammatory response through activation of toll-like Receptor-4 signaling. Biomaterials 2019, 203, 23–30. [Google Scholar] [CrossRef]
- Cordani, M.; Somoza, A. Targeting autophagy using metallic nanoparticles: A promising strategy for cancer treatment. Cell. Mol. Life Sci. 2019, 76, 1215–1242. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.C.; Luo, P.C.; Huang, C.W.; Leu, Y.L.; Wang, T.H.; Wei, K.C.; Wang, H.E.; Ma, Y.H. Augmented cellular uptake of nanoparticles using tea catechins: Effect of surface modification on nanoparticle-cell interaction. Nanoscale 2014, 6, 10297–10306. [Google Scholar] [CrossRef]
- Hua, X.; Tan, S.; Bandara, H.M.; Fu, Y.; Liu, S.; Smyth, H.D. Externally controlled triggered-release of drug from PLGA micro and nanoparticles. PLoS ONE 2014, 9, e114271. [Google Scholar] [CrossRef] [Green Version]
- Marcus, M.; Karni, M.; Baranes, K.; Levy, I.; Alon, N.; Margel, S.; Shefi, O. Iron oxide nanoparticles for neuronal cell applications: Uptake study and magnetic manipulations. J. Nanobiotechnol. 2016, 14, 37. [Google Scholar] [CrossRef] [Green Version]
- Domey, J.; Bergemann, C.; Bremer-Streck, S.; Krumbein, I.; Reichenbach, J.R.; Teichgraber, U.; Hilger, I. Long-term prevalence of NIRF-labeled magnetic nanoparticles for the diagnostic and intraoperative imaging of inflammation. Nanotoxicology 2016, 10, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunova, M.; Smolkova, B.; Uzhytchak, M.; Janouskova, K.Z.; Jirsa, M.; Egorova, D.; Kulikov, A.; Kubinova, S.; Dejneka, A.; Lunov, O. Light-induced modulation of the mitochondrial respiratory chain activity: Possibilities and limitations. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Pagano, R.E. Measurement of spontaneous transfer and transbilayer movement of BODIPY-labeled lipids in lipid vesicles. Biochemistry 1997, 36, 8840–8848. [Google Scholar] [CrossRef] [PubMed]
- Naguib, Y.M. A fluorometric method for measurement of peroxyl radical scavenging activities of lipophilic antioxidants. Anal. Biochem. 1998, 265, 290–298. [Google Scholar] [CrossRef]
- Drummen, G.P.C.; van Liebergen, L.C.M.; Op den Kamp, J.A.F.; Post, J.A. C11-BODIPY581/591, an oxidation-sensitive fluorescent lipid peroxidation probe: (Micro)spectroscopic characterization and validation of methodology. Free Radic. Biol. Med. 2002, 33, 473–490. [Google Scholar] [CrossRef]
- Cheloni, G.; Slaveykova, V.I. Optimization of the C11-BODIPY581/591 Dye for the Determination of Lipid Oxidation in Chlamydomonas reinhardtii by Flow Cytometry. Cytom. Part. A 2013, 83, 952–961. [Google Scholar]
- Pap, E.H.W.; Drummen, G.P.C.; Winter, V.J.; Kooij, T.W.A.; Rijken, P.; Wirtz, K.W.A.; Op den Kamp, J.A.F.; Hage, W.J.; Post, J.A. Ratio-fluorescence microscopy of lipid oxidation in living cells using C11-BODIPY581/591. FEBS Lett. 1999, 453, 278–282. [Google Scholar] [CrossRef] [Green Version]
- Vranic, S.; Boggetto, N.; Contremoulins, V.; Mornet, S.; Reinhardt, N.; Marano, F.; Baeza-Squiban, A.; Boland, S. Deciphering the mechanisms of cellular uptake of engineered nanoparticles by accurate evaluation of internalization using imaging flow cytometry. Part. Fibre Toxicol. 2013, 10, 2. [Google Scholar] [CrossRef]
- Jevprasesphant, R.; Penny, J.; Attwood, D.; D’Emanuele, A. Transport of dendrimer nanocarriers through epithelial cells via the transcellular route. J. Control. Release 2004, 97, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Hachani, R.; Birchall, M.A.; Lowdell, M.W.; Kasparis, G.; Tung, L.; Manshian, B.B.; Soenen, S.J.; Gsell, W.; Himmelreich, U.; Gharagouzloo, C.A.; et al. Assessing cell-nanoparticle interactions by high content imaging of biocompatible iron oxide nanoparticles as potential contrast agents for magnetic resonance imaging. Sci. Rep. 2017, 7, 7850. [Google Scholar] [CrossRef] [PubMed]
- Zuliani, T.; Duval, R.; Jayat, C.; Schnebert, S.; Andre, P.; Dumas, M.; Ratinaud, M.H. Sensitive and reliable JC-1 and TOTO-3 double staining to assess mitochondrial transmembrane potential and plasma membrane integrity: Interest for cell death investigations. Cytom. Part. A 2003, 54, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Lunov, O.; Zablotskii, V.; Churpita, O.; Lunova, M.; Jirsa, M.; Dejneka, A.; Kubinova, S. Chemically different non-thermal plasmas target distinct cell death pathways. Sci. Rep. 2017, 7, 600. [Google Scholar] [CrossRef] [Green Version]
- Torrano, A.A.; Blechinger, J.; Osseforth, C.; Argyo, C.; Reller, A.; Bein, T.; Michaelis, J.; Brauchle, C. A fast analysis method to quantify nanoparticle uptake on a single cell level. Nanomedicine 2013, 8, 1815–1828. [Google Scholar] [CrossRef] [Green Version]
- Blechinger, J.; Bauer, A.T.; Torrano, A.A.; Gorzelanny, C.; Brauchle, C.; Schneider, S.W. Uptake kinetics and nanotoxicity of silica nanoparticles are cell type dependent. Small 2013, 9, 3970–3980. [Google Scholar] [CrossRef]
- Hamilton, N. Quantification and its applications in fluorescent microscopy imaging. Traffic 2009, 10, 951–961. [Google Scholar] [CrossRef]
- Jonkman, J.; Brown, C.M.; Wright, G.D.; Anderson, K.I.; North, A.J. Tutorial: Guidance for quantitative confocal microscopy. Nat. Protoc. 2020. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kitaoka, M. A beginner’s guide to rigor and reproducibility in fluorescence imaging experiments. Mol. Biol. Cell 2018, 29, 1519–1525. [Google Scholar] [CrossRef]
- Dell, R.B.; Holleran, S.; Ramakrishnan, R. Sample size determination. ILAR J. 2002, 43, 207–213. [Google Scholar] [CrossRef]
- Estelrich, J.; Sanchez-Martin, M.J.; Busquets, M.A. Nanoparticles in magnetic resonance imaging: From simple to dual contrast agents. Int. J. Nanomed. 2015, 10, 1727–1741. [Google Scholar]
- Wei, H.; Bruns, O.T.; Kaul, M.G.; Hansen, E.C.; Barch, M.; Wisniowska, A.; Chen, O.; Chen, Y.; Li, N.; Okada, S.; et al. Exceedingly small iron oxide nanoparticles as positive MRI contrast agents. Proc. Natl. Acad. Sci. USA 2017, 114, 2325–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.L.; Thorling, C.A.; Liang, X.W.; Bridle, K.R.; Grice, J.E.; Zhu, Y.A.; Crawford, D.H.G.; Xu, Z.P.; Liu, X.; Roberts, M.S. Diagnostic imaging and therapeutic application of nanoparticles targeting the liver. J. Mater. Chem. B 2015, 3, 939–958. [Google Scholar] [CrossRef] [PubMed]
- Feliu, N.; Docter, D.; Heine, M.; Del Pino, P.; Ashraf, S.; Kolosnjaj-Tabi, J.; Macchiarini, P.; Nielsen, P.; Alloyeau, D.; Gazeau, F.; et al. In vivo degeneration and the fate of inorganic nanoparticles. Chem. Soc. Rev. 2016, 45, 2440–2457. [Google Scholar] [CrossRef] [Green Version]
- Docter, D.; Westmeier, D.; Markiewicz, M.; Stolte, S.; Knauer, S.K.; Stauber, R.H. The nanoparticle biomolecule corona: Lessons learned - challenge accepted? Chem. Soc. Rev. 2015, 44, 6094–6121. [Google Scholar] [CrossRef] [Green Version]
- Lunova, M.; Prokhorov, A.; Jirsa, M.; Hof, M.; Olzynska, A.; Jurkiewicz, P.; Kubinova, S.; Lunov, O.; Dejneka, A. Nanoparticle core stability and surface functionalization drive the mTOR signaling pathway in hepatocellular cell lines. Sci. Rep. 2017, 7, 16049. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Zheng, J. Clearance pathways and tumor targeting of imaging nanoparticles. ACS Nano 2015, 9, 6655–6674. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Liu, Y.; Huang, J.; Chen, K.; Huang, J.; Xiao, K. Uptake, distribution, clearance, and toxicity of iron oxide nanoparticles with different sizes and coatings. Sci. Rep. 2018, 8, 2082. [Google Scholar] [CrossRef]
- Hamm, B.; Staks, T.; Taupitz, M.; Maibauer, R.; Speidel, A.; Huppertz, A.; Frenzel, T.; Lawaczeck, R.; Wolf, K.J.; Lange, L. Contrast-enhanced MR imaging of liver and spleen: First experience in humans with a new superparamagnetic iron oxide. J. Magn. Reson. Imaging 1994, 4, 659–668. [Google Scholar] [CrossRef]
- Panariti, A.; Miserocchi, G.; Rivolta, I. The effect of nanoparticle uptake on cellular behavior: Disrupting or enabling functions? Nanotechnol. Sci. Appl. 2012, 5, 87–100. [Google Scholar]
- Mao, Z.; Xu, B.; Ji, X.; Zhou, K.; Zhang, X.; Chen, M.; Han, X.; Tang, Q.; Wang, X.; Xia, Y. Titanium dioxide nanoparticles alter cellular morphology via disturbing the microtubule dynamics. Nanoscale 2015, 7, 8466–8475. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Okada, Y. Ultrafast superresolution fluorescence imaging with spinning disk confocal microscope optics. Mol. Biol. Cell 2015, 26, 1743–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathore, B.; Sunwoo, K.; Jangili, P.; Kim, J.; Kim, J.H.; Huang, M.N.; Xiong, J.; Sharma, A.; Yang, Z.G.; Qu, J.L.; et al. Nanomaterial designing strategies related to cell lysosome and their biomedical applications: A review. Biomaterials 2019, 211, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Xu, X.; Bertrand, N.; Pridgen, E.; Swami, A.; Farokhzad, O.C. Interactions of nanomaterials and biological systems: Implications to personalized nanomedicine. Adv. Drug Deliv. Rev. 2012, 64, 1363–1384. [Google Scholar] [CrossRef]
- Nel, A.E.; Madler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [Green Version]
- Sorkin, A.; von Zastrow, M. Endocytosis and signalling: Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009, 10, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Rodriguez-Muela, N.; Hernandez-Pinto, A.M.; Serrano-Puebla, A.; Garcia-Ledo, L.; Latorre, S.H.; de la Rosa, E.J.; Boya, P. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 2015, 22, 476–487. [Google Scholar] [CrossRef] [Green Version]
- Song, X.B.; Liu, G.; Liu, F.; Yan, Z.G.; Wang, Z.Y.; Liu, Z.P.; Wang, L. Autophagy blockade and lysosomal membrane permeabilization contribute to lead-induced nephrotoxicity in primary rat proximal tubular cells. Cell Death Dis. 2017, 8, e2863. [Google Scholar] [CrossRef]
- Wang, F.; Salvati, A.; Boya, P. Lysosome-dependent cell death and deregulated autophagy induced by amine-modified polystyrene nanoparticles. Open Biol. 2018, 8, 170271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Man, S.M.; Malireddi, R.K.; Karki, R.; Lupfer, C.; Gurung, P.; Neale, G.; Guy, C.S.; Lamkanfi, M.; Kanneganti, T.D. Cathepsin B modulates lysosomal biogenesis and host defense against Francisella novicida infection. J. Exp. Med. 2016, 213, 2081–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cermak, S.; Kosicek, M.; Mladenovic-Djordjevic, A.; Smiljanic, K.; Kanazir, S.; Hecimovic, S. Loss of cathepsin B and L leads to lysosomal dysfunction, NPC-like cholesterol sequestration and accumulation of the key Alzheimer’s proteins. PLoS ONE 2016, 11, e0167428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, W.H.t.; Szymanski, C.J.; Payne, C.K. Endo-lysosomal vesicles positive for Rab7 and LAMP1 are terminal vesicles for the transport of dextran. PLoS ONE 2011, 6, e26626. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.T.; Xie, Y.X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.H. Characterization of LAMP1-labeled nondegradative lysosomal and endocytic compartments in neurons. J. Cell Biol. 2018, 217, 3127–3139. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Y.; Wei, S.S.; Li, Z.J.; Lin, C.; Zhu, Z.F.; Sun, D.S.; Bai, R.P.; Qian, J.; Gao, X.W.; Chen, G.D.; et al. Autophagic flux blockage in alveolar epithelial cells is essential in silica nanoparticle-induced pulmonary fibrosis. Cell Death Dis. 2019, 10, 127. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yu, Y.; Lu, K.; Yang, M.; Li, Y.; Zhou, X.; Sun, Z. Silica nanoparticles induce autophagy dysfunction via lysosomal impairment and inhibition of autophagosome degradation in hepatocytes. Int. J. Nanomed. 2017, 12, 809–825. [Google Scholar] [CrossRef] [Green Version]
- Dukhinova, M.S.; Prilepskii, A.Y.; Shtil, A.A.; Vinogradov, V.V. Metal oxide nanoparticles in therapeutic regulation of macrophage functions. Nanomaterials 2019, 9, 1631. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Kang, J.; Fu, C.Y. The independence of and associations among apoptosis, autophagy, and necrosis. Signal. Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Loos, C.; Syrovets, T.; Musyanovych, A.; Mailander, V.; Landfester, K.; Simmet, T. Amino-functionalized nanoparticles as inhibitors of mTOR and inducers of cell cycle arrest in leukemia cells. Biomaterials 2014, 35, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Hulea, L.; Markovic, Z.; Topisirovic, I.; Simmet, T.; Trajkovic, V. Biomedical potential of mTOR modulation by nanoparticles. Trends Biotechnol. 2016, 34, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Jeevan, D.; Neil, J.; Shannon, C.; Albert, L.; Murali, R. Deconstructing mTOR complexes in regulation of Glioblastoma Multiforme and its stem cells. Adv. Biol. Regul. 2013, 53, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y. mTOR goes to the nucleus. Cell Cycle 2010, 9, 868. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Clister, T.L.; Lowry, P.R.; Seldin, M.M.; Wong, G.W.; Zhang, J. Dynamic visualization of mTORC1 activity in living cells. Cell Rep. 2015, 10, 1767–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobashi, Y.; Suzuki, S.; Sato, E.; Hamada, Y.; Yanagawa, T.; Ooi, A. EGFR-dependent and independent activation of Akt/mTOR cascade in bone and soft tissue tumors. Mod. Pathol. 2009, 22, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Tsang, C.K.; Watkins, M.; Bertram, P.G.; Zheng, X.F. Nutrient regulates Tor1 nuclear localization and association with rDNA promoter. Nature 2006, 442, 1058–1061. [Google Scholar] [CrossRef]
- Audet-Walsh, E.; Dufour, C.R.; Yee, T.; Zouanat, F.Z.; Yan, M.; Kalloghlian, G.; Vernier, M.; Caron, M.; Bourque, G.; Scarlata, E.; et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Gene Dev. 2017, 31, 1228–1242. [Google Scholar] [CrossRef] [Green Version]
- Serpooshan, V.; Sheibani, S.; Pushparaj, P.; Wojcik, M.; Jang, A.Y.; Santoso, M.R.; Jang, J.H.; Huang, H.; Safavi-Sohi, R.; Haghjoo, N.; et al. Effect of cell sex on uptake of nanoparticles: The overlooked factor at the nanobio interface. ACS Nano 2018, 12, 2253–2266. [Google Scholar] [CrossRef]
- Lunov, O.; Syrovets, T.; Loos, C.; Beil, J.; Delacher, M.; Tron, K.; Nienhaus, G.U.; Musyanovych, A.; Mailander, V.; Landfester, K.; et al. Differential uptake of functionalized polystyrene nanoparticles by human macrophages and a monocytic cell line. ACS Nano 2011, 5, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Cagatay, T.; Ozturk, M. p53 mutation as a source of aberrant beta-catenin accumulation in cancer cells. Oncogene 2002, 21, 7971–7980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressac, B.; Galvin, K.M.; Liang, T.J.; Isselbacher, K.J.; Wands, J.R.; Ozturk, M. Abnormal structure and expression of p53 gene in human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 1990, 87, 1973–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolkova, B.; Lunova, M.; Lynnyk, A.; Uzhytchak, M.; Churpita, O.; Jirsa, M.; Kubinova, S.; Lunov, O.; Dejneka, A. Non-thermal plasma, as a new physicochemical source, to induce redox imbalance and subsequent cell death in liver cancer cell lines. Cell. Physiol. Biochem. 2019, 52, 119–140. [Google Scholar]
- Zhang, Z.Y.; Hong, D.; Nam, S.H.; Kim, J.M.; Paik, Y.H.; Joh, J.W.; Kwon, C.H.; Park, J.B.; Choi, G.S.; Jang, K.Y.; et al. SIRT1 regulates oncogenesis via a mutant p53-dependent pathway in hepatocellular carcinoma. J. Hepatol. 2015, 62, 121–130. [Google Scholar] [CrossRef]
- Agarwal, S.; Bell, C.M.; Taylor, S.M.; Moran, R.G. p53 deletion or hotspot mutations enhance mTORC1 activity by altering lysosomal dynamics of TSC2 and Rheb. Mol. Cancer Res. 2016, 14, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Ni, Z.; Wang, B.; Dai, X.; Ding, W.; Yang, T.; Li, X.; Lewin, S.; Xu, L.; Lian, J.; He, F. HCC cells with high levels of Bcl-2 are resistant to ABT-737 via activation of the ROS-JNK-autophagy pathway. Free Radic. Biol. Med. 2014, 70, 194–203. [Google Scholar] [CrossRef]
- Luo, D.; Cheng, S.C.; Xie, Y. Expression of Bcl-2 family proteins during chemotherapeutic agents-induced apoptosis in the hepatoblastoma HepG2 cell line. Br. J. Biomed. Sci. 1999, 56, 114–122. [Google Scholar]
- Li, C.H.; Li, W.Y.; Hsu, I.N.; Liao, Y.Y.; Yang, C.Y.; Taylor, M.C.; Liu, Y.F.; Huang, W.H.; Chang, H.H.; Huang, H.L.; et al. Recombinant aflatoxin-degrading F420H2-dependent reductase from mycobacterium smegmatis protects mammalian cells from aflatoxin toxicity. Toxins 2019, 11, 259. [Google Scholar] [CrossRef] [Green Version]
- Johansson, A.C.; Appelqvist, H.; Nilsson, C.; Kagedal, K.; Roberg, K.; Ollinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 2010, 15, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Mrschtik, M.; Ryan, K.M. Lysosomal proteins in cell death and autophagy. FEBS J. 2015, 282, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of chemotherapy resistance in triple-negative breast cancer-How we can rise to the challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentoor, I.; Engelbrecht, A.M.; van Jaarsveld, P.J.; Nell, T. Chemoresistance: Intricate interplay between breast tumor cells and adipocytes in the tumor microenvironment. Front. Endocrinol. 2018, 9, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kast, D.J.; Dominguez, R. The cytoskeleton-autophagy connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonneau, B.; Nougarede, A.; Prudent, J.; Popgeorgiev, N.; Peyrieras, N.; Rimokh, R.; Gillet, G. The Bcl-2 homolog Nrz inhibits binding of IP3 to its receptor to control calcium signaling during zebrafish epiboly. Sci. Signal. 2014, 7, ra14. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Zhou, X.; Hao, Q.; Lu, H. Mutant p53 in cancer therapy-the barrier or the path. J. Mol. Cell Biol. 2019, 11, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Vijayakumaran, R.; Tan, K.H.; Miranda, P.J.; Haupt, S.; Haupt, Y. Regulation of mutant p53 protein expression. Front. Oncol. 2015, 5, 284. [Google Scholar] [CrossRef] [Green Version]
- Oude Engberink, R.D.; van der Pol, S.M.; Dopp, E.A.; de Vries, H.E.; Blezer, E.L. Comparison of SPIO and USPIO for in vitro labeling of human monocytes: MR detection and cell function. Radiology 2007, 243, 467–474. [Google Scholar] [CrossRef]
- Erofeev, A.; Gorelkin, P.; Garanina, A.; Alova, A.; Efremova, M.; Vorobyeva, N.; Edwards, C.; Korchev, Y.; Majouga, A. Novel method for rapid toxicity screening of magnetic nanoparticles. Sci. Rep. 2018, 8, 7462. [Google Scholar] [CrossRef] [Green Version]
- Patil, R.M.; Thorat, N.D.; Shete, P.B.; Bedge, P.A.; Gavde, S.; Joshi, M.G.; Tofail, S.A.M.; Bohara, R.A. Comprehensive cytotoxicity studies of superparamagnetic iron oxide nanoparticles. Biochem. Biophys. Rep. 2018, 13, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Soenen, S.J.; Himmelreich, U.; Nuytten, N.; De Cuyper, M. Cytotoxic effects of iron oxide nanoparticles and implications for safety in cell labelling. Biomaterials 2011, 32, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Z.J.; Li, X.M.; Zhang, Y.; Yin, M.; Li, J.; Song, H.Y.; Shi, J.Y.; Ling, D.S.; Wang, L.H.; et al. Deciphering active biocompatibility of iron oxide nanoparticles from their intrinsic antagonism. Nano Res. 2018, 11, 2746–2755. [Google Scholar] [CrossRef]
- Lipski, A.M.; Pino, C.J.; Haselton, F.R.; Chen, I.W.; Shastri, V.P. The effect of silica nanoparticle-modified surfaces on cell morphology, cytoskeletal organization and function. Biomaterials 2008, 29, 3836–3846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falagan-Lotsch, P.; Grzincic, E.M.; Murphy, C.J. One low-dose exposure of gold nanoparticles induces long-term changes in human cells. Proc. Natl. Acad. Sci. USA 2016, 113, 13318–13323. [Google Scholar] [CrossRef] [Green Version]
- Denisenko, T.V.; Pivnyuk, A.D.; Zhivotovsky, B. p53-autophagy-metastasis link. Cancers 2018, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Paz Hernandez, M.; Oses, C.; Peña, D.; Criollo, A.; Morselli, E. Chapter 10—Mutant p53 located in the cytoplasm inhibits autophagy. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 189–203. [Google Scholar]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uzhytchak, M.; Smolková, B.; Lunova, M.; Jirsa, M.; Frtús, A.; Kubinová, Š.; Dejneka, A.; Lunov, O. Iron Oxide Nanoparticle-Induced Autophagic Flux Is Regulated by Interplay between p53-mTOR Axis and Bcl-2 Signaling in Hepatic Cells. Cells 2020, 9, 1015. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041015
Uzhytchak M, Smolková B, Lunova M, Jirsa M, Frtús A, Kubinová Š, Dejneka A, Lunov O. Iron Oxide Nanoparticle-Induced Autophagic Flux Is Regulated by Interplay between p53-mTOR Axis and Bcl-2 Signaling in Hepatic Cells. Cells. 2020; 9(4):1015. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041015
Chicago/Turabian StyleUzhytchak, Mariia, Barbora Smolková, Mariia Lunova, Milan Jirsa, Adam Frtús, Šárka Kubinová, Alexandr Dejneka, and Oleg Lunov. 2020. "Iron Oxide Nanoparticle-Induced Autophagic Flux Is Regulated by Interplay between p53-mTOR Axis and Bcl-2 Signaling in Hepatic Cells" Cells 9, no. 4: 1015. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041015