


Genes 2024, 15(5), 555; https://0-doi-org.brum.beds.ac.uk/10.3390/genes15050555 (registering DOI) - 26 Apr 2024
Abstract
The ionic toxicity induced by salinization has adverse effects on the growth and development of crops. However, researches on ionic toxicity and salt tolerance in plants have focused primarily on cations such as sodium ions (Na+), with very limited studies on
[...] Read more.
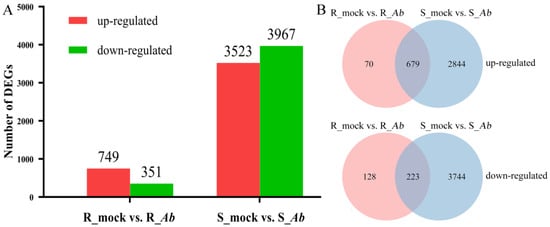
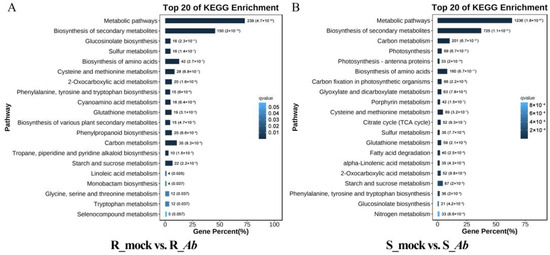
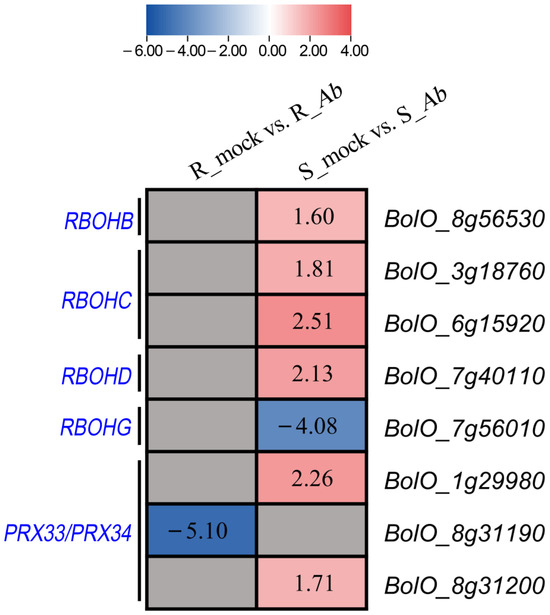
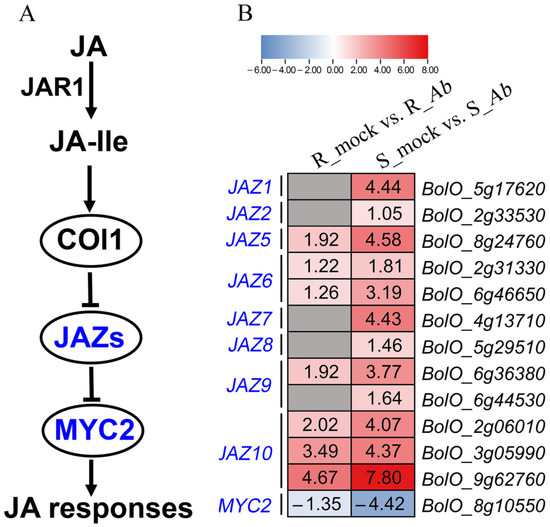
The ionic toxicity induced by salinization has adverse effects on the growth and development of crops. However, researches on ionic toxicity and salt tolerance in plants have focused primarily on cations such as sodium ions (Na+), with very limited studies on chloride ions (Cl−). Here, we cloned the homologous genes of Arabidopsis thaliana AtCLCc, GhCLCc-1A/D, from upland cotton (Gossypium hirsutum), which were significantly induced by NaCl or KCl treatments. Subcellular localization showed that GhCLCc-1A/D were both localized to the tonoplast. Complementation of Arabidopsis atclcc mutant with GhCLCc-1 rescued its salt-sensitive phenotype. In addition, the silencing of the GhCLCc-1 gene led to an increased accumulation of Cl− in the roots, stems, and leaves of cotton seedlings under salt treatments, resulting in compromised salt tolerance. And ectopic expression of the GhCLCc-1 gene in Arabidopsis reduced the accumulation of Cl− in transgenic lines under salt treatments, thereby enhancing salt tolerance. These findings elucidate that GhCLCc-1 positively regulates salt tolerance by modulating Cl− accumulation and could be a potential target gene for improving salt tolerance in plants.
Full article
(This article belongs to the Special Issue Cotton Genes, Genetics, and Genomics)


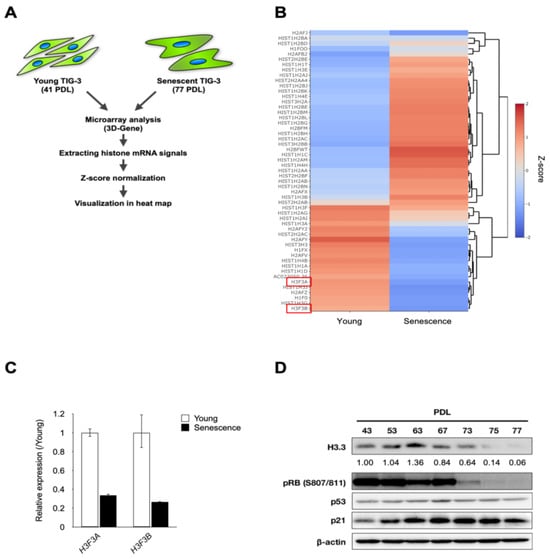
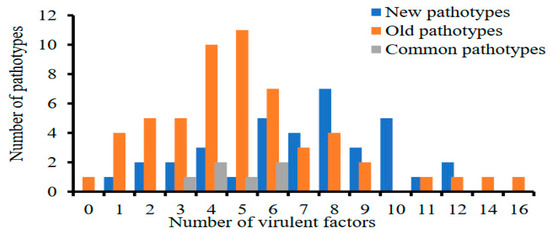
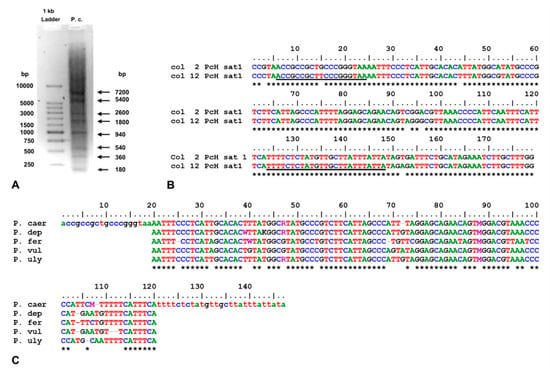
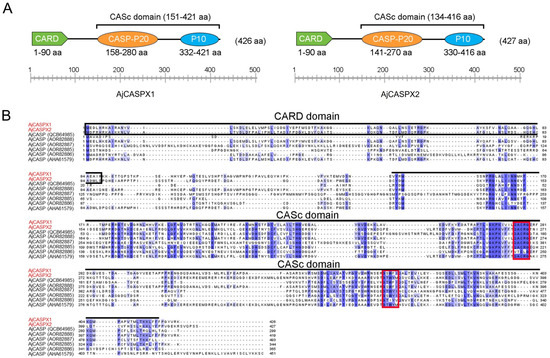





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}