Commensal and Pathogenic Members of the Dental Calculus Microbiome of Badia Pozzeveri Individuals from the 11th to 19th Centuries

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. History of Excavation Site and Sample Description
2.2. Sample Collection, DNA Extraction, and Avoidance of Contamination
2.3. 16S rRNA Gene High-Throughput Sequencing
2.4. 16S rRNA Gene Sequence Analyses
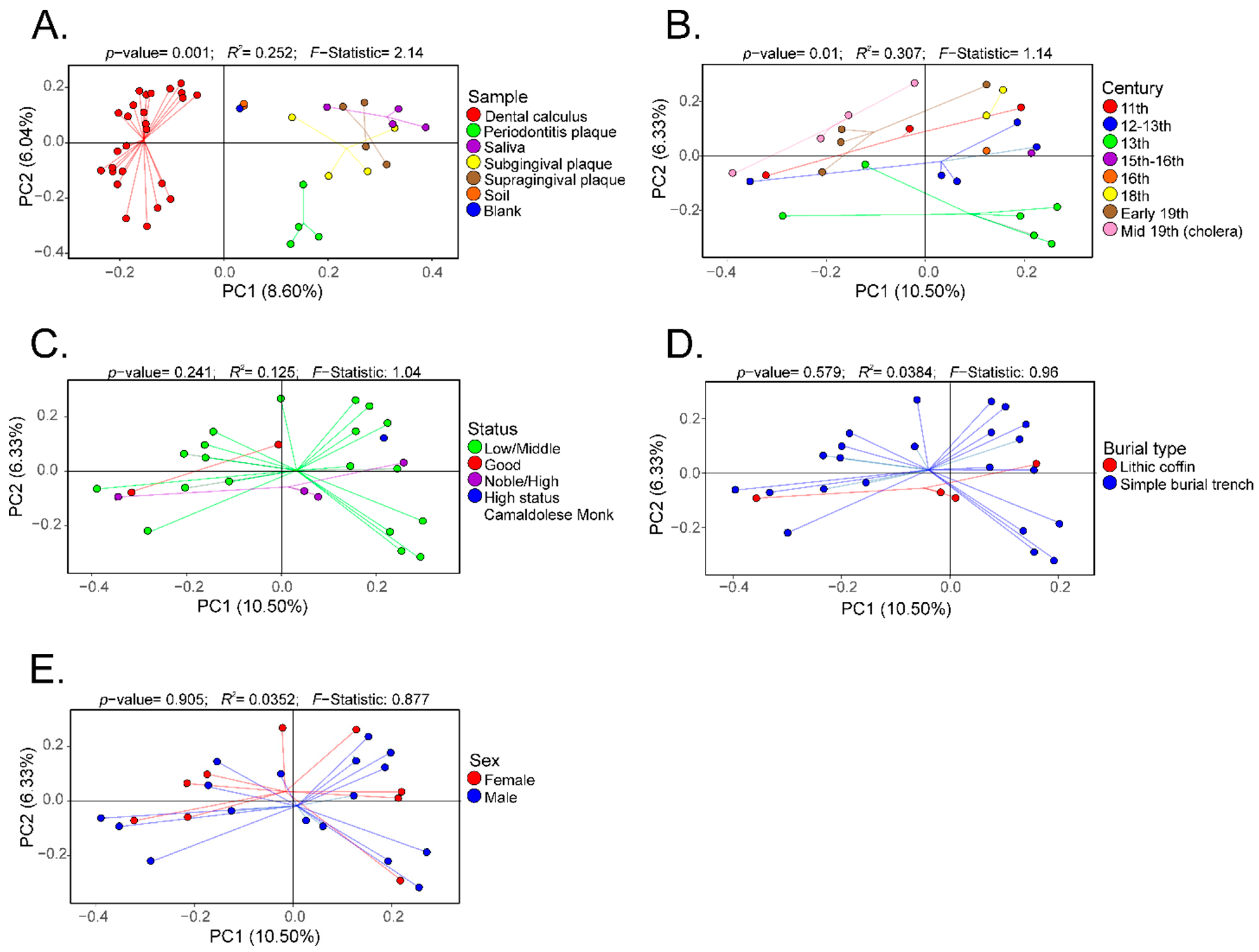
3. Results
4. Discussion
4.1. Environmental Microbiome of Badia Pozzeveri Dental Calculus Remains
4.2. Commensal and Pathogenic Microbiome in Badia Pozzeveri Remains
4.3. Dietary Habits from Badia Pozzeveri and Oral Microbiome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kilian, M.; Chapple, I.L.C.; Hannig, M.; Marsh, P.D.; Meuric, V.; Pedersen, A.M.L.; Tonetti, M.S.; Wade, W.G.; Zaura, E. The oral microbiome—An update for oral healthcare professionals. Br. Dent. J. 2016. [Google Scholar] [CrossRef]
- Verma, D.; Garg, P.K.; Dubey, A.K. Insights into the human oral microbiome. Arch. Microbiol. 2018, 200, 525–540. [Google Scholar] [CrossRef]
- Li, J.; Quinque, D.; Horz, H.P.; Li, M.; Rzhetskaya, M.; Raff, J.A.; Hayes, M.G.; Stoneking, M. Comparative analysis of the human saliva microbiome from different climate zones: Alaska, Germany, and Africa. BMC Microbiol. 2014. [Google Scholar] [CrossRef]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; Lander, O.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015. [Google Scholar] [CrossRef] [PubMed]
- Struzycka, I. The oral microbiome in dental caries. Pol. J. Microbiol. 2014, 63, 127–135. [Google Scholar]
- Burne, R.A.; Zeng, L.; Ahn, S.J.; Palmer, S.R.; Liu, Y.; Lefebure, T.; Stanhope, M.J.; Nascimento, M.M. Progress dissecting the oral microbiome in caries and health. Adv. Dent. Res. 2012, 24, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Faller, L.L.; Klitgord, N.; Mazumdar, V.; Ghodsi, M.; Sommer, D.D.; Gibbons, T.R.; Treangen, T.J.; Chang, Y.C.; Li, S.; et al. Deep sequencing of the oral microbiome reveals signatures of periodontal disease. PLoS ONE 2012. [Google Scholar] [CrossRef]
- Kolenbrander, P.E.; Palmer, R.J.; Periasamy, S.; Jakubovics, N.S. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 2010, 8, 471–480. [Google Scholar] [CrossRef]
- Mark Welch, J.L.; Rossetti, B.J.; Rieken, C.W.; Dewhirst, F.E.; Borisy, G.G. Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.N.; Snesrud, E.; Liu, J.; Ong, A.C.; Kilian, M.; Schork, N.J.; Bretz, W. The Dental Plaque Microbiome in Health and Disease. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Narganes-Storde, Y.; Chanlatte-Baik, L.; Toranzos, G.A.; Cano, R.J. Insights of the dental calculi microbiome of pre-Columbian inhabitants from Puerto Rico. PeerJ 2017. [Google Scholar] [CrossRef]
- Adler, C.J.; Dobney, K.; Weyrich, L.S.; Kaidonis, J.; Walker, A.W.; Haak, W.; Bradshaw, C.J.A.; Townsend, G.; Sołtysiak, A.; Alt, K.W.; et al. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the Neolithic and Industrial revolutions. Nat. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Warinner, C.; Rodrigues, J.F.M.; Vyas, R.; Trachsel, C.; Shved, N.; Grossmann, J.; Radini, A.; Hancock, Y.; Tito, R.Y.; Fiddyment, S.; et al. Pathogens and host immunity in the ancient human oral cavity. Nat. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, L.S.; Duchene, S.; Soubrier, J.; Arriola, L.; Llamas, B.; Breen, J.; Morris, A.G.; Alt, K.W.; Caramelli, D.; Dresely, V.; et al. Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 2017. [Google Scholar] [CrossRef] [PubMed]
- Fornaciari, A.; Francesco, C.; Cariboni, A.; Cavallini, L.; Farnocchia, A.; Testi, S.; Vercellotti, G. Lo scavo bioarcheologico di un monastero lungo la via Francigena. Not. della Soprintend. Archeol. della Toscana 2016, 11, 123–135. [Google Scholar]
- Lovejoy, C.O. Dental wear in the Libben population: Its functional pattern and role in the determination of adult skeletal age at death. Am. J. Phys. Anthropol. 1985. [Google Scholar] [CrossRef]
- Suchey, J.M.; Brooks, S. Skeletal age determination based on the os pubis: A comparison of the Acsádi-Nemeskéri and Suchey-Brooks methods. Hum. Evol. 1990. [Google Scholar] [CrossRef]
- Loth, S. Morphological assessment of age in the adult: The thoracic region. Age Markers Hum. Skelet. 1989, 105–136. [Google Scholar]
- Buikstra, J.E.; Ubelaker, D.H. Standards for Data Collection From Human Skeletal Remains. Arkansas Archaeol. Surv. Res. Ser. 1994, 44, 272. [Google Scholar] [CrossRef]
- Weyrich, L.S.; Dobney, K.; Cooper, A. Ancient DNA analysis of dental calculus. J. Hum. Evol. 2015. [Google Scholar] [CrossRef]
- Warinner, C.; Speller, C.; Collins, M.J. A new era in palaeomicrobiology: Prospects for ancient dental calculus as a long-term record of the human oral microbiome. Philos. Trans. R. Soc. B Biol. Sci. 2015. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Fornaciari, G.; Luciani, S.; Toranzos, G.A.; Marota, I.; Giuffra, V.; Cano, R.J. Gut microbiome and putative resistome of inca and italian nobility mummies. Genes 2017, 8, 310. [Google Scholar] [CrossRef]
- Weyrich, L.; Farrer, A.G.; Eisenhofer, R.; Arriola, L.A.; Young, J.; Selway, C.A.; Handsley-Davis, M.; Adler, C.; Breen, J.; Cooper, A. Laboratory contamination over time during low-biomass sample analysis. Mol. Ecol. Resour. 2019. [Google Scholar] [CrossRef]
- Chen, C.; Hemme, C.; Beleno, J.; Shi, Z.J.; Ning, D.; Qin, Y.; Tu, Q.; Jorgensen, M.; He, Z.; Wu, L.; et al. Oral microbiota of periodontal health and disease and their changes after nonsurgical periodontal therapy. ISME J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011. [Google Scholar] [CrossRef]
- Guettler, M.V.; Rumler, D.; Jain, M.K. Actinobacillus succinogenes sp. nov., a novel succinic-acid-producing strain from the bovine rumen. Int. J. Syst. Bacteriol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.D.; Rodrigues, U.M.; Pigott, N.E.; Facklam, R.R. Enterococcus dispar sp. nov. a new Enterococcus species from human sources. Lett. Appl. Microbiol. 1991. [Google Scholar] [CrossRef]
- Goodson, J.M.; Groppo, D.; Halem, S.; Carpino, E. Is obesity an oral bacterial disease? J. Dent. Res. 2009. [Google Scholar] [CrossRef]
- Fisher, R.G.; Denison, M.R. Veillonella parvula bacteremia without an underlying source. J. Clin. Microbiol. 1996, 34, 3235–3236. [Google Scholar] [PubMed]
- Nakatsu, C.H.; Hristova, K.; Hanada, S.; Meng, X.Y.; Hanson, J.R.; Scow, K.M.; Kamagata, Y. Methylibium petroleiphilum gen. nov., sp. nov., a novel methyl tert-butyl ether-degrading methylotroph of the Betaproteobacteria. Int. J. Syst. Evol. Microbiol. 2006. [Google Scholar] [CrossRef]
- Teshima, T.; Kabuki, T.; Seto, Y.; Bing Yao, L.; Nakajima, H.; Hai Hao, D.; Miyamoto, M.; Bo Sun, Y. Lactobacillus harbinensis sp. nov., consisted of strains isolated from traditional fermented vegetables ‘Suan cai’ in Harbin, Northeastern China and Lactobacillus perolens DSM 12745. Syst. Appl. Microbiol. 2005. [Google Scholar] [CrossRef]
- Barker, H.S.; Snyder, J.W.; Hicks, A.B.; Yanoviak, S.P.; Southern, P.; Dhakal, B.K.; Ghimire, G.R.; Couturier, M.R. First case reports of Ignatzschineria (Schineria) indica associated with myiasis. J. Clin. Microbiol. 2014. [Google Scholar] [CrossRef]
- Sass, H.; Overmann, J.; Rütters, H.; Babenzien, H.D.; Cypionka, H. Desulfosporomusa polytropa gen. nov., sp. nov., a novel sulfate-reducing bacterium from sediments of an oligotrophic lake. Arch. Microbiol. 2004. [Google Scholar] [CrossRef]
- Tessler, M.; Neumann, J.S.; Afshinnekoo, E.; Pineda, M.; Hersch, R.; Velho, L.F.M.; Segovia, B.T.; Lansac-Toha, F.A.; Lemke, M.; Desalle, R.; et al. Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Gao, L.; Xu, T.; Huang, G.; Jiang, S.; Gu, Y.; Chen, F. Oral microbiomes: more and more importance in oral cavity and whole body. Protein Cell 2018, 9, 488–500. [Google Scholar] [CrossRef]
- Wang, K.; Lu, W.; Tu, Q.; Ge, Y.; He, J.; Zhou, Y.; Gou, Y.; Van Nostrand, J.D.; Qin, Y.; Li, J.; et al. Preliminary analysis of salivary microbiome and their potential roles in oral lichen planus. Sci. Rep. 2016. [Google Scholar] [CrossRef]
- Stoyanova, M.; Pavlina, I.; Moncheva, P.; Bogatzevska, N. Biodiversity and incidence of Burkholderia species. Biotechnol. Biotechnol. Equip. 2007. [Google Scholar] [CrossRef]
- Bittar, F.; Leydier, A.; Bosdure, E.; Toro, A.; Reynaud-Gaubert, M.; Boniface, S.; Stremler, N.; Dubus, J.C.; Sarles, J.; Raoult, D.; et al. Inquilinus limosus and cystic fibrosis. Emerg. Infect. Dis. 2008, 14, 993–995. [Google Scholar] [CrossRef]
- Safaei, S.; Fatahi-Bafghi, M.; Pouresmaeil, O. Role of Tsukamurella species in human infections: First literature review. New Microbes New Infect. 2018, 22, 6–12. [Google Scholar] [CrossRef]
- Kovaleva, J.; Degener, J.E.; Van Der Mei, H.C. Methylobacterium and its role in health care-associated infection. J. Clin. Microbiol. 2014, 52, 1317–1321. [Google Scholar] [CrossRef]
- Gneiding, K.; Frodl, R.; Funke, G. Identities of Microbacterium spp. encountered in human clinical specimens. J. Clin. Microbiol. 2008. [Google Scholar] [CrossRef]
- Yassin, A.F.; Rainey, F.A.; Brzezinka, H.; Jahnke, K.D.; Weissbrodt, H.; Budzikiewicz, H.; Stackebrandt, E.; Schaal, K.P. Lentzea gen. nov., a new genus of the order Actinomycetales. Int. J. Syst. Bacteriol. 1995. [Google Scholar] [CrossRef]
- Tan, T.Y.; Ng, L.S.Y.; Eng, L.C. Five Clinical Cases of Necropsobacter rosorum Bacteremia. J. Clin. Microbiol. 2013. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, N.; He, L.; Wang, L.; Liu, Z.; You, M.; Guan, F. Arthrobacter scleromae sp. nov. isolated from human clinical specimens. J. Clin. Microbiol. 2005. [Google Scholar] [CrossRef]
- Brown, A.F.; Leech, J.M.; Rogers, T.R.; McLoughlin, R.M. Staphylococcus aureus colonization: Modulation of host immune response and impact on human vaccine design. Front. Immunol. 2014, 4, 507. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Sex | Age | Burial Typology | Chronology | Status |
|---|---|---|---|---|---|
| USK 3743 | F | 40–50 | Simple burial trench | 11th century | Good social status |
| USK 3746 | M | 30–40 | Simple burial trench | 11th century | Good social status |
| USK 2776 | M | 45–55 | Simple burial trench | 11th century | Middle/low status |
| USK 6073 | M | 25–30 | Lithic coffin | 12–13th century | Noble or high status |
| USK 4821 | M | 35–45 | Simple burial trench | 12–13th century | High-status Camaldolese monk |
| USK 3219 | M | 30–40 | Lithic coffin | 12–13th century | Noble or high status |
| USK 3395 | F | 30–40 | Lithic coffin | 12–13th century | Noble or high status |
| USK 3417 | M | 30–40 | Lithic coffin | 12–13th century | Noble or high status |
| USK 6082 | M | 20–25 | Simple burial trench | 13th century | Middle/low status |
| USK 6071 | M | 20–25 | Simple burial trench | 13th century | Middle/low status |
| USK 6054 | M | 35–45 | Simple burial trench | 13th century | Middle/low status |
| USK 6015 | M | 20–25 | Simple burial trench | 13th century | Middle/low status |
| USK 6065 | F | 40–50+ | Simple burial trench | 13th century | Middle/low status |
| USK 6072 | M | 40–50 | Simple burial trench | 13th century | Middle/low status |
| USK 3641 | F | 30–40 | Simple burial trench | 15th–16th century | Middle/low status |
| USK 3653 | M | 25–35 | Simple burial trench | 16th century | Middle/low status |
| USK 3391 | M | 30–40 | Simple burial trench | 18th century | Middle/low status |
| USK 3160 | M | 35–45 | Simple burial trench | 18th century | Middle/low status |
| USK 2299 | M | 25–30 | Simple burial trench | 1855 cholera | Middle/low status |
| USK 2339 | F | 60+ | Simple burial trench | 1855 cholera | Middle/low status |
| USK 2399 | M | 50–60 | Simple burial trench | 1855 cholera | Middle/low status |
| USK 2297 | F | 45–55 | Simple burial trench | 1855 cholera | Middle/low status |
| USK 2340 | M | 30–40 | Simple burial trench | First half 19th century | Middle/low status |
| USK 2383 | F | 30–40 | Simple burial trench | First half 19th century | Middle/low status |
| USK 2379 | F | 20–30 | Simple burial trench | First half 19th century | Middle/low status |
| USK 2170 | F | 20–30 | Simple burial trench | First half 19th century | Middle/low status |
| p-Value | FDR-Adjusted p | Dental Calculus | Periodontitis Plaque | Saliva | Subgingival Plaque | Supragingival Plaque | |
|---|---|---|---|---|---|---|---|
| Shared species | |||||||
| Rothia aeria | 4.96 × 10−1 | 9.94 × 10−1 | 0.02 | 0.01 | 0.03 | 1.13 | 0.14 |
| Rothia dentocariosa | 4.15 × 10−3 | 2.05 × 10−2 | 0.21 | 0.00 | 0.27 | 2.66 | 9.09 |
| Corynebacterium durum | 7.79 × 10−3 | 3.57 × 10−2 | 0.15 | 0.01 | 0.05 | 0.42 | 3.14 |
| Bacteroides endodontalis | 4.50 × 10−3 | 2.18 × 10−2 | 0.01 | 2.35 | 0.18 | 4.83 | 0.14 |
| Solobacterium moorei | 3.77 × 10−1 | 9.23 × 10−1 | 0.01 | 0.04 | 0.02 | 0.00 | 0.00 |
| Rothia mucilaginosa | 9.37 × 10−3 | 4.21 × 10−2 | 0.03 | 0.01 | 0.27 | 0.26 | 0.13 |
| Capnocytophaga ochracea | 1.41 × 10−3 | 8.74 × 10−3 | 0.02 | 0.51 | 0.27 | 2.62 | 2.2 |
| Steptococcus oralis | 3.58 × 10−1 | 8.93 × 10−1 | 0.01 | 0.00 | 0.00 | 0.00 | 0.03 |
| Haemophilus parainfluenzae | 2.29 × 10−5 | 4.93 × 10−4 | 0.03 | 0.02 | 9.09 | 0.91 | 1.98 |
| Campylobacter rectus | 3.71 × 10−2 | 1.40 × 10−1 | 0.04 | 1.43 | 0.02 | 0.02 | 0.17 |
| Aggregatibacter segnis | 2.56 × 10−1 | 6.57 × 10−1 | 0.05 | 0.18 | 0.06 | 1.14 | 0.14 |
| Treponema socranskii | 1.70 × 10−2 | 7.01 × 10−2 | 0.52 | 2.40 | 0.04 | 0.45 | 0.52 |
| Neisseria subflava | 3.49 × 10−2 | 1.33 × 10−1 | 0.03 | 0.00 | 1.74 | 0.13 | 0.02 |
| Micrococcus luteus | 7.50 × 10−1 | 9.94 × 10−1 | 0.01 | 0.00 | 0.00 | 0.00 | 0.01 |
| Argemone mexicana | 8.06 × 10−1 | 9.94 × 10−1 | 0.03 | 0.00 | 0.00 | 0.01 | 0.00 |
| Desulfomicrobium orale | 5.74 × 10−1 | 9.94 × 10−1 | 0.06 | 0.01 | 0.00 | 0.00 | 0.00 |
| Kocuria palustris | 7.99 × 10−1 | 9.94 × 10−1 | 0.01 | 0.00 | 0.00 | 0.00 | 0.01 |
| Unique species | |||||||
| Lentzea albidocapillata1 | 3.05 × 10−2 | 1.20 × 10−1 | 0.27 | 0.00 | 0.00 | 0.00 | 0.00 |
| Enterococcus cecorum2 | 9.60 × 10−1 | 9.94 × 10−1 | 0.05 | 0.00 | 0.00 | 0.00 | 0.00 |
| Enterococcus faecium2 | 9.94 × 10−1 | 9.94 × 10−1 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 |
| Burkholderia gladioli3 | 9.94 × 10−1 | 9.94 × 10−1 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 |
| Inquilinus limosus3 | 6.76 × 10−1 | 9.94 × 10−1 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 |
| Methylobacterium mesophilicum1 | 9.60 × 10−1 | 9.94 × 10−1 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 |
| Microbacterium oxydans1 | 1.56 × 10−3 | 8.94 × 10−3 | 0.17 | 0.00 | 0.00 | 0.00 | 0.00 |
| Tsukamurella pulmonis3 | 2.44 × 10−1 | 6.35 × 10−1 | 0.07 | 0.00 | 0.00 | 0.00 | 0.00 |
| Necropsobacter rosorum1 | 4.38 × 10−1 | 9.94 × 10−1 | 0.05 | 0.00 | 0.00 | 0.00 | 0.00 |
| Staphylococcus sciuri1 | 9.60 × 10−1 | 9.94 × 10−1 | 0.02 | 0.00 | 0.00 | 0.00 | 0.00 |
| Arthrobacter scleromae1 | 4.34 × 10−1 | 9.94 × 10−1 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiago-Rodriguez, T.M.; Fornaciari, A.; Fornaciari, G.; Luciani, S.; Marota, I.; Vercellotti, G.; Toranzos, G.A.; Giuffra, V.; Cano, R.J. Commensal and Pathogenic Members of the Dental Calculus Microbiome of Badia Pozzeveri Individuals from the 11th to 19th Centuries. Genes 2019, 10, 299. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10040299
Santiago-Rodriguez TM, Fornaciari A, Fornaciari G, Luciani S, Marota I, Vercellotti G, Toranzos GA, Giuffra V, Cano RJ. Commensal and Pathogenic Members of the Dental Calculus Microbiome of Badia Pozzeveri Individuals from the 11th to 19th Centuries. Genes. 2019; 10(4):299. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10040299
Chicago/Turabian StyleSantiago-Rodriguez, Tasha M., Antonio Fornaciari, Gino Fornaciari, Stefania Luciani, Isolina Marota, Giuseppe Vercellotti, Gary A. Toranzos, Valentina Giuffra, and Raul J. Cano. 2019. "Commensal and Pathogenic Members of the Dental Calculus Microbiome of Badia Pozzeveri Individuals from the 11th to 19th Centuries" Genes 10, no. 4: 299. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10040299