Enhancing Terminal Deoxynucleotidyl Transferase Activity on Substrates with 3′ Terminal Structures for Enzymatic De Novo DNA Synthesis

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Construction
2.2. Protein Expression and Purification of MBP-Fused TdT
2.3. Determination of Enzyme Melting Temperatures
2.4. Extension of Oligonucleotides by Free 2′,3′-Dideoxyribonucleoside-5′-Triphosphates (ddNTPs) Using TdT Variants
2.5. Preparation of TdT-dTTP Conjugates
2.6. Extension of Oligonucleotides Using TdT-dTTP Conjugates
2.7. Capillary Electrophoresis and Data Analysis
3. Results
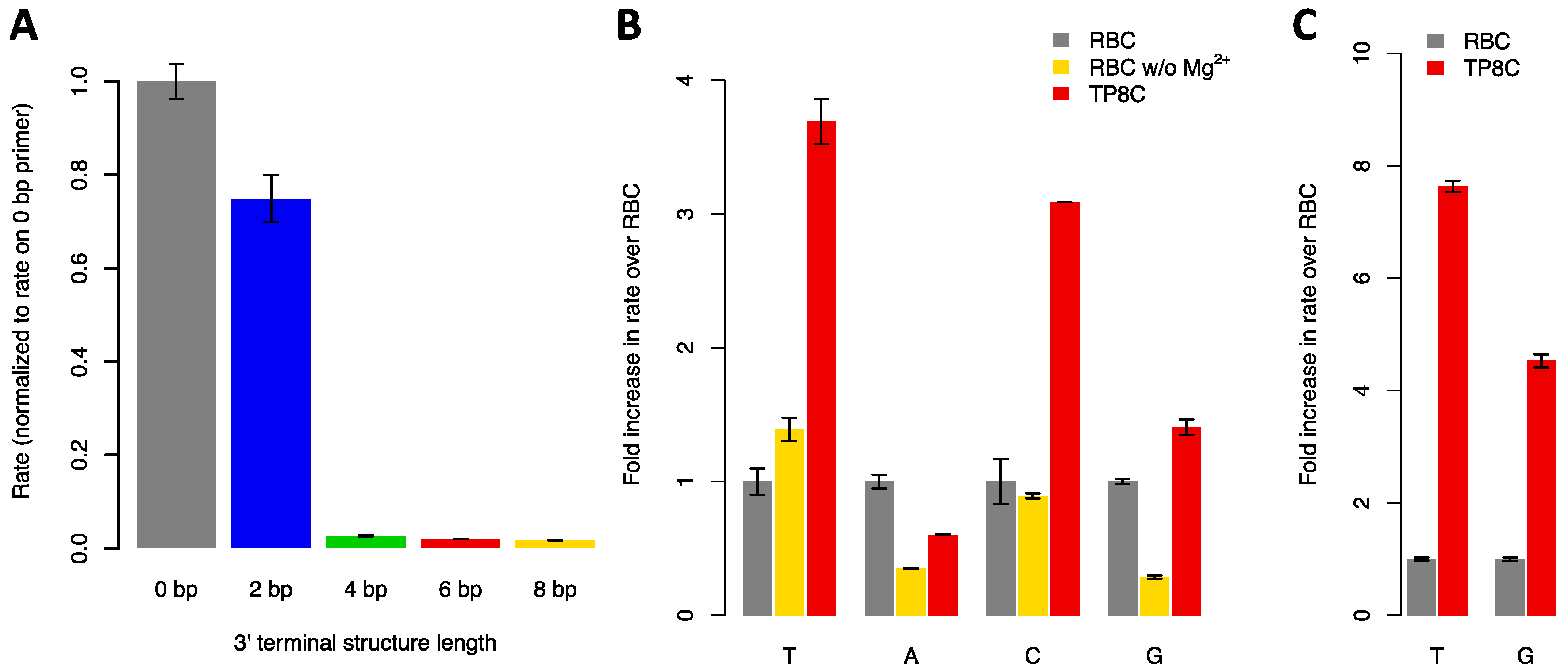
3.1. Formation of 3′ Terminal DNA Structures Inhibits Substrate Elongation by TdT
3.2. Substitution of Mg2+ Cofactor with Co2+ Improves TdT Activity on Hairpin Substrates
3.3. Computational Protein Design Yields a TdT Variant with Enhanced Thermostability
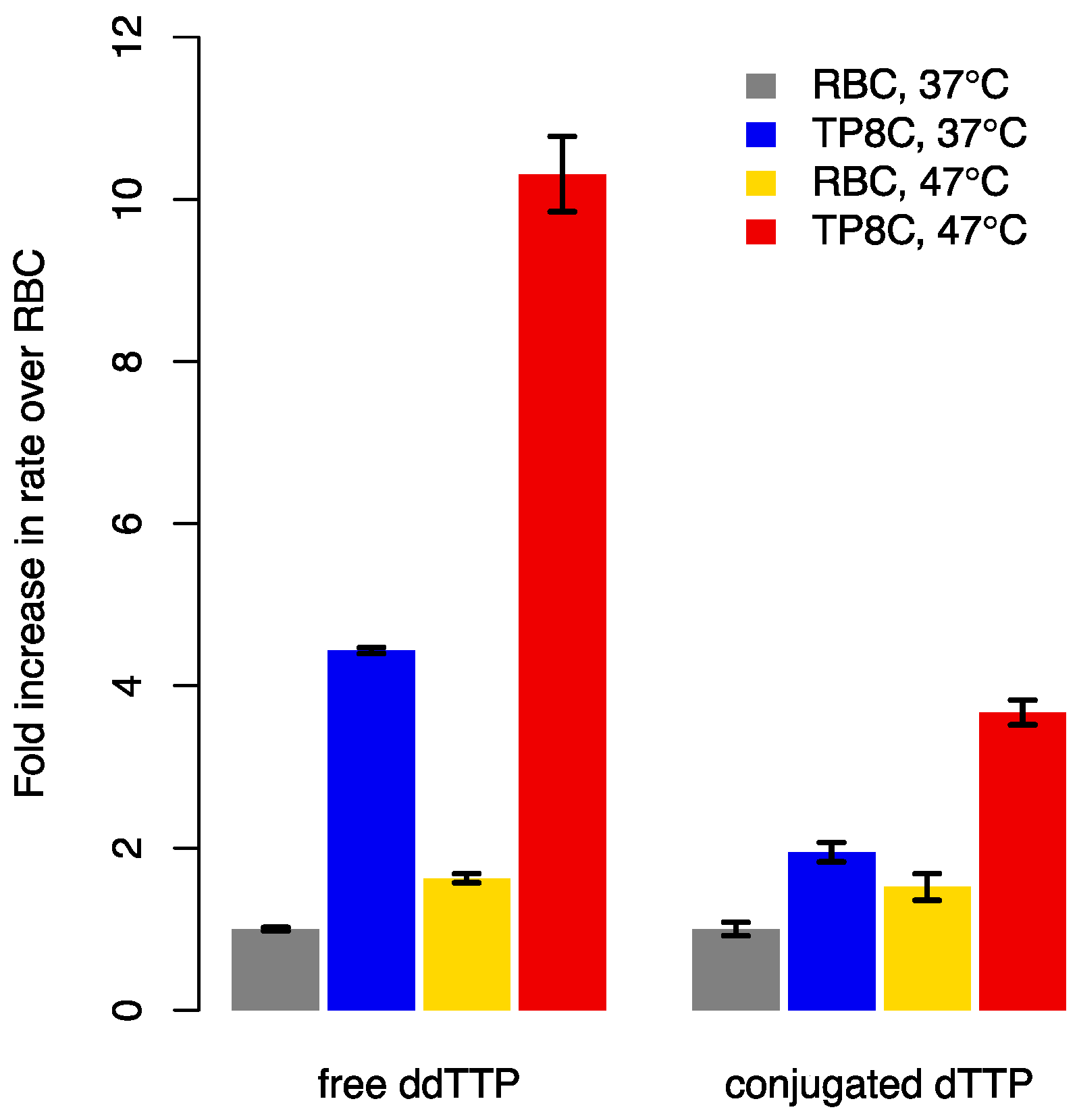
3.4. Elevated Reaction Temperature and Optimized Divalent Ion Concentrations Synergistically Accelerate Incorporation of Free and Conjugated Nucleotides into a Hairpin Primer
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Church, G.M.; Gao, Y.; Kosuri, S. Next-Generation Digital Information Storage in DNA. Science 2012, 337, 1628. [Google Scholar] [CrossRef] [Green Version]
- Caruthers, M.H. The Chemical Synthesis of DNA/RNA: Our Gift to Science. J. Biol. Chem. 2013, 288, 1420–1427. [Google Scholar] [CrossRef] [Green Version]
- Menezes, A.A.; Cumbers, J.; Hogan, J.A.; Arkin, A.P. Towards synthetic biological approaches to resource utilization on space missions. J. R. Soc. Interface 2015, 12, 20140715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, A.S.; Yang, H.; Montemagno, C. 3′-O-Caged 2′-Deoxynucleoside Triphosphates for Light-Mediated, Enzyme-Catalyzed, Template-Independent DNA Synthesis. Curr. Protoc. Nucleic Acid Chem. 2017, 71, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Palluk, S.; Arlow, D.H.; de Rond, T.; Barthel, S.; Kang, J.S.; Bector, R.; Baghdassarian, H.M.; Truong, A.N.; Kim, P.W.; Singh, A.K.; et al. De novo DNA synthesis using polymerase-nucleotide conjugates. Nat. Biotechnol. 2018, 36, 645. [Google Scholar] [CrossRef] [PubMed]
- Mathews, A.S.; Yang, H.; Montemagno, C. Photo-cleavable nucleotides for primer free enzyme mediated DNA synthesis. Org. Biomol. Chem. 2016, 14, 8278–8288. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Kalhor, R.; Goela, N.; Bolot, J.; Church, G.M. Terminator-free template-independent enzymatic DNA synthesis for digital information storage. Nat. Commun. 2019, 10, 2383. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.A.; Griffin, P.B.; Davis, R.W. Free-running enzymatic oligonucleotide synthesis for data storage applications. bioRxiv 2018, 355719. [Google Scholar] [CrossRef] [Green Version]
- Bollum, F.J. 5. Terminal Deoxynucleotidyl Transferase. In The Enzymes; Boyer, P.D., Ed.; Academic Press: Cambridge, MA, USA, 1974; Volume 10, pp. 145–171. [Google Scholar]
- Bednar, D.; Beerens, K.; Sebestova, E.; Bendl, J.; Khare, S.; Chaloupkova, R.; Prokop, Z.; Brezovsky, J.; Baker, D.; Damborsky, J. FireProt: Energy- and Evolution-Based Computational Design of Thermostable Multiple-Point Mutants. PLOS Comput. Biol. 2015, 11, e1004556. [Google Scholar] [CrossRef]
- Musil, M.; Stourac, J.; Bendl, J.; Brezovsky, J.; Prokop, Z.; Zendulka, J.; Martinek, T.; Bednar, D.; Damborsky, J. FireProt: Web server for automated design of thermostable proteins. Nucleic Acids Res. 2017, 45, W393–W399. [Google Scholar] [CrossRef]
- Gouge, J.; Rosario, S.; Romain, F.; Beguin, P.; Delarue, M. Structures of Intermediates along the Catalytic Cycle of Terminal Deoxynucleotidyltransferase: Dynamical Aspects of the Two-Metal Ion Mechanism. J. Mol. Biol. 2013, 425, 4334–4352. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Partch, C.L. Current Protocols in Protein Science: Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 2015, 79, 28.29.1–28.29.14. [Google Scholar] [CrossRef] [PubMed]
- Bollum, F.J. Thermal Conversion of Nonpriming Deoxyribonucleic Acid to Primer. J. Biol. Chem. 1959, 234, 2733–2734. [Google Scholar] [PubMed]
- Chang, L.M.; Bollum, F.J. Multiple roles of divalent cation in the terminal deoxynucleotidyltransferase reaction. J. Biol. Chem. 1990, 265, 17436–17440. [Google Scholar] [PubMed]
- Robbins, D.J.; Barkley, M.D.; Coleman, M.S. Interaction of terminal transferase with single-stranded DNA. J. Biol. Chem. 1987, 262, 9494–9502. [Google Scholar]
- Yang, B.; Gathy, K.N.; Coleman, M.S. Mutational analysis of residues in the nucleotide binding domain of human terminal deoxynucleotidyl transferase. J. Biol. Chem. 1994, 269, 11859–11868. [Google Scholar]
- Romain, F.; Barbosa, I.; Gouge, J.; Rougeon, F.; Delarue, M. Conferring a template-dependent polymerase activity to terminal deoxynucleotidyltransferase by mutations in the Loop1 region. Nucleic Acids Res. 2009, 37, 4642–4656. [Google Scholar] [CrossRef] [Green Version]
- Roychoudhury, R.; Jay, E.; Wu, R. Terminal labeling and addition of homopolymer tracts to duplex DNA fragments by terminal deoxynucleotidyl transferase. Nucleic Acids Res. 1976, 3, 863–878. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.-i.; Gonalves, J.M.; Houts, G.E.; Bollum, F.J. Deoxynucleotide-polymerizing Enzymes of Calf Thymus Gland: II. Properties of the terminal deoxynucleotidyltransferase. J. Biol. Chem. 1967, 242, 2780–2789. [Google Scholar]
- Eichhorn, G.L. Metal Ions as Stabilizers or Destabilizers of the Deoxyribonucleic Acid Structure. Nature 1962, 194, 474–475. [Google Scholar] [CrossRef]
- Every, A.E.; Russu, I.M. Influence of Magnesium Ions on Spontaneous Opening of DNA Base Pairs. J. Phys. Chem. B 2008, 112, 7689–7695. [Google Scholar] [CrossRef] [PubMed]
- Boule, J.B.; Rougeon, F.; Papanicolaou, C. Comparison of the two murine terminal deoxynucleotidyltransferase isoforms. J Biol. Chem. 2000, 275, 28984–28988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delarue, M.; Boulé, J.B.; Lescar, J.; Expert-Bezançon, N.; Jourdan, N.; Sukumar, N.; Rougeon, F.; Papanicolaou, C. Crystal structures of a template-independent DNA polymerase: Murine terminal deoxynucleotidyltransferase. EMBO J. 2002, 21, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Owczarzy, R.; Moreira, B.G.; You, Y.; Behlke, M.A.; Walder, J.A. Predicting Stability of DNA Duplexes in Solutions Containing Magnesium and Monovalent Cations. Biochemistry 2008, 47, 5336–5353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barthel, S.; Palluk, S.; Hillson, N.J.; Keasling, J.D.; Arlow, D.H. Enhancing Terminal Deoxynucleotidyl Transferase Activity on Substrates with 3′ Terminal Structures for Enzymatic De Novo DNA Synthesis. Genes 2020, 11, 102. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010102
Barthel S, Palluk S, Hillson NJ, Keasling JD, Arlow DH. Enhancing Terminal Deoxynucleotidyl Transferase Activity on Substrates with 3′ Terminal Structures for Enzymatic De Novo DNA Synthesis. Genes. 2020; 11(1):102. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010102
Chicago/Turabian StyleBarthel, Sebastian, Sebastian Palluk, Nathan J. Hillson, Jay D. Keasling, and Daniel H. Arlow. 2020. "Enhancing Terminal Deoxynucleotidyl Transferase Activity on Substrates with 3′ Terminal Structures for Enzymatic De Novo DNA Synthesis" Genes 11, no. 1: 102. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010102