Schizosaccharomyces pombe Assays to Study Mitotic Recombination Outcomes

1
Biology Program, The Ohio State University, Marion, OH 43302, USA
2
The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210, USA
3
Department of Molecular Genetics, The Ohio State University, Marion, OH 43302, USA
*
Author to whom correspondence should be addressed.
†
Denotes equal contribution.
Genes 2020, 11(1), 79; https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010079
Submission received: 20 December 2019
/
Revised: 7 January 2020
/
Accepted: 7 January 2020
/
Published: 10 January 2020
(This article belongs to the Special Issue Recent Advances in Molecular Genetics Technologies: A Themed Issue Honouring Professor Werner Arber on the Occasion of his 90th Birthday)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The fission yeast—Schizosaccharomyces pombe—has emerged as a powerful tractable system for studying DNA damage repair. Over the last few decades, several powerful in vivo genetic assays have been developed to study outcomes of mitotic recombination, the major repair mechanism of DNA double strand breaks and stalled or collapsed DNA replication forks. These assays have significantly increased our understanding of the molecular mechanisms underlying the DNA damage response pathways. Here, we review the assays that have been developed in fission yeast to study mitotic recombination.
1. Introduction
An accurate DNA damage response (DDR) is fundamentally important for cellular homeostasis. The DDR involves first detection of the damage and activation of the DNA damage checkpoints, then recruitment of the repair machinery and repair of the damage. In humans, defects in DDR can lead to a variety of syndromes and diseases [1].
The DNA double strand break (DSB) constitutes severing of the chromosome into two parts. Threatening the viability of a cell, DSBs can lead to loss of essential chromosomal regions. In multicellular organisms, unrepaired breaks may cause cells to undergo apoptosis and new cells can be regenerated [2,3]. However, inappropriate repair of breaks may give rise to translocations, deletions, duplications and inversions [4,5] which have been identified in cancer cells [6].
DSBs may be produced by endogenous or exogeneous sources but are generally repaired by the same mechanisms [7]. Most endogenous breaks occur as a consequence of replication stress. DNA replication forks can stall or collapse as they pass through heterochromatin regions [8,9], collide with RNA polymerases [10] or pass through other fragile sites characterized by repetitive elements or euchromatin to chromatin boundaries [11,12]. The recombination machinery evolved to rescue stalled or collapsed replication forks [13].
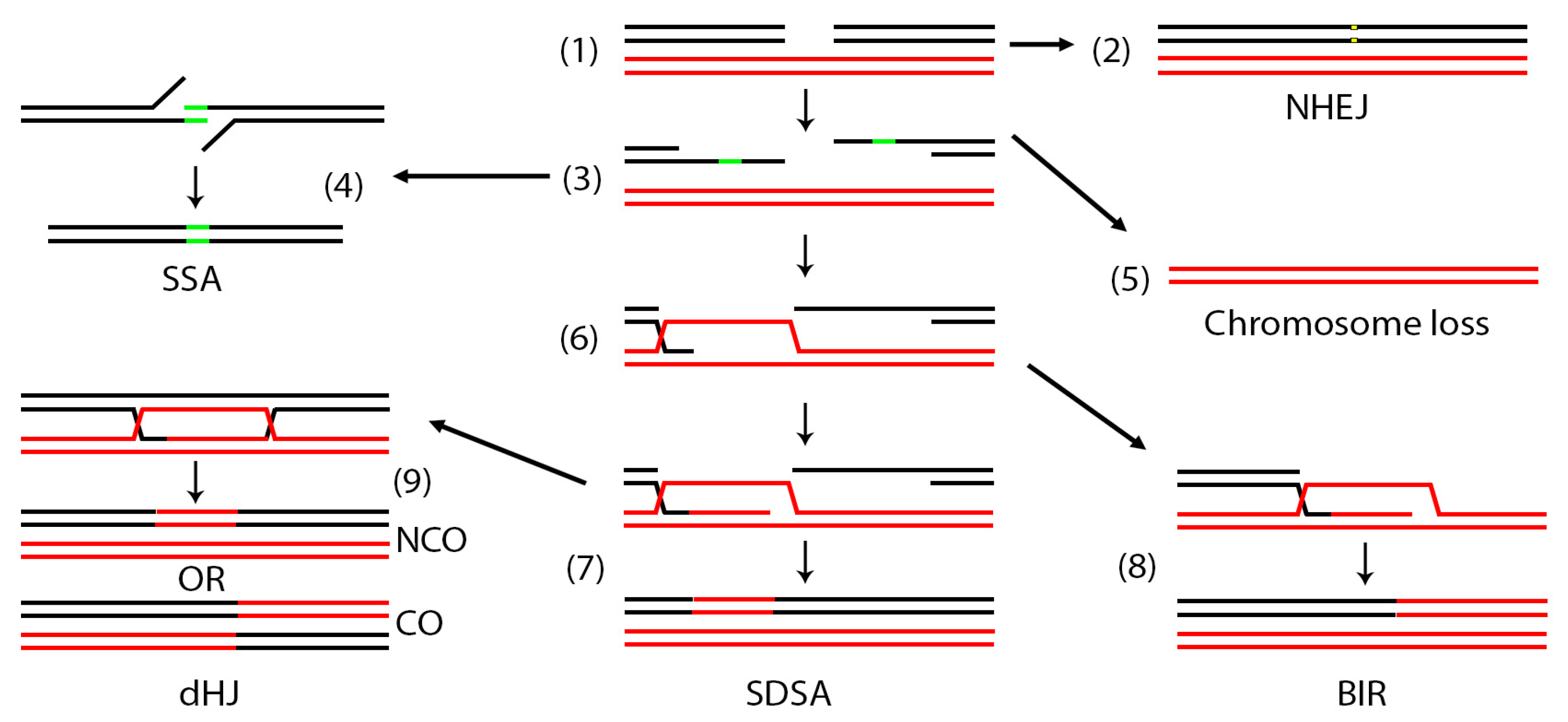
The multitude of repair mechanisms that eukaryotic cells evolved to deal with these breaks have been previously extensively reviewed [7,10,14,15,16,17,18,19] and can be generally broken into two major pathways, non-homologous end joining (NHEJ) and homologous recombination (HR). NHEJ involves localized repair of breaks with no major sequence rearrangements. Repair can result in small deletions or alterations of the sequences neighboring the break so NHEJ is considered an error-prone form of repair [20]. HR can be subdivided into several mechanisms that are both genetically and biochemically related such as single strand annealing (SSA), break induced replication (BIR) and the two closely related gene conversion (GC) mechanisms: synthesis dependent strand annealing (SDSA) and double Holliday junction (dHJ) (Figure 1) [7]. HR has been traditionally considered error proof because it uses an intact template sequence to copy the missing or broken region although some pathways of HR can be quite mutagenic [21].
Schizosaccharomyces pombe (fission yeast) diverged from Saccharomyces cerevisiae (budding yeast) approximately a billion years ago [22,23]. Many genes have been identified in S. pombe that show similarity to genes involved in human disease [24]. This is comparable to the level of conservation between S. cerevisiae and humans [25]. However, S. pombe appears to show higher conservations in chromosome structure and function genes [26] making S. pombe a great model system for studying chromosomal dynamics.
Specific to the study of chromosomal double strand break repair, fission yeast is great for several reasons. First, DDR genes are highly conserved from yeast to humans [27,28]. Second, in fission yeast, repetitive DNA elements, which are often a reason for endogenous breaks in human cells [29], are found only at centromeres and telomeres [30]. The overall structure of centromeres is conserved between yeast and humans. We and others have previously shown that replication trough the centromere does lead to chromosome breakage [31,32,33,34,35]. The presence of these repeats in only a few well characterized regions allows the better monitoring of the events that govern DSB repair. Third, haploid fission yeast has only three chromosomes [36] making it easier to track translocations and other aberrations resulting from inappropriate break repair. Fourth, fission yeast is as well suited for genetic screens as baker’s yeast [37]. Finally, working with fission yeast is inexpensive and easy to learn.
A plethora of assays have been developed to study DNA damage in various systems and some are described in several recent elegant reviews [38,39,40,41]. Here, we focus on S. pombe and review primarily the in vivo assays that have been developed in this system to study mitotic recombination. We briefly summarize these assays and direct the reader to the original publications for more details.
2. Mini-Chromosome Assays
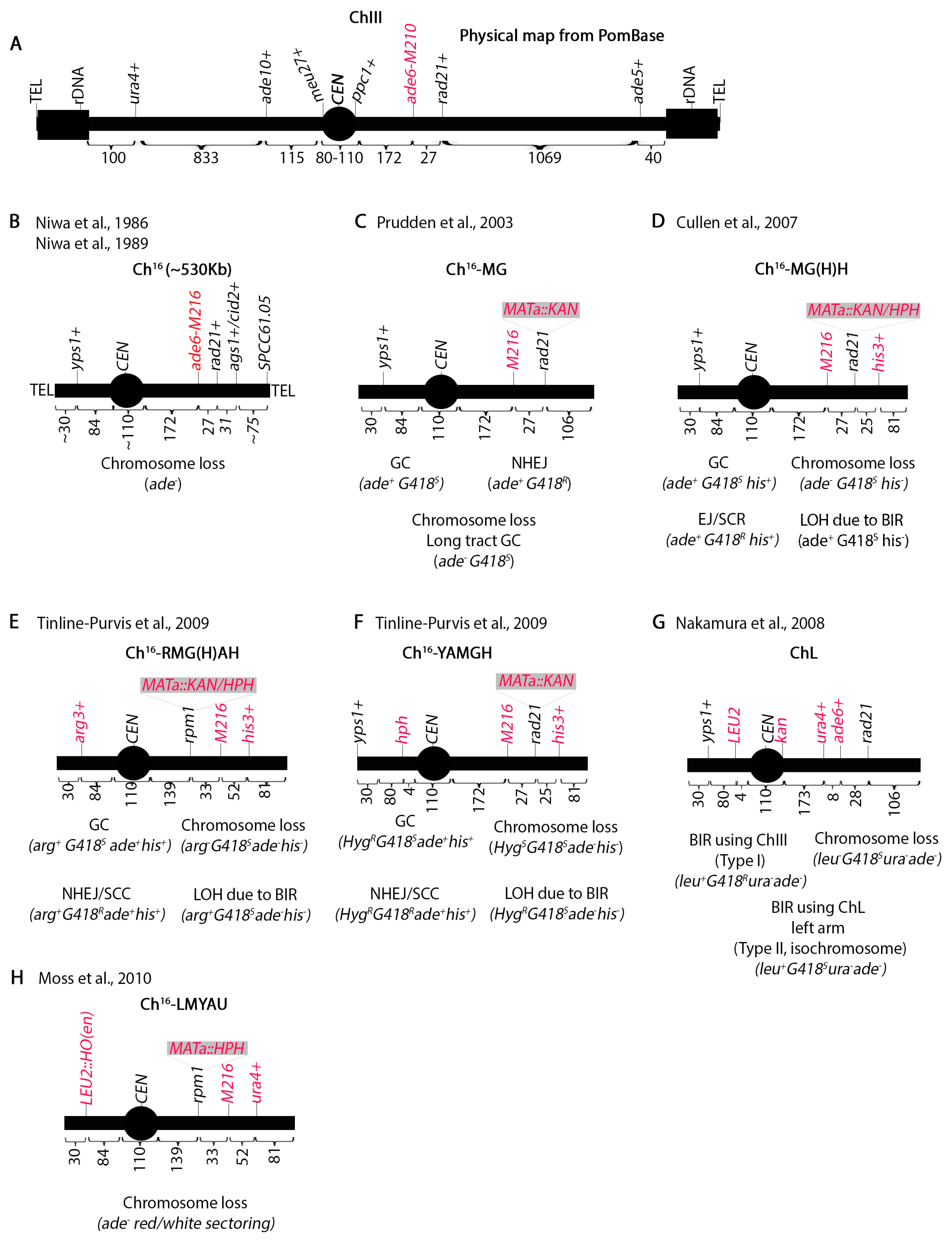
Mini-chromosome assays have been instrumental in elucidating many recombination pathways. Chromosome III (Ch.III), the smallest S. pombe chromosome, houses rDNA repeats at both ends next to the telomeres and has the longest centromere [36] (Figure 2A). Using γ irradiation, Niwa et al. [42,43] isolated a truncated Ch.III (termed Ch16) in haploid yeast (Figure 2B). Unlike S. cerevisiae which has point centromeres [44], S. pombe centromeres are characterized by repetitive elements resembling higher metazoans [30]. Since both the centromere and the rDNA repeats fluctuate in number between strains, the size of Ch.III cannot be determined precisely making it approximately 3.5 Mb. Initial genetic maps estimated the centromere to be 100 Kb [45,46,47] but latter structural analysis identified a 110 Kb centromere [48]. The fission yeast genome sequence [24] set the centromere size at 68 Kb with the caveat that seven 6760 bp repeats were missing. The addition of these repeats to the 68 Kb region brings the size of CEN III to 110 Kb which is what PomBase (the scientific database for fission yeast) reports [49,50]. The centromere spans between the meu27+ and ppc1+ loci (Figure 2A).
The truncated mini-chromosome Ch16 should retain an intact Ch.III centromere but not have rDNA repeats. Pulse field gel electrophoresis shows that this mini-chromosome is about 530 Kb [42,43,51]. The chromosome can be maintained in haploid yeast by intra-allelic complementation between ade6-M210 (Ch.III) and ade6-M216 (Ch16) (Figure 2A,B). Genetic mapping by Niwa et al. [42] showed that the left arm does not include the ade10+ locus but the yps1+ locus is present [51]. There are approximately 30 Kb between yps1+ and ade10+ and 84 Kb between yps1+ and meu27+, so it appears that the left arm is about 110 Kb. On the right arm, ade6+ is 172 Kb from ppc1+. Genetic mapping shows the presence of the ags1+ locus (formerly tps16+ [52]) 58 Kb telomere proximal from ade6+ but not tps14+ which is approximately 200 Kb from ags1+ [42]. cid2+ is within 5 Kb of ags1+. Southern blotting has shown the presence of the SPCC61.05 locus 75 Kb from ags1+ [53]. Thus, the right arm is about 305 Kb.
Niwa et al. determined that Ch16 is stable in mitotically dividing haploid cells at one copy per cell but unstable at two copies [42]. Remarkably, cells were able to simultaneously stably propagate both Ch16 and a 100 Kb shorter derivative (Ch16D1) suggesting that yeast cells may be able to determine ploidy by the size of the chromosome. Ch16 loss monitored by appearance of adenine auxotrophs occurred at a frequency of 1 in 104 cells.
The Humphrey lab has produced several derivatives of Ch16 to study recombination outcomes in vivo. They initially placed the S. cerevisiae homothallic endonuclease (HO) restriction site (MATa) marked with the kanamycin antibiotic resistance gene (KAN) at the rad21+ locus 27 Kb telomere distal of ade6-M216 producing Ch16-MG (Figure 2C) [54]. KAN confers resistance to G418 (Geneticin). Upon induction of a single DSB, gene conversion of the KAN locus can be monitored by screening for adenine prototrophic and G418 sensitive (ade+G418S) colonies while chromosome loss will lose both markers. Long track gene conversion produces the same phenotype as chromosome loss, so it was necessary to distinguish between the two by pulse field gel electrophoresis (PFGE). Similarly, because an ade+G418R phenotype could result from repair by NHEJ or failure of HO cutting, it was necessary to sequence across the DSB site.
Ch16-MG was improved by adding the his3+ marker at the cid2+ locus approximately 25 Kb from the MATa site creating Ch16-MGH (Figure 2D) [53]. The advantage of this is that BIR can be investigated by monitoring the loss of both KAN and his+ markers but retention of the adenine prototrophy (ade+G418Shis-). In BIR, the break is repaired by copying the missing information from Ch.III which is his3−. PFGE followed by southern blotting showed both a larger mini-chromosome termed Chx and the presence of the ade5+ marker, which could have only been transferred onto the Ch16 from Ch.III by BIR. A mini-chromosome with MATa-HPH marker (Ch16-MHH) was also constructed which behaved identically to Ch16-MGH. HPH confers resistance to hygromycin abbreviated as HYG in this paper. Bioneer has generated a deletion mutant library marked with KanMX [55] and this Ch16-MHH chromosome is a powerful tool in screening genes that affect repair.
A third marker (arg3+) was introduced on the left arm of Ch16-MGH at the yps1+ locus 84 Kb from the meu27+ locus. An additional modification was moving the MATa site 33 Kb left of the ade6-M216 marker. This created Ch16-RMGAH (Figure 2E) [51] which can monitor extensive BIR that results in loss of all markers on the right arm but retention of the arg3+ marker. Thus, the arg3+ marker distinguishes.
Extensive BIR from chromosome loss. To ensure that the new placement of the MATa and arg3+ marker on the left arm did not cause a locus-specific form of repair, they generated chromosome Ch16-YAMGH where the MATa site was left at the rad21+ locus and an HPH marker was put 4 Kb from the centromere (Figure 2F). Remarkably, PFGE analysis of the arg+G418Sade-his- or HYGRG418Sade-his- colonies showed that the two outcomes were identical suggesting that the mechanism of repair was independent of the break position.
Further analysis showed that following break induction, telomere distal sequences of the break are lost while extensive resection of the centromere proximal sequences occurs. Inverted centromeric repeats facilitate formation of an isochromosome which results from invasion and duplication of both the centromere and the left arm of the chromosome onto the right [51]. Nakamura et al. [33] constructed the ChL mini-chromosome (Figure 2G) to study these isochromosomes and they found that they can occur in the absence of a break. Both groups showed that Rad51 suppresses isochromosome formation [35,51]. Using the ChL chromosome, we have also shown that perturbation of the replication machinery or the centromere heterochromatin increases these isochromosome events. This suggests that isochromosomes originate from improperly repaired random breaks that occur during replication [31]. Remarkably, we also identified some more drastic copy number variations in double chromatin and replication mutants which entirely lost Ch.III and acquired other smaller chromosome fragments. The structure and mechanism of these smaller chromosome fragments remains to be elucidated.
Finally, Ch16-LMYAU was generated to screen Bioneer mutants for chromosome loss phenotypes (Figure 2H) [56]. Previously, the HO endonuclease was propagated on a plasmid but here the investigators introduced it at the yps1+ locus marked with the S. cerevisiae LEU2. The ura4+ marker was placed at the cid2+ locus. Chromosome loss was assayed by red/white sectoring in response to break induction. Detailed experimental procedures for using these assays were published [57]. Although we do not discuss it here, a chromosome loss assay in diploid cells has also been designed [58]. In addition to chromosome loss, this assay also monitors intra-homologue recombination.
3. Recombination at Repetitive Elements
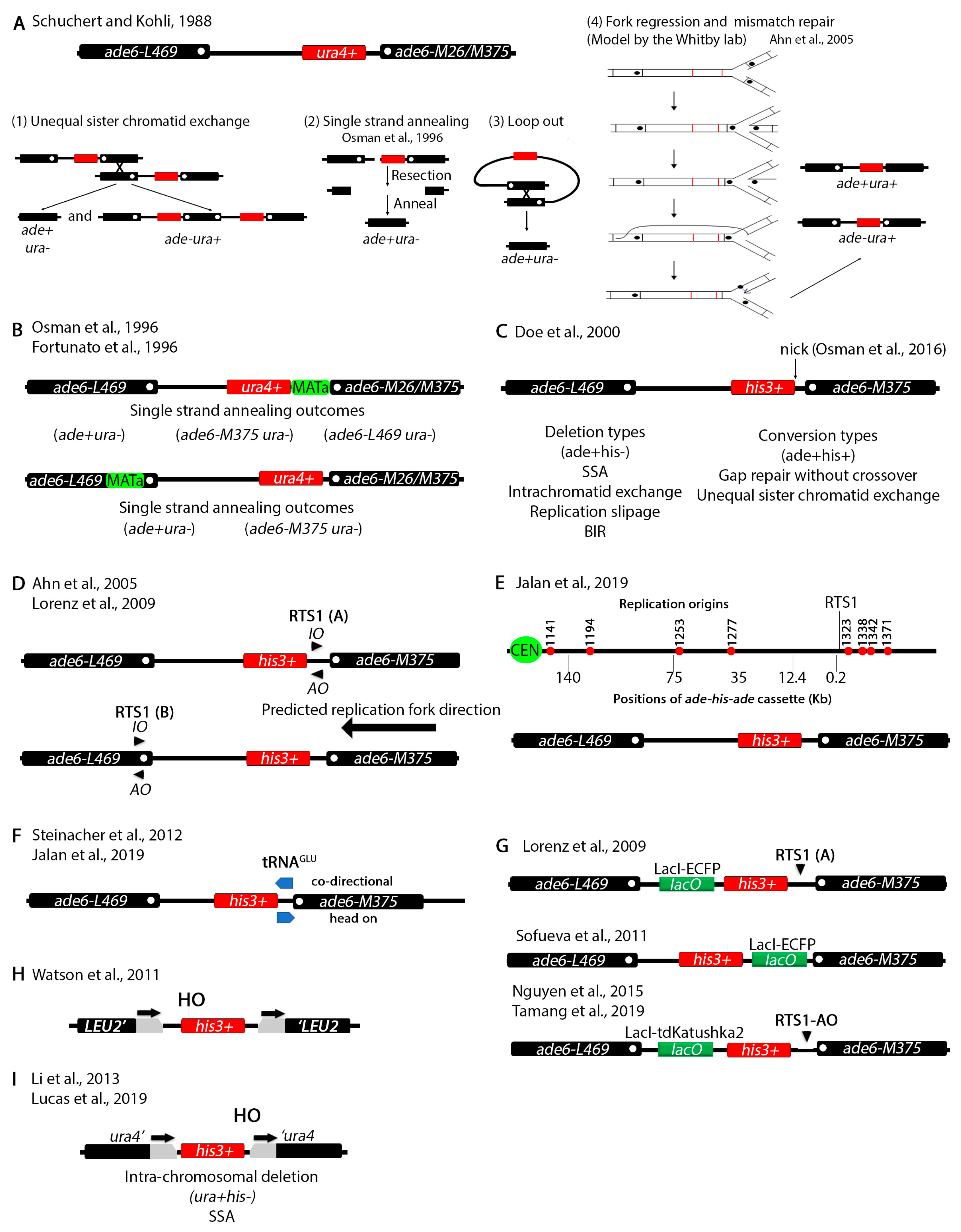
Another series of assays have been devised in S. pombe to study chromosomal recombination at non-tandem repeats (Figure 3). Repetitive elements had been previously shown to cause deletions, inversions or duplications [59,60]. It was known from studies in several species including S. cerevisiae that certain genetic mutations act as hotspots for meiotic recombination [61,62]. The ade6-M26 (G135T) allele in S. pombe creates such a hotspot [63]. Remarkably, the ade6-M375 (G132T) and the ade6-L469 (C1467T) as well as several other alleles did not serve as recombination hotspots. The G135T, G132T and C1467T mutations inactivate the gene because they introduce stop codons.
Schuchert and Kohli designed a clever non-tandem repeat assay to study crossover frequency at the ade6-M26 locus (Figure 3A) [64]. They positioned the ade6-L469 3′ end mutation on the left side and the ade6-M26 or ade6-M375 5′ end mutation on the right side while placing the functional ura4+ gene in between. Then, the investigators assayed for deletion or conversion. Deletion (ade+ura-) can be predicted to arise by several mechanisms. Unequal sister chromatid exchange should generate both ade-ura+ and ade+ura- phenotypes (Figure 3A(1)). A break between the two ade6 alleles, followed by resection in both directions past the mutations and annealing can also generate a deletion through SSA (Figure 3A(2)). Since both ade6 mutations are proximal to the ura4+ gene, it is possible to reconstitute a wildtype ade6+ allele. Deletion may also occur through an intra-chromosomal crossover and loopout (Figure 3A(3)). An ade+ura+ phenotype arises by replacing the mutation in one allele with the wildtype region of the other allele. The mechanism by which this happened remained elusive until later when the Whitby lab proposed a fork regression and mismatch repair model (see below).
To study mitotic recombination induced by a double strand break the Subramani group introduced the MATa site either in the unique sequence between ura4+ and ade6-M26/M375 or within the ade6-L469 allele (Figure 3B) [65,66]. Upon induction of the DSB, two outcomes predominated (ade+ura- and ade-ura-) which the authors explained to arise by SSA. Subsequent genotyping by restriction digestion or backcrossing identified the exact allele in the ade-ura- phenotypes. ade+ura+ and ade-ura+ recombinants (not shown) also arose at a much lower rate and only when the HO restriction site was placed within the ade6-L469 allele suggesting that the position of the break determines the repair mechanism. These assays were used to analyze the role of several recombination genes in DSB repair [65].
The Whitby lab has subsequently been influential in designing several variations of the tandem repeat assay and have been able to explain some of the more complex outcomes. These assays have been used to identify and characterize genes involved in DNA damage repair [67,68,69,70,71,72,73,74,75,76,77,78]. The investigators initially designed a system similar to the Schuchert and Kholi assay except that instead of ura4+ they placed a his3+ marker between the ade6 alleles (Figure 3C) [79]. This assay was used to show that the ura+his+ recombination outcomes may arise due to recombination dependent restart of stalled replication forks usually through BIR. Inactivation of various cellular processes such as Holliday junction resolution or the checkpoint changes the recombination outcomes [70,79,80,81,82]. In a more recent report [83], the Whitby lab introduced a nicking site for the gpII M13 phage enzyme. This enzyme makes a nick that is converted to a DSB by the replication fork which approaches from the right (see Figure 3D,E) [84,85]. This assay was used to study restart of stalled replication forks due to the DSB produced by this nick.
To further investigate the effect of replication fork stalling on recombination outcomes, the Whitby lab modified the assay by placing RTS1, the naturally occurring replication fork termination site at the mating type loci [86], either between the his3+ and the ade6-M375 alleles or within the ade6-L469 allele (Figure 3D) [87,88]. RTS1 is polar meaning that it can only stall forks in one direction, so strains were constructed with different RTS1 orientations, RTS1-AO (active orientation) and RTS1-IO (inactive orientation). A cluster of origins of replication are found to the right of the ade6-M375 allele and the replication fork is predicted to approach from the right (Figure 3E). Two-dimensional gel electrophoresis showed that the RTS1-AO efficiently stalls replication forks [89]. The investigators used this assay to study the function of various recombination genes in fork restart and to show that rescue of collapsed replication forks can cause BIR dependent template switching that can generate chromosomal rearrangements. [76,87,88,90,91]. A protocol was published with extensive details on the use of these elegant assays [92].
By inserting the ade-his-ade cassette at different distances from the RTS1 pause site (Figure 3E) the investigators showed that template switching can occur up to 75 Kb from collapsed forks [93]. Collisions between Pol III which transcribes tRNA and replication can cause chromosomal rearrangements [94]. A tRNAGLU gene was inserted between the his3+ and the ade6-M375 allele to show that collisions between Pol III and replication machinery increases the frequency of recombination (Figure 3F) [91,93]. An increase in template switching was observed when both RTS1 and tRNAGLU were introduced in the same construct and the orientation of transcription of tRNAGLU faced RTS1 head on [93]. To monitor interaction of fluorescently tagged recombination genes (rad52+, rad51+, etc.), the LacO array was also introduced at different positions within the ade-his-ade repeat (Figure 3G) [88,89,95,96].
An assay to study SSA in S. pombe was designed by Watson et al. (Figure 3H) [97]. Two S. cerevisiae LEU2 fragments with overlapping regions were placed on either side of a functional his3+ gene. The MATa sequence was cloned at the 5′ end of his3+ right before the start codon. Using this assay, the investigators showed that SSA is rad52+ dependent, confirming previous findings. In their report Watson and colleagues also designed an elegant system that allows fast transcriptional induction in S. pombe. Historically, transcriptional induction in S. pombe relied on the nmt1 promoter which is repressed by thiamine. Removal of thiamine de-represses the promoter but it takes anywhere between 14–20 h for full induction [98,99]. The new system which is based on upregulation of the urg1 promoter allows induction within 30 min mirroring the S. cerevisiae GAL induction system [100]. The urg1 system was optimized in a subsequent publication [101]. Other systems for faster induction of gene expression in S. pombe that we do not discuss here have also been engineered more recently [102,103].
4. Chromosomal Rearrangements Caused by Stalled or Collapsed Replication Forks at Inverted Repeats
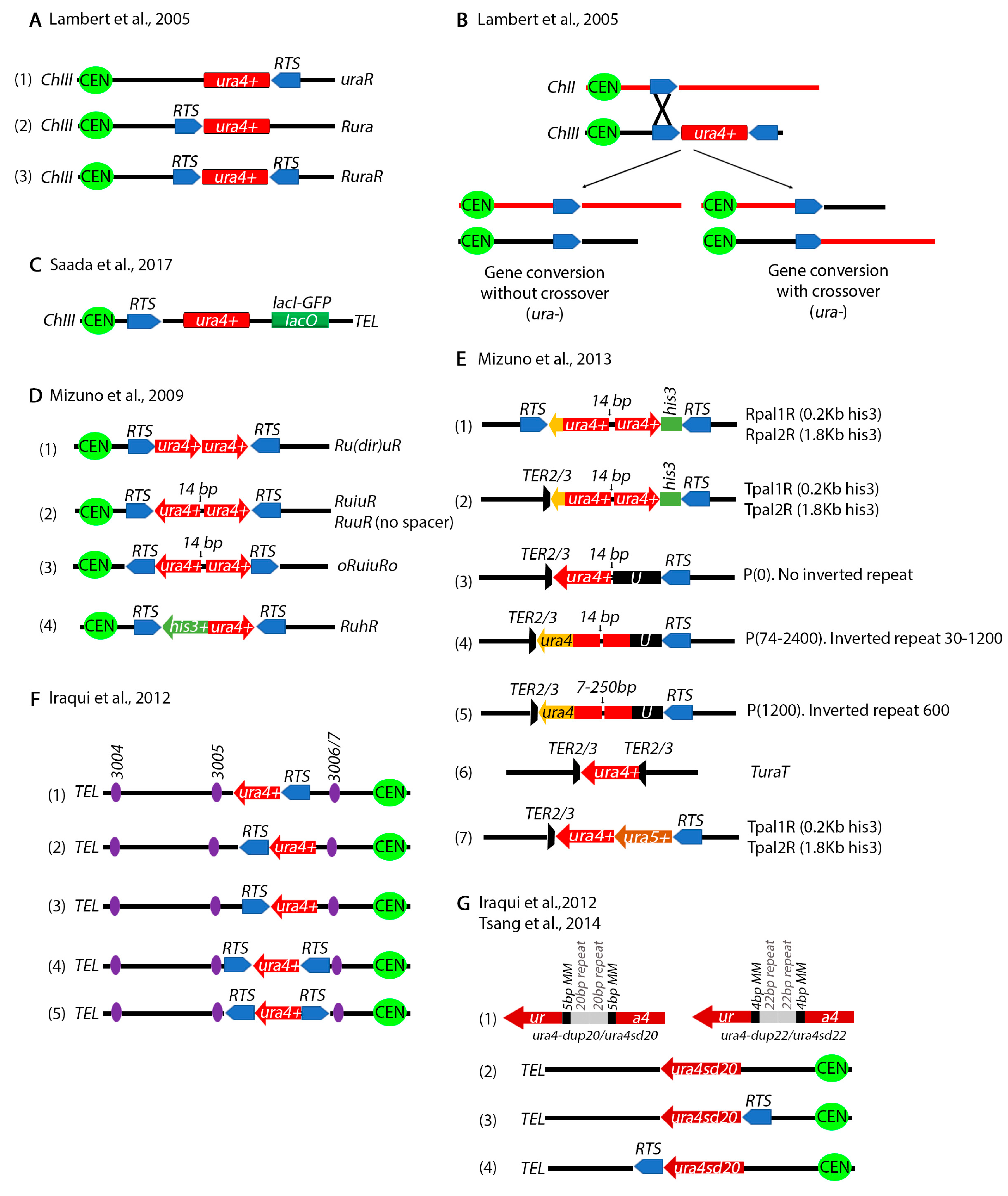
A series of other assays have been designed to study chromosomal rearrangements resulting from stalled replication forks by the Carr, Murray and Lambert labs. Carr and colleagues placed the RTS1 on either side of ura4+ gene on Ch.III (Figure 4A) [105]. Using 2-D gel electrophoresis they showed that these constructs can efficiently stall forks in the vicinity of the ura4+ gene. Deletion of several recombination genes including rad51+ decreases cell viability suggesting that homologous recombination is required for rescue of stalled forks. PFGE and PCR showed that some of the outcomes resulted in ura4+ loss through gene conversion without crossover, while others through a crossover between Ch.III and Ch.II produce a reciprocal translocation (Figure 4B). In all cases, information was exchanged between Ch.III RTS1 and the endogenous Ch.II RTS1. To monitor anaphase bridges by microscopy, the Lambert lab also placed a LacO array next to the RTS1 pause site (Figure 4C) [106].
Mizuno et al. engineered several repeat constructs flanked by RTS1 sites (Figure 4D) [107]. Using these constructs, the investigators showed that recombination dependent rescue of stalled replication forks at inverted repeats can produce dicentric and acentric isochromosomes. In subsequent and even more sophisticated studies, the investigators generated a series of more complex constructs to precisely analyze the mechanisms of chromosomal rearrangements at inverted repeats (Figure 4E) [108]. The length of the repeats as well as the gap between the repeats was varied, unique sequences were introduced at either side of the repeats, RTS1 sites were placed at different distances from the repeats and the TER2/3 ribosomal fork barrier was also tested. Their findings showed that inverted repeats cause forks to turn around or “execute a U-turn” and generate gross chromosomal rearrangements. Remarkably, the DNA damage checkpoint does not appear to detect the recombination intermediates that cause these rearrangements, at least in the cell cycle in which they occur [109].
The position of the RTS1 was varied relative to several Ch.III replication origions (Figure 4F) [110,111] to show that when a replication fork approaching from one of these origins collides with the RTS1 site, it causes deletions in addition to gross chromosomal translocations. To test for replication fork slippage, the investigators designed a construct where the ura4+ gene was interrupted by 20 bp repeats flanked by 5 bp of microhomology sequences (Figure 4G) [77,110]. A slightly modified construct has 22 bp repeats flanked by 4 bp of microhomology (Figure 4G). Both constructs inactivate the ura4+ gene. Microhomology mediated repair results in a functional ura4+. Using these constructs, it was shown that fork slippage, but not translesion synthesis or mismatch repair, is responsible for the restoration of the functional ura4+ cassette. The intra-S phase DNA damage checkpoint (Rad3) represses fork slippage and microhomology mediated repair at stalled forks [77]. Further, the NHEJ factor Ku appears to regulate recombination at arrested forks by controlling end resection [112].
5. Mating Type Loci Serve as a Natural Site for Studying Collapsed Replication Forks
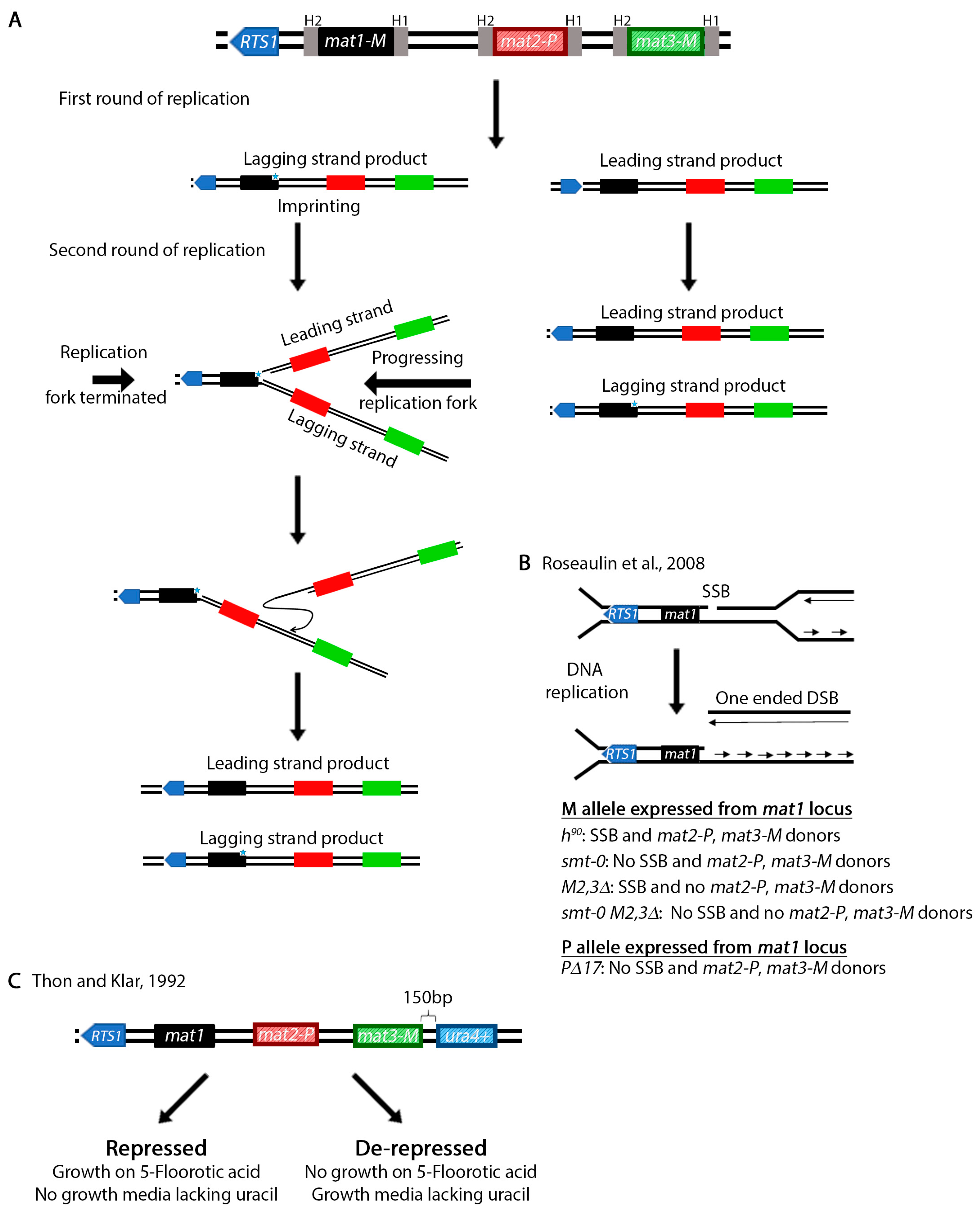
The mating type of a S. pombe cell is determined by the allele present at the mat1 cassette (Figure 5A) [113]. This allele can be either mat1M (M cell, M stands for minus) or mat1P (P cell, P stands for plus) [114,115]. Switching between mat1M and mat1P is accomplished by copying information from the silent mat2-P and mat3-M cassettes [116,117,118,119]. In addition to the allele present at the mat1 locus, the mating type of a population of cells is also determined by the ability to switch and the alleles present at the silent mat2-P and mat3-M regions. Wild type h90 cells can switch information at the mat1 locus and are mixture of M and P cells [120,121,122,123]. A population that has the M allele at the mat1 locus (mat1M) and lost the ability to switch is h-. A population may also be h- if it lost the mat2-P locus and repairs the mat1M with the same information (e.g., from mat3-M). Alternatively, a population of cells are h+ if the P allele is expressed from the mat1 locus (mat1P) and the strain is unable to switch (e.g., mat1PΔ17).
In wild type populations, switching occurs in only 25% of the cells [124]. This is because unlike in S. cerevisiae where switching is initiated by a double strand break generated by the HO endonuclease [125], in S. pombe switching begins with an imprint in the lagging strand during an initial round of DNA replication. Two concurrent models exist to explain the nature of the imprint. The imprint may be caused by the introduction of one or two ribonuclotides in the DNA sequence [126,127,128] or the generation of a single strand break [129] or possibly both at the same time. Regardless, the imprint is converted to a one-ended double strand break in the next round of replication [129,130,131,132,133]. However, only one of the replication forks may convert the imprint into a DSB. To ensure that the other replication fork does not pass through the imprint, replication is terminated by the above mentioned RTS1 site [86,134]. This process allows unidirectional transfer of information from the two mat2-P and mat3-M cassettes onto mat1 in only one of the four cells.
Investigation of the mating type loci in S. pombe, which increased our understanding of replication, recombination and gene silencing has been reviewed previously [120,135]. This natural system is a remarkable assay for the study of the role of DNA recombination in restart of collapsed replication forks. Perhaps even more important is the fact that the system produces one-ended DSBs which more accurately resemble the type of substrates resulting from collapsed replication forks [136]. Using wild type cells or several mating type defective mutants (Figure 5B) [130,137,138] the role of various DNA damage response genes have been investigated [136,139,140]. Work has also led to the identification of several recombination and replication genes, particularly the swi (switching) genes [117,118,141,142]. Additionally, various mechanisms of gene silencing were identified (reviewed in reference [143]). For example, unlike the centromeres, the mating type loci are silenced through an RNAi independent mechanism [144,145,146]. Work on silencing led to the identification of the clr (cryptic loci regulator) genes [147,148,149]. To investigate silencing, Thon and Klar placed the ura4+ gene 150 bp from the mat3 cassette [147] (Figure 5C). Finally, as Klar et al. point out in their review [135], the asymmetric mating type switching in S. pombe may also explain how genes are differentially regulated during development of higher eukaryotic organisms.
6. Other Fluorescence and Biochemical Assays
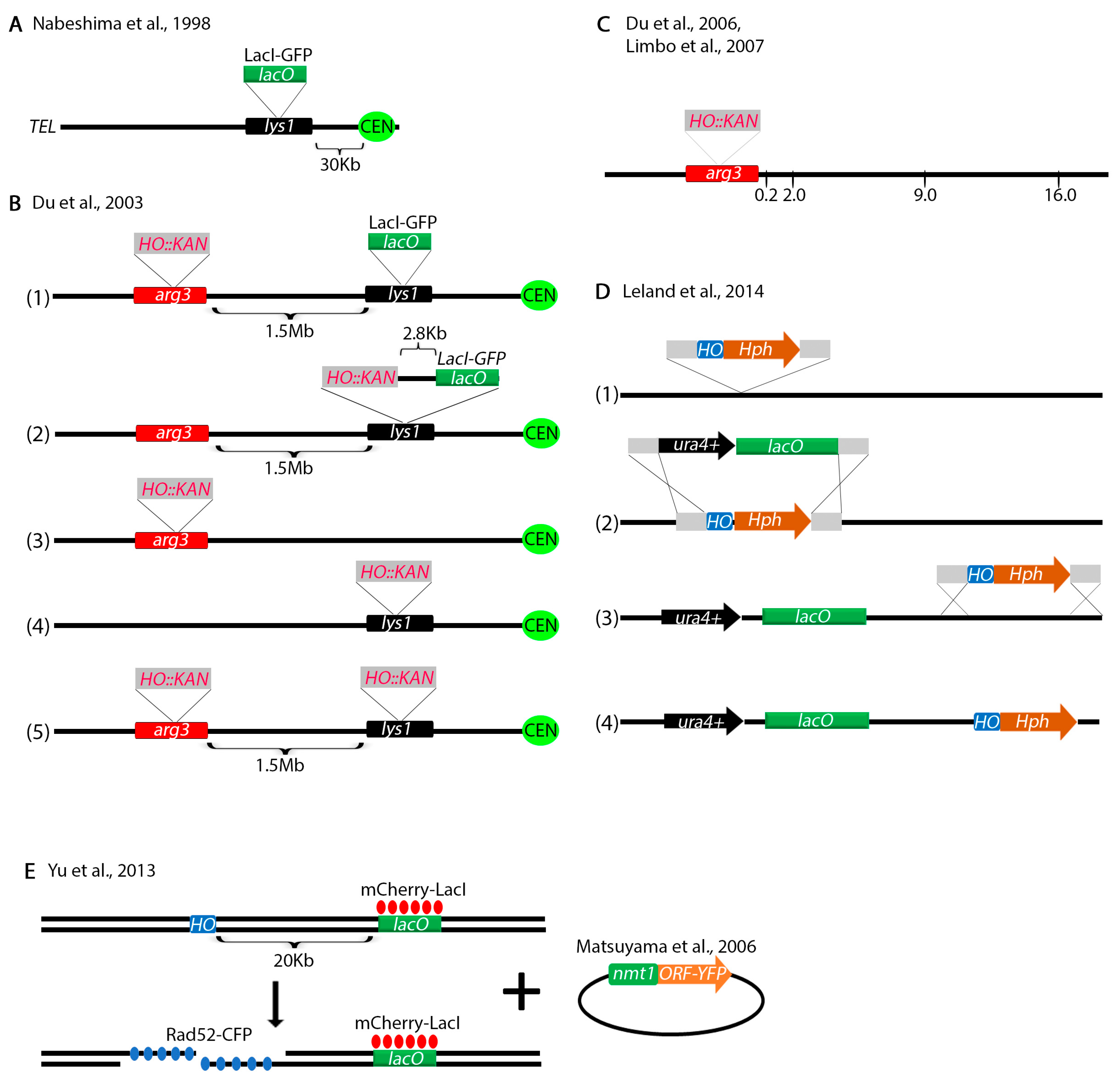
To investigate centromere dynamics, Nabeshima et al. placed a LacO array at the lys1+ locus, 30 Kb from the centromere (Figure 6A) [150]. The Russell lab modified this assay by placing the HO endonuclease restriction site 1.5 Mb away at the arg3+ locus within 2.8 Kb of the LacO array (Figure 6B) [151]. Using these assays, they showed that Crb2-YFP (yellow fluorescent protein) co-localizes with the LacI-GFP (green fluorescent protein) bound to the LacO array suggesting that Crb2 interacts with the chromosome at the site of the DSB. When the break was at the arg3+ locus, no co-localization was observed because the Crb2-YFP interacts with a DNA sequence too far from the LacO array. Several other constructs were made without the LacO array to investigate foci of various fluorescent-tagged proteins in response to a DSB (Figure 6B). For finer analysis of repair proteins interacting with the break, the investigators next turned to Chromatin IP. Primers were designed to amplify sequences by PCR at 0.2, 2.0, 9.0 and 16.0 Kb and the interaction of a variety of proteins with the break were tested biochemically (Figure 6C) [152,153]. Using these assays, the investigators have unraveled the role of various DNA damage checkpoint and repair proteins in processing of DSBs [136,152,154,155,156,157,158,159,160]. Not discussed here are some other biochemical assays that the Russell lab engineered, including some assays to study resection [161] with some adaptations from S. cerevisiae [162].
Another ingenious assay was designed by Leland and King to introduce a LacO array and the HO restriction site anywhere in the genome (Figure 6D) [163]. This technique requires several steps. First, the HO restriction site marked with the HPH marker and flanked by two homology regions is placed at a desired location in the genome (Figure 6D(1)). Then, a sequence harboring the LacO array marked with ura4+ and flanked by the same homologous sequences as the HO restriction site is integrated at the HO locus (Figure 6D(2)). The HO restriction site is re-integrated at a location neighboring the LacO array (Figure 6D(3)). This produces a construct with an HO restriction site next to the LacO array (Figure 6C(4)) similar to the Du et al. assay (Figure 6B). The advantage of this technique is that the LacO and the HO restriction site can be introduced anywhere in the genome.
Yu et al. has designed an assay to screen for proteins interacting with a DSB (Figure 6E) [164]. They engineered the HO endonuclease restriction site 20 Kb from a LacO array that binds LacI-Cherry. The strain also expresses Rad52-CFP (cyan fluorescent protein) which interacts with DSBs. Upon DSB induction, co-localization of red and blue foci indicates that Rad52-CFP interacted with the DSB. When they transformed a library of S. pombe YFP tagged proteins [165], they were able to screen for other proteins that interact with the DSB by monitoring co-localization of all three foci (red, blue, yellow).
7. Non-Homologous Repair
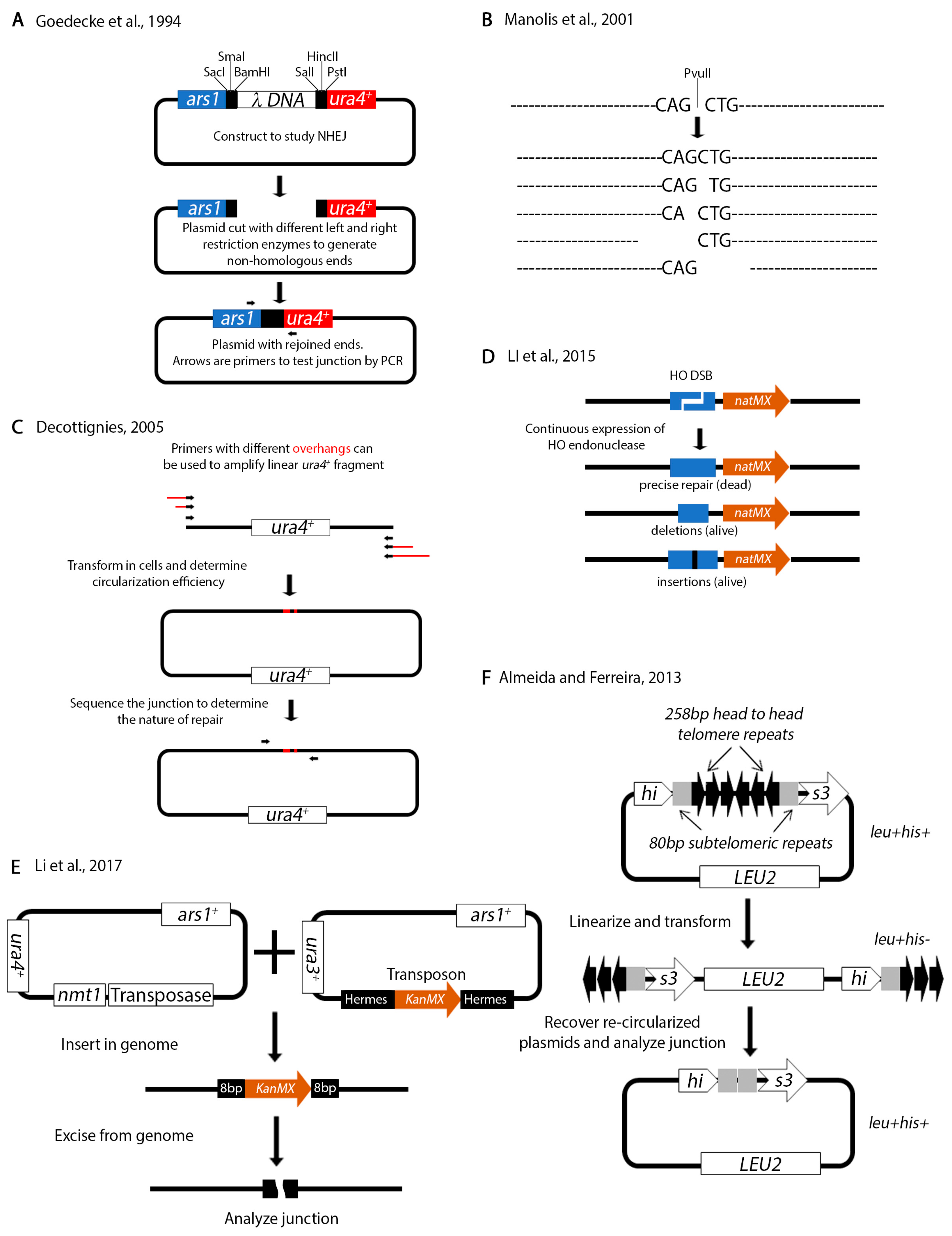
Non-homologous repair can occur in the absence of homologous regions. Several assays have been designed to test non-homologous repair in S. pombe. In an NHEJ assay designed by Goedecke and colleagues, a plasmid is linearized with various restriction sites to produce non-homologous ends (Figure 7A) [166]. The restriction sites used to generate one of the ends are different from those used to generate the other end, thus ensuring that the ends cannot be re-ligated. The linear fragment is transformed into living cells and allowed to re-circularize. The junction is amplified by PCR and sequenced. Using this assay, the investigators showed that the ends are resected up to 14 bps prior to rejoining. A similar assay by Manolis et al. relies on transforming linearized sequences with just one restriction enzyme (PvuII) (Figure 7B) [167]. Subsequent sequence analysis of rejoined products identified various sequence alterations at the junction. The investigators also showed that in S. pombe rad50+, mre11+ and the DNA damage checkpoint is not required for NHEJ. These plasmid type NHEJ assays were instrumental in demonstrating that key components of NHEJ are conserved form yeast to humans [168,169].
In a variation of these assays, Anabelle Decottignies used PCR to amplify a linear fragment (Figure 7C) [170]. The fragment was transformed and allowed to re-circularize in living cells followed by junction analysis. Remarkably, she identified mitochondrial DNA sequences at the junctions. As she points out, the advantage of this assay is that primers with different microhomology overhangs can be engineered to test recircularization of different ends. This assay was successfully used by the Du lab to identify the XRCC4 NHEJ protein in a genome wide screen [171]. The Du lab also designed an assay that relies on the HO restriction enzyme (Figure 7D) [172] similarly to their previous assays (Figure 6B). In this assay, the 24 base pair HO restriction site marked with the natMX cassette (nat confers resistance to nourseothricin) was cloned at the arg3+ locus on Ch.I. The nmt1-HO endonuclease was integrated at ars1+. Because the cells were grown under continuous expression of the HO endonuclease (no thiamine in the media), only those cells with imprecise repair of the junction (either deletions or insertions) could survive. These cells had destroyed the HO endonuclease restriction site, in effect inactivating the function of the enzyme. The junction of surviving cells was amplified by PCR and sequenced either by the Sanger methods or by Illumina sequencing. This assay is a useful tool for studying break repair that does not rely on homologous recombination.
A different NHEJ assay was designed by Li et al. that relies on analyzing the genome insertion and excision of a transposon (Figure 7E) [173]. Two plasmids, one encoding the transposase and the other the transposon are co-transformed in the cells. De-repression of the transposase causes random insertion of the transposon. The role of NHEJ factors can be investigated by monitoring the efficiency of insertion. Alternatively, re-expression of the transposase will cause excision of the transposon from the genome. The transposon is characterized by 8 bp of homology on either end. Analysis of the repaired junction can determine the nature of the microhomology mediated repair.
Finally, we want to mention an assay to study chromosome end fusions due to telomere erosion (Figure 7F) [174]. The his3+ is one of the genes in S. pombe that has introns. The investigators introduced head to head telomeric repeats in the second intron of his3+. This insertion does not affect the function of his3+. The plasmid is linearized between the telomeric repeats and transformed into living cells. The construct may be propagated as a linear fragment because it has telomeres and can be selected for on media lacking leucine. However, the linear fragment interrupts transcription of the his3+ gene. Deletion of telomerase causes telomere attrition and re-circularization of the fragment which can be selected for on media lacking histidine because it reconstitutes the function of the his3+ gene. The plasmid can be recovered and the junction analyzed by sequencing.
8. Concluding Remarks
Although none of the assays mentioned here can independently unravel the function of every repair pathway, when the data are put together, a clearer picture emerges. This is particularly true if combined with results from other systems such as S. cerevisiae. Nevertheless, the work done in S. pombe has not just been complementary to the work done in other model systems but instrumental in discovering and defining new mechanisms of DSB repair.
Nevertheless, there are many unanswered questions that remain. For example, the role of chromatin remodeling in modulating DSB repair requires further investigation. The fact that histone modifications play a role in biasing repair towards different pathways is clear. However, how these modifications choreograph repair is not well understood [175]. This is because histone modifications are transient and hard to capture. Experiments using some of the assays described here that either knock out histone modifying enzymes or substitute modifiable histone residues for unmodifiable ones have provided some insight but these are likely to cause genome wide changes which complicates interpretation. Some recent experiments have also shown that RNA plays a major role in DSB repair [176] but the exact dynamics are poorly understood. With advances in technology, such as CRISPR, it is almost certain that in the next few years we will see much more complex assays that are likely to elucidate some of these questions.
Author Contributions
H.M.H. and B.E.L. performed an extensive literature search, summarized the papers read and made some figures. R.C.P. wrote the first draft. H.M.H. and B.E.L. revised the draft. All authors contributed equally to the revisions to address referees’ points. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by the National Institutes of Health (grant number RO3 CA223545-01). Other support to RCP from The Ohio State University James Comprehensive Cancer Center. Bailey E. Lucas was supported in part by an Ohio State University Comprehensive Cancer Center Pelotonia Undergraduate Fellowship.
Acknowledgments
We thank James and Ellen Bazzoli for their generous donation to sponsor our laboratory.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kerzendorfer, C.; O’Driscoll, M. Human DNA damage response and repair deficiency syndromes: Linking genomic instability and cell cycle checkpoint proficiency. DNA Repair 2009, 8, 1139–1152. [Google Scholar] [CrossRef]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [Green Version]
- Jeggo, P.A.; Lobrich, M. How cancer cells hijack DNA double-strand break repair pathways to gain genomic instability. Biochem. J. 2015, 471, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Lasko, D.; Cavenee, W.; Nordenskjold, M. Loss of constitutional heterozygosity in human cancer. Annu. Rev. Genet. 1991, 25, 281–314. [Google Scholar] [CrossRef] [PubMed]
- Ortega, V.; Chaubey, A.; Mendiola, C.; Ehman, W., Jr.; Vadlamudi, K.; Dupont, B.; Velagaleti, G. Complex Chromosomal Rearrangements in B-Cell Lymphoma: Evidence of Chromoanagenesis? A Case Report. Neoplasia 2016, 18, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [Green Version]
- Nikolov, I.; Taddei, A. Linking replication stress with heterochromatin formation. Chromosoma 2016, 125, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Tabancay, A.P., Jr.; Forsburg, S.L. Eukaryotic DNA replication in a chromatin context. Curr. Top. Dev. Biol. 2006, 76, 129–184. [Google Scholar] [CrossRef]
- Prado, F.; Aguilera, A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005, 24, 1267–1276. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, S.; Freudenreich, C.H. The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosomes Cancer 2019, 58, 270–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irony-Tur Sinai, M.; Kerem, B. Genomic instability in fragile sites-still adding the pieces. Genes Chromosomes Cancer 2019, 58, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Cavalier-Smith, T. Origins of the machinery of recombination and sex. Heredity 2002, 88, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Piazza, A.; Heyer, W.D. Homologous Recombination and the Formation of Complex Genomic Rearrangements. Trends. Cell. Biol. 2019, 29, 135–149. [Google Scholar] [CrossRef]
- West, S.C.; Blanco, M.G.; Chan, Y.W.; Matos, J.; Sarbajna, S.; Wyatt, H.D. Resolution of Recombination Intermediates: Mechanisms and Regulation. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prado, F. Homologous recombination maintenance of genome integrity during DNA damage tolerance. Mol. Cell. Oncol. 2014, 1, e957039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [Green Version]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef]
- Daley, J.M.; Kwon, Y.; Niu, H.; Sung, P. Investigations of homologous recombination pathways and their regulation. Yale J. Biol. Med. 2013, 86, 453–461. [Google Scholar]
- Pannunzio, N.R.; Li, S.; Watanabe, G.; Lieber, M.R. Non-homologous end joining often uses microhomology: Implications for alternative end joining. DNA Repair 2014, 17, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Malkova, A.; Haber, J.E. Mutations arising during repair of chromosome breaks. Annu. Rev. Genet. 2012, 46, 455–473. [Google Scholar] [CrossRef] [PubMed]
- Hedges, S.B. The origin and evolution of model organisms. Nat. Rev. Genet. 2002, 3, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Heckman, D.S.; Geiser, D.M.; Eidell, B.R.; Stauffer, R.L.; Kardos, N.L.; Hedges, S.B. Molecular evidence for the early colonization of land by fungi and plants. Science 2001, 293, 1129–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, V.; Gwilliam, R.; Rajandream, M.A.; Lyne, M.; Lyne, R.; Stewart, A.; Sgouros, J.; Peat, N.; Hayles, J.; Baker, S.; et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002, 415, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Rubin, G.M.; Yandell, M.D.; Wortman, J.R.; Gabor Miklos, G.L.; Nelson, C.R.; Hariharan, I.K.; Fortini, M.E.; Li, P.W.; Apweiler, R.; Fleischmann, W.; et al. Comparative genomics of the eukaryotes. Science 2000, 287, 2204–2215. [Google Scholar] [CrossRef] [Green Version]
- Forsburg, S.L. The yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe: Models for cell biology research. Gravit. Space Biol. Bull. 2005, 18, 3–9. [Google Scholar]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef]
- Madireddy, A.; Gerhardt, J. Replication Through Repetitive DNA Elements and Their Role in Human Diseases. DNA Replication Old Princ. New Discov. 2017, 1042, 549–581. [Google Scholar] [CrossRef]
- Forsburg, S.L. The CINs of the centromere. Biochem. Soc. Trans. 2013, 41, 1706–1711. [Google Scholar] [CrossRef] [Green Version]
- Li, P.C.; Petreaca, R.C.; Jensen, A.; Yuan, J.P.; Green, M.D.; Forsburg, S.L. Replication fork stability is essential for the maintenance of centromere integrity in the absence of heterochromatin. Cell Rep. 2013, 3, 638–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onaka, A.T.; Toyofuku, N.; Inoue, T.; Okita, A.K.; Sagawa, M.; Su, J.; Shitanda, T.; Matsuyama, R.; Zafar, F.; Takahashi, T.S.; et al. Rad51 and Rad54 promote noncrossover recombination between centromere repeats on the same chromatid to prevent isochromosome formation. Nucleic Acids Res. 2016, 44, 10744–10757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Okamoto, A.; Katou, Y.; Yadani, C.; Shitanda, T.; Kaweeteerawat, C.; Takahashi, T.S.; Itoh, T.; Shirahige, K.; Masukata, H.; et al. Rad51 suppresses gross chromosomal rearrangement at centromere in Schizosaccharomyces pombe. EMBO J. 2008, 27, 3036–3046. [Google Scholar] [CrossRef] [Green Version]
- Forsburg, S.L.; Shen, K.F. Centromere Stability: The Replication Connection. Genes-Basel 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- He, H.J.; Gonzalez, M.; Zhang, F.; Li, F. DNA replication components as regulators of epigenetic inheritance-lesson from fission yeast centromere. Protein Cell 2014, 5, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Mizuguchi, T.; Barrowman, J.; Grewal, S.I.S. Chromosome domain architecture and dynamic organization of the fission yeast genome. Febs Lett. 2015, 589, 2975–2986. [Google Scholar] [CrossRef] [Green Version]
- Forsburg, S.L. The art and design of genetic screens: Yeast. Nat. Rev. Genet. 2001, 2, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, H.; Huang, Y.; Wang, Y.; Liu, Y.; Chen, X. Pathways and assays for DNA double-strand break repair by homologous recombination. Acta Biochim. Biophys. Sin. 2019, 51, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, H.L.; Bacinskaja, G.; Che, J.; Cheblal, A.; Elango, R.; Epshtein, A.; Fitzgerald, D.M.; Gomez-Gonzalez, B.; Khan, S.R.; Kumar, S.; et al. Guidelines for DNA recombination and repair studies: Cellular assays of DNA repair pathways. Microb. Cell 2019, 6, 1–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, H.L.; Ang, K.K.H.; Arkin, M.R.; Beckwitt, E.C.; Chang, Y.H.; Fan, J.; Kwon, Y.; Morten, M.J.; Mukherjee, S.; Pambos, O.J.; et al. Guidelines for DNA recombination and repair studies: Mechanistic assays of DNA repair processes. Microb. Cell 2019, 6, 65–101. [Google Scholar] [CrossRef]
- Kai, M.; Taricani, L.; Wang, T.S. Methods for studying mutagenesis and checkpoints in Schizosaccharomyces pombe. Methods Enzymol. 2006, 409, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Niwa, O.; Matsumoto, T.; Yanagida, M. Construction of a Minichromosome by Deletion and Its Mitotic and Meiotic Behavior in Fission Yeast. Mol. Gen. Genet. 1986, 203, 397–405. [Google Scholar] [CrossRef]
- Niwa, O.; Matsumoto, T.; Chikashige, Y.; Yanagida, M. Characterization of Schizosaccharomyces pombe minichromosome deletion derivatives and a functional allocation of their centromere. EMBO J. 1989, 8, 3045–3052. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Costanzo, V. Centromere Structure and Function. Prog. Mol. Subcell. Biol. 2017, 56, 515–539. [Google Scholar] [CrossRef]
- Kohli, J.; Hottinger, H.; Munz, P.; Strauss, A.; Thuriaux, P. Genetic-Mapping in Schizosaccharomyces-Pombe by Mitotic and Meiotic Analysis and Induced Haploidization. Genetics 1977, 87, 471–489. [Google Scholar]
- Steiner, N.C.; Hahnenberger, K.M.; Clarke, L. Centromeres of the Fission Yeast Schizosaccharomyces-Pombe Are Highly Variable Genetic-Loci. Mol. Cell. Biol. 1993, 13, 4578–4587. [Google Scholar] [CrossRef] [Green Version]
- Gygax, A.; Thuriaux, P. A Revised Chromosome Map of the Fission Yeast Schizosaccharomyces-Pombe. Curr. Genet. 1984, 8, 85–92. [Google Scholar] [CrossRef]
- Murakami, S.; Matsumoto, T.; Niwa, O.; Yanagida, M. Structure of the fission yeast centromere cen3: Direct analysis of the reiterated inverted region. Chromosoma 1991, 101, 214–221. [Google Scholar] [CrossRef]
- Lock, A.; Rutherford, K.; Harris, M.A.; Wood, V. PomBase: The Scientific Resource for Fission Yeast. Eukaryot. Genom. Databases Methods Protoc. 2018, 1757, 49–68. [Google Scholar] [CrossRef] [Green Version]
- PomBase. The Scientific Resource for Fission Yeast. Available online: https://www.pombase.org/ (accessed on 15 December 2019).
- Tinline-Purvis, H.; Savory, A.P.; Cullen, J.K.; Dave, A.; Moss, J.; Bridge, W.L.; Marguerat, S.; Bahler, J.; Ragoussis, J.; Mott, R.; et al. Failed gene conversion leads to extensive end processing and chromosomal rearrangements in fission yeast. EMBO J. 2009, 28, 3400–3412. [Google Scholar] [CrossRef] [Green Version]
- Cromie, G.A.; Rubio, C.A.; Hyppa, R.W.; Smith, G.R. A natural meiotic DNA break site in Schizosaccharomyces pombe is a hotspot of gene conversion, highly associated with crossing over. Genetics 2005, 169, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, J.K.; Hussey, S.P.; Walker, C.; Prudden, J.; Wee, B.Y.; Dave, A.; Findlay, J.S.; Savory, A.P.; Humphrey, T.C. Break-induced loss of heterozygosity in fission yeast: Dual roles for homologous recombination in promoting translocations and preventing de novo telomere addition. Mol. Cell. Biol. 2007, 27, 7745–7757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudden, J.; Evans, J.S.; Hussey, S.P.; Deans, B.; O’Neill, P.; Thacker, J.; Humphrey, T. Pathway utilization in response to a site-specific DNA double-strand break in fission yeast. EMBO J. 2003, 22, 1419–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.U.; Hayles, J.; Kim, D.; Wood, V.; Park, H.O.; Won, M.; Yoo, H.S.; Duhig, T.; Nam, M.; Palmer, G.; et al. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 2010, 28, 617–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, J.; Tinline-Purvis, H.; Walker, C.A.; Folkes, L.K.; Stratford, M.R.; Hayles, J.; Hoe, K.L.; Kim, D.U.; Park, H.O.; Kearsey, S.E.; et al. Break-induced ATR and Ddb1-Cul4(Cdt)(2) ubiquitin ligase-dependent nucleotide synthesis promotes homologous recombination repair in fission yeast. Genes Dev. 2010, 24, 2705–2716. [Google Scholar] [CrossRef] [Green Version]
- Pai, C.C.; Blaikley, E.; Humphrey, T.C. DNA Double-Strand Break Repair Assay. Cold Spring Harb. Protoc. 2018, 2018. [Google Scholar] [CrossRef]
- Hartsuiker, E.; Vaessen, E.; Carr, A.M.; Kohli, J. Fission yeast Rad50 stimulates sister chromatid recombination and links cohesion with repair. EMBO J. 2001, 20, 6660–6671. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.A.; Fink, G.R. Gene Conversion between Duplicated Genetic Elements in Yeast. Nature 1981, 292, 306–311. [Google Scholar] [CrossRef]
- Rothstein, R.; Helms, C.; Rosenberg, N. Concerted Deletions and Inversions Are Caused by Mitotic Recombination between Delta-Sequences in Saccharomyces-Cerevisiae. Mol. Cell. Biol. 1987, 7, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, M.V.; Whitehouse, H.L.K. A Buff Spore Color Mutant in Sordaria-Brevicollis Showing High-Frequency Conversion: 2. Loss of the High-Frequency Conversion. Genet. Res. 1983, 41, 155–163. [Google Scholar] [CrossRef]
- Pukkila, P.J.; Stephens, M.D.; Binninger, D.M.; Errede, B. Frequency and Directionality of Gene Conversion Events Involving the Cyc7-H3 Mutation in Saccharomyces-Cerevisiae. Genetics 1986, 114, 347–361. [Google Scholar] [PubMed]
- Gutz, H. Site Specific Induction of Gene Conversion in schizosaccharomyces pombe. Genetics 1971, 69, 317–337. [Google Scholar] [PubMed]
- Schuchert, P.; Kohli, J. The Ade6-M26 Mutation of Schizosaccharomyces-Pombe Increases the Frequency of Crossing Over. Genetics 1988, 119, 507–515. [Google Scholar] [PubMed]
- Fortunato, E.A.; Osman, F.; Subramani, S. Analysis of spontaneous and double-strand break-induced recombination in rad mutants of S. pombe. Mutat. Res. 1996, 364, 14–60. [Google Scholar] [CrossRef]
- Osman, F.; Fortunato, E.A.; Subramani, S. Double-strand break-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Genetics 1996, 142, 341–357. [Google Scholar] [PubMed]
- Ahamad, N.; Verma, S.K.; Ahmed, S. Activation of Checkpoint Kinase Chk1 by Reactive Oxygen Species Resulting from Disruption of wat1/pop3 in Schizosaccharomyces pombe. Genetics 2016, 204, 1397–1406. [Google Scholar] [CrossRef]
- Bellini, A.; Girard, P.M.; Lambert, S.; Tessier, L.; Sage, E.; Francesconi, S. Stress activated protein kinase pathway modulates homologous recombination in fission yeast. PLoS ONE 2012, 7, e47987. [Google Scholar] [CrossRef] [Green Version]
- Nandi, S.; Whitby, M.C. The ATPase activity of Fml1 is essential for its roles in homologous recombination and DNA repair. Nucleic Acids Res. 2012, 40, 9584–9595. [Google Scholar] [CrossRef]
- Osman, F.; Tsaneva, I.R.; Whitby, M.C.; Doe, C.L. UV irradiation causes the loss of viable mitotic recombinants in Schizosaccharomyces pombe cells lacking the G(2)/M DNA damage checkpoint. Genetics 2002, 160, 891–908. [Google Scholar]
- Bass, K.L.; Murray, J.M.; O’Connell, M.J. Brc1-dependent recovery from replication stress. J. Cell Sci. 2012, 125, 2753–2764. [Google Scholar] [CrossRef] [Green Version]
- Osman, F.; Bjoras, M.; Alseth, I.; Morland, I.; McCready, S.; Seeberg, E.; Tsaneva, I. A new Schizosaccharomyces pombe base excision repair mutant, nth1, reveals overlapping pathways for repair of DNA base damage. Mol. Microbiol. 2003, 48, 465–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommariva, E.; Pellny, T.K.; Karahan, N.; Kumar, S.; Huberman, J.A.; Dalgaard, J.Z. Schizosaccharomyces pombe Swi1, Swi3, and Hsk1 are components of a novel S-phase response pathway to alkylation damage. Mol. Cell. Biol. 2005, 25, 2770–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, M.; Inoue, H.; Ishii, C.; Murakami, Y. The novel gene mus7(+) is involved in the repair of replication-associated DNA damage in fission yeast. DNA Repair 2007, 6, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Tomita, K.; Matsuura, A.; Nakagawa, T.; Masukata, H.; Uritani, M.; Ushimaru, T.; Ueno, M. A novel allele of fission yeast rad11 that causes defects in DNA repair and telomere length regulation. Nucleic Acids Res. 2003, 31, 7141–7149. [Google Scholar] [CrossRef] [PubMed]
- Steinacher, R.; Osman, F.; Lorenz, A.; Bryer, C.; Whitby, M.C. Slx8 Removes Pli1-Dependent Protein-SUMO Conjugates Including SUMOylated Topoisomerase I to Promote Genome Stability. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, E.; Miyabe, I.; Iraqui, I.; Zheng, J.; Lambert, S.A.; Carr, A.M. The extent of error-prone replication restart by homologous recombination is controlled by Exo1 and checkpoint proteins. J. Cell Sci. 2014, 127, 2983–2994. [Google Scholar] [CrossRef] [Green Version]
- Octobre, G.; Lorenz, A.; Loidl, J.; Kohli, J. The Rad52 homologs Rad22 and Rti1 of Schizosaccharomyces pombe are not essential for meiotic interhomolog recombination, but are required for meiotic intrachromosomal recombination and mating-type-related DNA repair. Genetics 2008, 178, 2399–2412. [Google Scholar] [CrossRef] [Green Version]
- Doe, C.L.; Dixon, J.; Osman, F.; Whitby, M.C. Partial suppression of the fission yeast rqh1(-) phenotype by expression of a bacterial Holliday junction resolvase. EMBO J. 2000, 19, 2751–2762. [Google Scholar] [CrossRef] [Green Version]
- Doe, C.L.; Osman, F.; Dixon, J.; Whitby, M.C. The Holliday junction resolvase SpCCE1 prevents mitochondrial DNA aggregation in Schizosaccharomyces pombe. Mol. Gen. Genet. 2000, 263, 889–897. [Google Scholar] [CrossRef]
- Doe, C.L.; Whitby, M.C. The involvement of Srs2 in post-replication repair and homologous recombination in fission yeast. Nucleic Acids Res. 2004, 32, 1480–1491. [Google Scholar] [CrossRef] [Green Version]
- Doe, C.L.; Osman, F.; Dixon, J.; Whitby, M.C. DNA repair by a Rad22-Mus81-dependent pathway that is independent of Rhp51. Nucleic Acids Res. 2004, 32, 5570–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, F.; Ahn, J.S.; Lorenz, A.; Whitby, M.C. The RecQ DNA helicase Rqh1 constrains Exonuclease 1-dependent recombination at stalled replication forks. Sci. Rep. 2016, 6, 22837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef] [Green Version]
- Asano, S.; Higashitani, A.; Horiuchi, K. Filamentous phage replication initiator protein gpII forms a covalent complex with the 5’ end of the nick it introduced. Nucleic Acids Res. 1999, 27, 1882–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalgaard, J.Z.; Klar, A.J. A DNA replication-arrest site RTS1 regulates imprinting by determining the direction of replication at mat1 in S. pombe. Genes Dev. 2001, 15, 2060–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.S.; Osman, F.; Whitby, M.C. Replication fork blockage by RTS1 at an ectopic site promotes recombination in fission yeast. EMBO J. 2005, 24, 2011–2023. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, A.; Osman, F.; Folkyte, V.; Sofueva, S.; Whitby, M.C. Fbh1 limits Rad51-dependent recombination at blocked replication forks. Mol. Cell. Biol. 2009, 29, 4742–4756. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.O.; Jalan, M.; Morrow, C.A.; Osman, F.; Whitby, M.C. Recombination occurs within minutes of replication blockage by RTS1 producing restarted forks that are prone to collapse. Elife 2015, 4, e04539. [Google Scholar] [CrossRef]
- Sun, W.; Nandi, S.; Osman, F.; Ahn, J.S.; Jakovleska, J.; Lorenz, A.; Whitby, M.C. The FANCM Ortholog Fml1 Promotes Recombination at Stalled Replication Forks and Limits Crossing Over during DNA Double-Strand Break Repair. Mol. Cell. 2008, 32, 118–128. [Google Scholar] [CrossRef]
- Steinacher, R.; Osman, F.; Dalgaard, J.Z.; Lorenz, A.; Whitby, M.C. The DNA helicase Pfh1 promotes fork merging at replication termination sites to ensure genome stability. Gene Dev. 2012, 26, 594–602. [Google Scholar] [CrossRef] [Green Version]
- Osman, F.; Whitby, M.C. Monitoring homologous recombination following replication fork perturbation in the fission yeast Schizosaccharomyces pombe. Methods Mol. Biol. 2009, 521, 535–552. [Google Scholar] [CrossRef] [PubMed]
- Jalan, M.; Oehler, J.; Morrow, C.A.; Osman, F.; Whitby, M.C. Factors affecting template switch recombination associated with restarted DNA replication. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, R.J.; Whitehall, S.K. tRNA genes in eukaryotic genome organization and reorganization. Cell Cycle 2009, 8, 3102–3106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofueva, S.; Osman, F.; Lorenz, A.; Steinacher, R.; Castagnetti, S.; Ledesma, J.; Whitby, M.C. Ultrafine anaphase bridges, broken DNA and illegitimate recombination induced by a replication fork barrier. Nucleic Acids Res. 2011, 39, 6568–6584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamang, S.; Kishkevich, A.; Morrow, C.A.; Osman, F.; Jalan, M.; Whitby, M.C. The PCNA unloader Elg1 promotes recombination at collapsed replication forks in fission yeast. Elife 2019, 8, e47277. [Google Scholar] [CrossRef]
- Watson, A.T.; Werler, P.; Carr, A.M. Regulation of gene expression at the fission yeast Schizosaccharomyces pombe urg1 locus. Gene 2011, 484, 76–86. [Google Scholar] [CrossRef]
- Forsburg, S.L. Comparison of Schizosaccharomyces-Pombe Expression Systems. Nucleic Acids Res. 1993, 21, 2955–2956. [Google Scholar] [CrossRef] [Green Version]
- Maundrell, K. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 1993, 123, 127–130. [Google Scholar] [CrossRef]
- Muller, S.; Sandal, T.; Kamp-Hansen, P.; Dalboge, H. Comparison of expression systems in the yeasts Saccharomyces cerevisiae, Hansenula polymorpha, Klyveromyces lactis, Schizosaccharomyces pombe and Yarrowia lipolytica. Cloning of two novel promoters from Yarrowia lipolytica. Yeast 1998, 14, 1267–1283. [Google Scholar] [CrossRef]
- Watson, A.T.; Daigaku, Y.; Mohebi, S.; Etheridge, T.J.; Chahwan, C.; Murray, J.M.; Carr, A.M. Optimisation of the Schizosaccharomyces pombe urg1 Expression System. PLoS ONE 2013, 8, e83800. [Google Scholar] [CrossRef] [Green Version]
- Sunder, S.; Greeson-Lott, N.T.; Runge, K.W.; Sanders, S.L. A new method to efficiently induce a site-specific double-strand break in the fission yeast Schizosaccharomyces pombe. Yeast 2012, 29, 275–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilio, N.; Wehrkamp-Richter, S.; Boddy, M.N. A new versatile system for rapid control of gene expression in the fission yeast Schizosaccharomyces pombe. Yeast 2012, 29, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Lucas, B.E.; McPherson, M.T.; Hawk, T.M.; Wilson, L.N.; Kroh, J.M.; Hickman, K.G.; Fitzgerald, S.R.; Disbennett, W.M.; Rollins, P.D.; Hylton, H.M.; et al. An Assay to Study Intra-Chromosomal Deletions in Yeast. Methods Protoc. 2019, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, S.; Watson, A.; Sheedy, D.M.; Martin, B.; Carr, A.M. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 2005, 121, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saada, A.A.; Teixeira-Silva, A.; Iraqui, I.; Costes, A.; Hardy, J.; Paoletti, G.; Freon, K.; Lambert, S.A.E. Unprotected Replication Forks Are Converted into Mitotic Sister Chromatid Bridges. Mol. Cell 2017, 66, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, K.; Lambert, S.; Baldacci, G.; Murray, J.M.; Carr, A.M. Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Gene Dev. 2009, 23, 2876–2886. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, K.; Miyabe, I.; Schalbetter, S.A.; Carr, A.M.; Murray, J.M. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature 2013, 493, 246–249. [Google Scholar] [CrossRef] [Green Version]
- Mohebi, S.; Mizuno, K.; Watson, A.; Carr, A.M.; Murray, J.M. Checkpoints are blind to replication restart and recombination intermediates that result in gross chromosomal rearrangements. Nat. Commun. 2015, 6, 6357. [Google Scholar] [CrossRef] [Green Version]
- Iraqui, I.; Chekkal, Y.; Jmari, N.; Pietrobon, V.; Freon, K.; Costes, A.; Lambert, S.A.E. Recovery of Arrested Replication Forks by Homologous Recombination Is Error-Prone. Plos Genetics 2012, 8, e1002976. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.; Mizuno, K.; Blaisonneau, J.; Martineau, S.; Chanet, R.; Freon, K.; Murray, J.M.; Carr, A.M.; Baldacci, G. Homologous Recombination Restarts Blocked Replication Forks at the Expense of Genome Rearrangements by Template Exchange. Mol. Cell 2010, 39, 346–359. [Google Scholar] [CrossRef]
- Teixeira-Silva, A.; Ait Saada, A.; Hardy, J.; Iraqui, I.; Nocente, M.C.; Freon, K.; Lambert, S.A.E. The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nat. Commun. 2017, 8, 1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcangioli, B.; Roseaulin, L.; Holmes, A. Mating-Type Switching in S. Pombe. In Molecular Genetics of Recombination; Springer: Berlin, Germany, 2007; pp. 95–133. [Google Scholar]
- Leupold, U. Studies on recombination in Schizosaccharomyces pombe. Cold Spring Harb. Symp. Quant. Biol. 1958, 23, 161–170. [Google Scholar] [CrossRef]
- Egel, R. Genes involved in mating type expression of fission yeast. Mol. Gen. Genet. 1973, 122, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Egel, R.; Gutz, H. Gene activation by copy transposition in mating-type switching of a homothallic fission yeast. Curr. Genet. 1981, 3, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Egel, R.; Beach, D.H.; Klar, A.J. Genes required for initiation and resolution steps of mating-type switching in fission yeast. Proc. Natl Acad. Sci. USA 1984, 81, 3481–3485. [Google Scholar] [CrossRef] [Green Version]
- Egel, R. Two tightly linked silent cassettes in the mating-type region of Schizosaccharomyces pombe. Curr. Genet. 1984, 8, 199–203. [Google Scholar] [CrossRef]
- Beach, D.H.; Klar, A.J. Rearrangements of the transposable mating-type cassettes of fission yeast. EMBO J. 1984, 3, 603–610. [Google Scholar] [CrossRef]
- Klar, A.J. Lessons learned from studies of fission yeast mating-type switching and silencing. Annu. Rev. Genet. 2007, 41, 213–236. [Google Scholar] [CrossRef]
- Thon, G.; Maki, T.; Haber, J.E.; Iwasaki, H. Mating-type switching by homology-directed recombinational repair: A matter of choice. Curr. Genet. 2019, 65, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Hanson, S.J.; Wolfe, K.H. An Evolutionary Perspective on Yeast Mating-Type Switching. Genetics 2017, 206, 9–32. [Google Scholar] [CrossRef] [Green Version]
- Gutz, H.; Doe, F.J. Two Different h Mating Types in schizosaccharomyces pombe. Genetics 1973, 74, 563–569. [Google Scholar] [PubMed]
- Egel, R. Frequency of mating-type switching in homothallic fission yeast. Nature 1977, 266, 172–174. [Google Scholar] [CrossRef]
- Lee, C.S.; Haber, J.E. Mating-type Gene Switching in Saccharomyces cerevisiae. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef]
- Vengrova, S.; Dalgaard, J.Z. RNase-sensitive DNA modification(s) initiates S. pombe mating-type switching. Gene Dev. 2004, 18, 794–804. [Google Scholar] [CrossRef] [Green Version]
- Vengrova, S.; Dalgaard, J.Z. The wild-type Schizosaccharomyces pombe mat1 imprint consists of two ribonucleotides. EMBO Rep. 2006, 7, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Bisht, K.K.; Upadhyay, U.; Kushwaha, A.C.; Nanda, J.S.; Srivastava, S.; Saini, J.K.; Klar, A.J.S.; Singh, J. Role of Cdc23/Mcm10 in generating the ribonucleotide imprint at the mat1 locus in fission yeast. Nucleic Acids Res. 2019, 47, 3422–3433. [Google Scholar] [CrossRef] [PubMed]
- Kaykov, A.; Holmes, A.M.; Arcangioli, B. Formation, maintenance and consequences of the imprint at the mating-type locus in fission yeast. EMBO J. 2004, 23, 930–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalgaard, L.Z.; Klar, A.J.S. Orientation of DNA replication establishes mating-type switching pattern in S-pombe. Nature 1999, 400, 181–184. [Google Scholar] [CrossRef]
- Arcangioli, B. A site- and strand-specific DNA break confers asymmetric switching potential in fission yeast. EMBO J. 1998, 17, 4503–4510. [Google Scholar] [CrossRef] [Green Version]
- Arcangioli, B.; de Lahondes, R. Fission yeast switches mating type by a replication-recombination coupled process. Embo J. 2000, 19, 1389–1396. [Google Scholar] [CrossRef] [Green Version]
- Holmes, A.M.; Kaykov, A.; Arcangioli, B. Molecular and cellular dissection of mating-type switching steps in Schizosaccharomyces pombe. Mol. Cell. Biol. 2005, 25, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vengrova, S.; Codlin, S.; Dalgaard, J.Z. RTS1—an eukaryotic terminator of replication. Int. J. Biochem. Cell. B 2002, 34, 1031–1034. [Google Scholar] [CrossRef]
- Klar, A.J.S.; Ishikawa, K.; Moore, S. A Unique DNA Recombination Mechanism of the Mating/Cell-type Switching of Fission Yeasts: A Review. Microbiol. Spectr. 2014, 2, 5. [Google Scholar] [CrossRef]
- Roseaulin, L.; Yamada, Y.; Tsutsui, Y.; Russell, P.; Iwasaki, H.; Arcangioli, B. Mus81 is essential for sister chromatid recombination at broken replication forks. EMBO J. 2008, 27, 1378–1387. [Google Scholar] [CrossRef] [Green Version]
- Styrkarsdottir, U.; Egel, R.; Nielsen, O. The smt-0 mutation which abolishes mating-type switching in fission yeast is a deletion. Curr. Genet. 1993, 23, 184–186. [Google Scholar] [CrossRef]
- Klar, A.J.; Miglio, L.M. Initiation of meiotic recombination by double-strand DNA breaks in S. pombe. Cell 1986, 46, 725–731. [Google Scholar] [CrossRef]
- Zhu, M.; Zhao, H.; Limbo, O.; Russell, P. Mre11 complex links sister chromatids to promote repair of a collapsed replication fork. Proc. Natl. Acad. Sci. USA 2018, 115, 8793–8798. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhu, M.; Limbo, O.; Russell, P. RNase H eliminates R-loops that disrupt DNA replication but is nonessential for efficient DSB repair. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Gutz, H.; Schmidt, H. Switching genes in Schizosaccharomyces pombe. Curr. Genet. 1985, 9, 325–331. [Google Scholar] [CrossRef]
- Inagawa, T.; Yamada-Inagawa, T.; Eydmann, T.; Mian, I.S.; Wang, T.S.; Dalgaard, J.Z. Schizosaccharomyces pombe Rtf2 mediates site-specific replication termination by inhibiting replication restart. Proc. Natl. Acad. Sci. USA 2009, 106, 7927–7932. [Google Scholar] [CrossRef] [Green Version]
- Allshire, R.C.; Ekwall, K. Epigenetic Regulation of Chromatin States in Schizosaccharomyces pombe. Cold Spring Harb. Perspect. Biol. 2015, 7, a018770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpe, T.A.; Kidner, C.; Hall, I.M.; Teng, G.; Grewal, S.I.; Martienssen, R.A. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 2002, 297, 1833–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, S.; Noma, K.; Grewal, S.I. RNAi-independent heterochromatin nucleation by the stress-activated ATF/CREB family proteins. Science 2004, 304, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Verdel, A.; Jia, S.; Gerber, S.; Sugiyama, T.; Gygi, S.; Grewal, S.I.; Moazed, D. RNAi-mediated targeting of heterochromatin by the RITS complex. Science 2004, 303, 672–676. [Google Scholar] [CrossRef] [Green Version]
- Thon, G.; Klar, A.J. The clr1 locus regulates the expression of the cryptic mating-type loci of fission yeast. Genetics 1992, 131, 287–296. [Google Scholar]
- Ekwall, K.; Ruusala, T. Mutations in rik1, clr2, clr3 and clr4 genes asymmetrically derepress the silent mating-type loci in fission yeast. Genetics 1994, 136, 53–64. [Google Scholar]
- Thon, G.; Cohen, A.; Klar, A.J. Three additional linkage groups that repress transcription and meiotic recombination in the mating-type region of Schizosaccharomyces pombe. Genetics 1994, 138, 29–38. [Google Scholar]
- Nabeshima, K.; Nakagawa, T.; Straight, A.F.; Murray, A.; Chikashige, Y.; Yamashita, Y.M.; Hiraoka, Y.; Yanagida, M. Dynamics of centromeres during metaphase-anaphase transition in fission yeast: Dis1 is implicated in force balance in metaphase bipolar spindle. Mol. Biol. Cell 1998, 9, 3211–3225. [Google Scholar] [CrossRef] [Green Version]
- Du, L.L.; Nakamura, T.M.; Moser, B.A.; Russell, P. Retention but not recruitment of Crb2 at double-strand breaks requires Rad1 and Rad3 complexes. Mol. Cell. Biol. 2003, 23, 6150–6158. [Google Scholar] [CrossRef] [Green Version]
- Du, L.L.; Nakamura, T.M.; Russell, P. Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev. 2006, 20, 1583–1596. [Google Scholar] [CrossRef] [Green Version]
- Sofueva, S.; Du, L.L.; Limbo, O.; Williams, J.S.; Russell, P. BRCT domain interactions with phospho-histone H2A target Crb2 to chromatin at double-strand breaks and maintain the DNA damage checkpoint. Mol. Cell. Biol. 2010, 30, 4732–4743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.M.; Moser, B.A.; Du, L.L.; Russell, P. Cooperative control of Crb2 by ATM family and Cdc2 kinases is essential for the DNA damage checkpoint in fission yeast. Mol. Cell. Biol. 2005, 25, 10721–10730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limbo, O.; Chahwan, C.; Yamada, Y.; de Bruin, R.A.M.; Wittenberg, C.; Russell, P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell 2007, 28, 134–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limbo, O.; Porter-Goff, M.E.; Rhind, N.; Russell, P. Mre11 Nuclease Activity and Ctp1 Regulate Chk1 Activation by Rad3(ATR) and Tel1(ATM) Checkpoint Kinases at Double-Strand Breaks. Mol. Cell. Biol. 2011, 31, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limbo, O.; Moiani, D.; Kertokalio, A.; Wyman, C.; Tainer, J.A.; Russell, P. Mre11 ATLD17/18 mutation retains Tel1/ATM activity but blocks DNA double-strand break repair. Nucleic Acids Res. 2012, 40, 11435–11449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Wang, H.T.; Zhai, Y.; Russell, P.; Du, L.L. Mdb1, a Fission Yeast Homolog of Human MDC1, Modulates DNA Damage Response and Mitotic Spindle Function. PLoS ONE 2014, 9, e97028. [Google Scholar] [CrossRef]
- Limbo, O.; Yamada, Y.; Russell, P. Mre11-Rad50-dependent activity of ATM/Tel1 at DNA breaks and telomeres in the absence of Nbs1. Mol. Biol. Cell 2018, 29, 1389–1399. [Google Scholar] [CrossRef]
- Langerak, P.; Mejia-Ramirez, E.; Limbo, O.; Russell, P. Release of Ku and MRN from DNA Ends by Mre11 Nuclease Activity and Ctp1 Is Required for Homologous Recombination Repair of Double-Strand Breaks. PLoS Genet. 2011, 7, e1002271. [Google Scholar] [CrossRef] [Green Version]
- Zierhut, C.; Diffley, J.F.X. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 2008, 27, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Leland, B.A.; King, M.C. Using LacO Arrays to Monitor DNA Double-Strand Break Dynamics in Live Schizosaccharomyces pombe Cells. In Cancer Genomics and Proteomics: Methods and Protocols, 2nd ed.; Humana Press: New York, NY, USA, 2014; Volume 1176, pp. 127–141. [Google Scholar]
- Yu, Y.; Ren, J.Y.; Zhang, J.M.; Suo, F.; Fang, X.F.; Wu, F.; Du, L.L. A proteome-wide visual screen identifies fission yeast proteins localizing to DNA double-strand breaks. DNA Repair 2013, 12, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, A.; Arai, R.; Yashiroda, Y.; Shirai, A.; Kamata, A.; Sekido, S.; Kobayashi, Y.; Hashimoto, A.; Hamamoto, M.; Hiraoka, Y.; et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 2006, 24, 841–847. [Google Scholar] [CrossRef]
- Goedecke, W.; Pfeiffer, P.; Vielmetter, W. Nonhomologous DNA end joining in Schizosaccharomyces pombe efficiently eliminates DNA double-strand-breaks from haploid sequences. Nucleic Acids Res. 1994, 22, 2094–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolis, K.G.; Nimmo, E.R.; Hartsuiker, E.; Carr, A.M.; Jeggo, P.A.; Allshire, R.C. Novel functional requirements for non-homologous DNA end joining in Schizosaccharomyces pombe. EMBO J. 2001, 20, 210–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulton, S.J.; Jackson, S.P. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996, 15, 5093–5103. [Google Scholar] [CrossRef] [PubMed]
- Hentges, P.; Ahnesorg, P.; Pitcher, R.S.; Bruce, C.K.; Kysela, B.; Green, A.J.; Bianchi, J.; Wilson, T.E.; Jackson, S.P.; Doherty, A.J. Evolutionary and functional conservation of the DNA non-homologous end-joining protein, XLF/cernunnos. J. Biol. Chem. 2006, 281, 37517–37526. [Google Scholar] [CrossRef] [Green Version]
- Decottignies, A. Capture of extranuclear DNA at fission yeast double-strand breaks. Genetics 2005, 171, 1535–1548. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yu, Y.; Suo, F.; Sun, L.L.; Zhao, D.; Du, L.L. Genome-wide Screens for Sensitivity to Ionizing Radiation Identify the Fission Yeast Nonhomologous End Joining Factor Xrc4. G3-Genes Genom Genet. 2014, 4, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Li, J.; Li, M.; Dou, K.; Zhang, M.J.; Suo, F.; Du, L.L. Multiple end joining mechanisms repair a chromosomal DNA break in fission yeast. DNA Repair 2012, 11, 120–130. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Zhou, G.; Lajeunesse, M.; Le, N.; Stawicki, B.N.; Corcino, Y.L.; Berkner, K.L.; Runge, K.W. Nonhomologous End-Joining with Minimal Sequence Loss Is Promoted by the Mre11-Rad50-Nbs1-Ctp1 Complex in Schizosaccharomyces pombe. Genetics 2017, 206, 481–496. [Google Scholar] [CrossRef] [Green Version]
- Almeida, H.; Godinho Ferreira, M. Spontaneous telomere to telomere fusions occur in unperturbed fission yeast cells. Nucleic Acids Res. 2013, 41, 3056–3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van, H.T.; Santos, M.A. Histone modifications and the DNA double-strand break response. Cell Cycle 2018, 17, 2399–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, A.S.; Hawley, B.R.; Wilczynska, A.; Bushell, M. The roles of RNA in DNA double-strand break repair. Br. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Cellular pathways of mitotic DNA double strand break repair. In a diploid cell, a DSB may occur in one of the two homologous chromosomes (1). The DSB may be repaired by non-homologous end joining (NHEJ) (2). When repair occurs by homologous recombination (HR), the DSB is first resected (3) to expose areas of single stranded DNA. If direct repeats (green areas) exist on the same chromosome, the break may be repaired by single strand annealing (SSA) (4). If homology is not found, the chromosome may be entirely lost (5). When homology is found elsewhere, the broken ends may invade this region (6). In synthesis dependent strand annealing (SDSA) (7) the invading strand may copy a small region then release and re-anneal. In break induced replication (BIR) (8) the invading strand may copy to the end of the red chromosome. In this case the right part of the broken black chromosome is lost. Occasionally, a more complex double Holiday junction (dHJ) may be established (9), the resolution of which can result in crossovers (CO) or non-crossovers (NCO).
Figure 1.
Cellular pathways of mitotic DNA double strand break repair. In a diploid cell, a DSB may occur in one of the two homologous chromosomes (1). The DSB may be repaired by non-homologous end joining (NHEJ) (2). When repair occurs by homologous recombination (HR), the DSB is first resected (3) to expose areas of single stranded DNA. If direct repeats (green areas) exist on the same chromosome, the break may be repaired by single strand annealing (SSA) (4). If homology is not found, the chromosome may be entirely lost (5). When homology is found elsewhere, the broken ends may invade this region (6). In synthesis dependent strand annealing (SDSA) (7) the invading strand may copy a small region then release and re-anneal. In break induced replication (BIR) (8) the invading strand may copy to the end of the red chromosome. In this case the right part of the broken black chromosome is lost. Occasionally, a more complex double Holiday junction (dHJ) may be established (9), the resolution of which can result in crossovers (CO) or non-crossovers (NCO).

Figure 2.
S. pombe mini-chromosome derivatives of Ch.III to study recombination. (A) Diagram of Chromosome III indicating the physical position of some genes. The numbers indicate kilobases. ade6-M210 provides intra-allelic complementation with ade6-M216. Ch.III is approximately 3.5 Mb. (B) The Ch16 mini-chromosome isolated by Niwa et al. Ch16 is 0.5 Mb and lacks rDNA repeats. Chromosome loss can be monitored by appearance of adenine auxotrophs. (C) Ch16-MG designed by Prudden et al. with the HO endonuclease restriction site at the rad21 locus. This chromosome can monitor, gene conversion, NHEJ and chromosome loss. (D) Modified Ch16-MG to include the his3+ marker close to the right telomere which can monitor LOH due to BIR. (E) The arg3+ marker was placed on the left arm of Ch16-MGH to produce Ch16-RMGAH. The arg3+ marker allows monitoring of extensive BIR that leads to loss of all markers on the right arm but retention of the left arm. (F) This mini-chromosome was generated to show that the outcomes of the assay in E were not dependent on break position. (G) A mini-chromosome designed to investigate recombination at centromeric repeats. (H) A mini-chromosome to investigate chromosome loss by red/white sectoring. The HO endonuclease is integrated in the left arm of the chromosome and marked with LEU2.
Figure 2.
S. pombe mini-chromosome derivatives of Ch.III to study recombination. (A) Diagram of Chromosome III indicating the physical position of some genes. The numbers indicate kilobases. ade6-M210 provides intra-allelic complementation with ade6-M216. Ch.III is approximately 3.5 Mb. (B) The Ch16 mini-chromosome isolated by Niwa et al. Ch16 is 0.5 Mb and lacks rDNA repeats. Chromosome loss can be monitored by appearance of adenine auxotrophs. (C) Ch16-MG designed by Prudden et al. with the HO endonuclease restriction site at the rad21 locus. This chromosome can monitor, gene conversion, NHEJ and chromosome loss. (D) Modified Ch16-MG to include the his3+ marker close to the right telomere which can monitor LOH due to BIR. (E) The arg3+ marker was placed on the left arm of Ch16-MGH to produce Ch16-RMGAH. The arg3+ marker allows monitoring of extensive BIR that leads to loss of all markers on the right arm but retention of the left arm. (F) This mini-chromosome was generated to show that the outcomes of the assay in E were not dependent on break position. (G) A mini-chromosome designed to investigate recombination at centromeric repeats. (H) A mini-chromosome to investigate chromosome loss by red/white sectoring. The HO endonuclease is integrated in the left arm of the chromosome and marked with LEU2.

Figure 3.
Recombination at tandem repeats. (A) An assay to study recombination at non-tandem direct repeats. The ade6-L469 and ade6-M26 or ade6-M375 inactivating alleles were placed on either side of a functional ura4+ gene. White dots represent the positions of the mutations in the ade6 alleles. The strain is adenine auxotrophic and uracil prototrophic (ade-ura+). Different repair pathways lead to various phenotypic outcomes. Unequal sister chromatid exchange (1) produces both ade+ura- and ade-ura+ phenotypes while single strand annealing (2) or loop out (3) produces only ade+ura-. ade+ura+ phenotypes also arose for which the Whitby lab proposed a fork regression and mismatch repair model. (B) An assay similar to (A) but with the S. cerevisiae HO endonuclease restriction site (MATa) either between the ura4+ and the ade6-M26/375 alleles or in the ade6-L469 allele. Shown are phenotypes that may arise by single strand annealing. (C) An assay similar to (A) except that ura4+ was replaced with his3+. The assay was further modified by Osman et al., 2016 to introduce a nick site between the his3+ and ade6-M375 repeat. The nick is created by the M13 bacteriophage gpII enzyme. A replication fork which approaches from the right converts this nick into a DSB. (D) The assay in (C) was modified by introducing the RTS1 pause site (black arrows) in between the his3+ and ade6-M375 or within the ade6-L469 allele. Because RTS1 is polar, two different strains were made for RTS1(A) and RTS1(B) each with a different orientation AO = active orientation, IO = inactive orientation) of the pause site. (E) The RTS1 was cloned near ori1323 while the ade-his-ade cassette was placed at different distances (indicated in kilobases) from the RTS1. The relative positions of endogenous replication origins are also shown. (F) An assay to test collisions of replication machinery with RNA Pol III. The tRNAGLU gene was placed in either of the two orientations between the his3+ and the ade6-M375 allele while the RTS1 pause site was placed on the right side of ade6-M375. (G) Three LacI arrays using the ade-his-ade cassette to allow microscopic visualization of repair dynamics. (H) An assay to study single strand annealing. Two LEU2 fragments with overlapping regions (black arrows) were placed on either side of a functional his3+ gene. The MATa site was placed on the 5′ end of the his3+ gene. (I) An assay similar to (H) except that his3+ is flanked by ura4 with overlapping regions (arrows) instead of LEU2. The MATa site was placed on the 3′ end of his3+.
Figure 3.
Recombination at tandem repeats. (A) An assay to study recombination at non-tandem direct repeats. The ade6-L469 and ade6-M26 or ade6-M375 inactivating alleles were placed on either side of a functional ura4+ gene. White dots represent the positions of the mutations in the ade6 alleles. The strain is adenine auxotrophic and uracil prototrophic (ade-ura+). Different repair pathways lead to various phenotypic outcomes. Unequal sister chromatid exchange (1) produces both ade+ura- and ade-ura+ phenotypes while single strand annealing (2) or loop out (3) produces only ade+ura-. ade+ura+ phenotypes also arose for which the Whitby lab proposed a fork regression and mismatch repair model. (B) An assay similar to (A) but with the S. cerevisiae HO endonuclease restriction site (MATa) either between the ura4+ and the ade6-M26/375 alleles or in the ade6-L469 allele. Shown are phenotypes that may arise by single strand annealing. (C) An assay similar to (A) except that ura4+ was replaced with his3+. The assay was further modified by Osman et al., 2016 to introduce a nick site between the his3+ and ade6-M375 repeat. The nick is created by the M13 bacteriophage gpII enzyme. A replication fork which approaches from the right converts this nick into a DSB. (D) The assay in (C) was modified by introducing the RTS1 pause site (black arrows) in between the his3+ and ade6-M375 or within the ade6-L469 allele. Because RTS1 is polar, two different strains were made for RTS1(A) and RTS1(B) each with a different orientation AO = active orientation, IO = inactive orientation) of the pause site. (E) The RTS1 was cloned near ori1323 while the ade-his-ade cassette was placed at different distances (indicated in kilobases) from the RTS1. The relative positions of endogenous replication origins are also shown. (F) An assay to test collisions of replication machinery with RNA Pol III. The tRNAGLU gene was placed in either of the two orientations between the his3+ and the ade6-M375 allele while the RTS1 pause site was placed on the right side of ade6-M375. (G) Three LacI arrays using the ade-his-ade cassette to allow microscopic visualization of repair dynamics. (H) An assay to study single strand annealing. Two LEU2 fragments with overlapping regions (black arrows) were placed on either side of a functional his3+ gene. The MATa site was placed on the 5′ end of the his3+ gene. (I) An assay similar to (H) except that his3+ is flanked by ura4 with overlapping regions (arrows) instead of LEU2. The MATa site was placed on the 3′ end of his3+.

Figure 4.
Assays to study gross chromosomal rearrangements at inverted repeats. (A) An assay to study fork stalling at repetitive elements. (1) The RTS1 pause site was placed on the right of a functional ura4+ gene (uraR), (2) left (Rura) or (3) both sides (RuraR). Fork stalling was assayed using pulse field gel electrophoresis (PFGE). (B) Chromosomal rearrangements of constructs in (A) can occur by two mechanisms: gene conversion without crossover and gene conversion with crossover. In both cases there is a recombination event between the RTS1 site next to the ura4+ cassette and the endogenous RTS1 at the MAT locus on Ch.II. Both types of events result in loss of ura4+. (C) Modification of construct in (A) to monitor chromosomal dynamics by fluorescence microscopy. A LacO array was placed on the telomere side of the RTS1/ura4+ cassettes. (D) Repetitive elements constructs to study chromosomal rearrangements. (1) RTS1 cassettes were placed on both sides of two directional ura4+ repeats (Ru(dir)uR). (2) RTS1 was placed on both sides of inverted ura4+ repeats spaced by 14 bp of unique sequence (RuiuR). A construct without the 14bp spacer was also made (RuuR) (3) In oRuiuR the RTS1 is in opposite direction from the RuiuR construct. (4) In the RuhR construct, one of the ura4+ repeats was replaced with his3+. (E) Several constructs to test the effect of repeat length and the position of the replication pause sites in producing rearrangements. (1) Two ura4+ inverted repeats spaced by 14 bp of unique sequence were engineered with 0.2 Kb (Rpal1R) or 1.8 Kb (Rpal2R) his3+ sequence between the right ura4+ repeat and the RTS1 pause site. An additional short ura4+ was placed between the left RTS1 and the ura4+ repeat (yellow arrow). (2) Tpal1R and Tpal2R constructs are similar to constructs in (1) except that the left RTS1 was replaced with three TER2/3 sequences. (3) In this construct the right ura4+ repeat was replaced by a unique sequence. (4) Each of the ura4+ repeat sizes were varied between 30–1200 base pairs. (5) The spacer between the inverted repeats was varied between 7–250 base pairs. (6) ura4+ cassette flanked by the TER2/3 termination sites. (7). Direct repeats but one of the ura4+ repeats was replaced with a ura5+ repeat. (F) The ura4+ and RTS1 pause sites were placed next to known origins of replication (3004, 3005, 3006/7). (1) Co-directional ura4+ and RTS1 with RTS1 placed either after (1) or in front (2) of the ura4+ repeat. (3) Construct similar to (A) (2) and (4) construct similar to (A) (3) placed between the 3005 and 3006/7 replication origins. (5) Construct similar to (4) but with RTS1 in the opposite orientation. (G) Constructs to test microhomology mediate repair caused by rescued replication forks. (1) Two different microhomology constructs. ura4-dup20/ura4sd20 was engineered by placing two 20 bp (grey boxes) repeats flanked by 5 bp of microhomology sequences (black boxes) in the middle of the ura4+ gene. ura4-dup22/ura4sd22 is has 22 bp repeats flanked by 4 bp of homology. Microhomology mediated repair was investigated either in the absence of a pause site (2) or with the pause site placed on the left (3) or right (4) of the construct. Only ura4sd20 is shown.
Figure 4.
Assays to study gross chromosomal rearrangements at inverted repeats. (A) An assay to study fork stalling at repetitive elements. (1) The RTS1 pause site was placed on the right of a functional ura4+ gene (uraR), (2) left (Rura) or (3) both sides (RuraR). Fork stalling was assayed using pulse field gel electrophoresis (PFGE). (B) Chromosomal rearrangements of constructs in (A) can occur by two mechanisms: gene conversion without crossover and gene conversion with crossover. In both cases there is a recombination event between the RTS1 site next to the ura4+ cassette and the endogenous RTS1 at the MAT locus on Ch.II. Both types of events result in loss of ura4+. (C) Modification of construct in (A) to monitor chromosomal dynamics by fluorescence microscopy. A LacO array was placed on the telomere side of the RTS1/ura4+ cassettes. (D) Repetitive elements constructs to study chromosomal rearrangements. (1) RTS1 cassettes were placed on both sides of two directional ura4+ repeats (Ru(dir)uR). (2) RTS1 was placed on both sides of inverted ura4+ repeats spaced by 14 bp of unique sequence (RuiuR). A construct without the 14bp spacer was also made (RuuR) (3) In oRuiuR the RTS1 is in opposite direction from the RuiuR construct. (4) In the RuhR construct, one of the ura4+ repeats was replaced with his3+. (E) Several constructs to test the effect of repeat length and the position of the replication pause sites in producing rearrangements. (1) Two ura4+ inverted repeats spaced by 14 bp of unique sequence were engineered with 0.2 Kb (Rpal1R) or 1.8 Kb (Rpal2R) his3+ sequence between the right ura4+ repeat and the RTS1 pause site. An additional short ura4+ was placed between the left RTS1 and the ura4+ repeat (yellow arrow). (2) Tpal1R and Tpal2R constructs are similar to constructs in (1) except that the left RTS1 was replaced with three TER2/3 sequences. (3) In this construct the right ura4+ repeat was replaced by a unique sequence. (4) Each of the ura4+ repeat sizes were varied between 30–1200 base pairs. (5) The spacer between the inverted repeats was varied between 7–250 base pairs. (6) ura4+ cassette flanked by the TER2/3 termination sites. (7). Direct repeats but one of the ura4+ repeats was replaced with a ura5+ repeat. (F) The ura4+ and RTS1 pause sites were placed next to known origins of replication (3004, 3005, 3006/7). (1) Co-directional ura4+ and RTS1 with RTS1 placed either after (1) or in front (2) of the ura4+ repeat. (3) Construct similar to (A) (2) and (4) construct similar to (A) (3) placed between the 3005 and 3006/7 replication origins. (5) Construct similar to (4) but with RTS1 in the opposite orientation. (G) Constructs to test microhomology mediate repair caused by rescued replication forks. (1) Two different microhomology constructs. ura4-dup20/ura4sd20 was engineered by placing two 20 bp (grey boxes) repeats flanked by 5 bp of microhomology sequences (black boxes) in the middle of the ura4+ gene. ura4-dup22/ura4sd22 is has 22 bp repeats flanked by 4 bp of homology. Microhomology mediated repair was investigated either in the absence of a pause site (2) or with the pause site placed on the left (3) or right (4) of the construct. Only ura4sd20 is shown.

Figure 5.
Mating type switching in S. pombe. (A) Diagram of the process of mating type switching. The mating type region has three cassettes mat1, mat2-P(plus) and mat3-M(minus). Only mat1 is expressed with either P or M while mat2-P and mat3-M are silenced. The shading of mat2-P and mat3-M diagrams represents this silencing. Information switches from mat2-P and mat3-M onto mat1 so that mat1 expresses either P or M. All three cassettes are flanked by homologous regions (H1, H2) that facilitate recombination. In this diagram, we begin with a hypothetical example where mat1 expresses the M information. After one round of replication, an imprint consisting of insertion of one or two ribonucleotides or a single strand nick is made next to the mat1-M cassette on the lagging strand (blue star). For simplicity, the top diagram does not have the imprint. In the second round of replication, the leading strand collides with the imprint and produces a blunt one-ended DSB. To ensure that only the leading strand passes through the imprint, the replication fork from the other direction is paused and terminated by the RTS1 site. The one-ended break is repaired by copying information from the mat2-P or mat3-M regions. Since in our example the M information was expressed at mat1, the break may be repaired using mat2-P and the information at mat1 switches. Note that the imprint is always placed on the lagging strand with each DNA replication. (B) Exploiting the mating type loci to study rescue of collapsed forks. Collision of the leading strand with the imprint produces a blunt one-ended break that can be studied in WT and other control strains. h90 is a wild type strain capable of switching that represents a mix of M and P cells. The smt-0 has a deletion that eliminates the imprint while M2,3Δ have deletions in the mat2-M and mat3-P regions. smt-0 M2,3Δ is a double mutant. The PΔ17 mutant is similar to smt-0 except that in this strain the mat1 locus expresses the P information. (C) An assay to study silencing at the mating type loci. The functional ura4+ gene was places 150 base pairs from mat3-M. If repressed, the strain can grow on 5-fluoroorotic acid (5-FOA) which negatively selects against uracil prototrophs and die on minimal media lacking uracil. If de-repressed, the strain will die on 5-FOA and grow on minimal media lacking uracil.
Figure 5.
Mating type switching in S. pombe. (A) Diagram of the process of mating type switching. The mating type region has three cassettes mat1, mat2-P(plus) and mat3-M(minus). Only mat1 is expressed with either P or M while mat2-P and mat3-M are silenced. The shading of mat2-P and mat3-M diagrams represents this silencing. Information switches from mat2-P and mat3-M onto mat1 so that mat1 expresses either P or M. All three cassettes are flanked by homologous regions (H1, H2) that facilitate recombination. In this diagram, we begin with a hypothetical example where mat1 expresses the M information. After one round of replication, an imprint consisting of insertion of one or two ribonucleotides or a single strand nick is made next to the mat1-M cassette on the lagging strand (blue star). For simplicity, the top diagram does not have the imprint. In the second round of replication, the leading strand collides with the imprint and produces a blunt one-ended DSB. To ensure that only the leading strand passes through the imprint, the replication fork from the other direction is paused and terminated by the RTS1 site. The one-ended break is repaired by copying information from the mat2-P or mat3-M regions. Since in our example the M information was expressed at mat1, the break may be repaired using mat2-P and the information at mat1 switches. Note that the imprint is always placed on the lagging strand with each DNA replication. (B) Exploiting the mating type loci to study rescue of collapsed forks. Collision of the leading strand with the imprint produces a blunt one-ended break that can be studied in WT and other control strains. h90 is a wild type strain capable of switching that represents a mix of M and P cells. The smt-0 has a deletion that eliminates the imprint while M2,3Δ have deletions in the mat2-M and mat3-P regions. smt-0 M2,3Δ is a double mutant. The PΔ17 mutant is similar to smt-0 except that in this strain the mat1 locus expresses the P information. (C) An assay to study silencing at the mating type loci. The functional ura4+ gene was places 150 base pairs from mat3-M. If repressed, the strain can grow on 5-fluoroorotic acid (5-FOA) which negatively selects against uracil prototrophs and die on minimal media lacking uracil. If de-repressed, the strain will die on 5-FOA and grow on minimal media lacking uracil.

Figure 6.
Fluorescence and biochemical assays. (A) An assay designed by Nabeshima et al. to study centromere dynamics. A LacO array was placed 30 Kb from the centromere of Ch.I. LacI-GFP binding allows visualization of centromeres. (B) Assays developed by Du et al. to study repair of a DSB. (1) The HO endonuclease restriction site marked with KAN was placed 1.5 Mb away from the LacO array at the arg3+ locus. (2) HO::KAN was placed 2.8 Kb from the LacO array. Constructs without a LacO array with the HO restriction site at arg3+ (3), lys1+ (4) or both arg3+ and lys1+ (5) were also engineered. These constructs can be used to study foci of fluorescently tagged DNA damage repair proteins. (C) The construct in (B) (3) was adapted for Chromatin Immunoprecipitation. PCR primers were engineered at 0.2, 2.0, 9.0 and 16.0 Kb from the break. Upon induction of the break, multiplex PCR can analyze binding of various proteins at all four locations at once. (D) An assay to integrate the LacO array and the HO restriction site anywhere in the yeast genome. Please see text for details. (E) An assay to study YFP tagged proteins interacting with a DSB. A lacO array that interacts with LacI-mCherry was placed 30 Kb from the HO endonuclease restriction site. The strain also contains Rad52 fused with cyan fluorescent protein (Rad52-CFP). A library of YFP tagged proteins was transformed in the cells and colocalization of all three foci (red, blue, yellow) was monitored.
Figure 6.
Fluorescence and biochemical assays. (A) An assay designed by Nabeshima et al. to study centromere dynamics. A LacO array was placed 30 Kb from the centromere of Ch.I. LacI-GFP binding allows visualization of centromeres. (B) Assays developed by Du et al. to study repair of a DSB. (1) The HO endonuclease restriction site marked with KAN was placed 1.5 Mb away from the LacO array at the arg3+ locus. (2) HO::KAN was placed 2.8 Kb from the LacO array. Constructs without a LacO array with the HO restriction site at arg3+ (3), lys1+ (4) or both arg3+ and lys1+ (5) were also engineered. These constructs can be used to study foci of fluorescently tagged DNA damage repair proteins. (C) The construct in (B) (3) was adapted for Chromatin Immunoprecipitation. PCR primers were engineered at 0.2, 2.0, 9.0 and 16.0 Kb from the break. Upon induction of the break, multiplex PCR can analyze binding of various proteins at all four locations at once. (D) An assay to integrate the LacO array and the HO restriction site anywhere in the yeast genome. Please see text for details. (E) An assay to study YFP tagged proteins interacting with a DSB. A lacO array that interacts with LacI-mCherry was placed 30 Kb from the HO endonuclease restriction site. The strain also contains Rad52 fused with cyan fluorescent protein (Rad52-CFP). A library of YFP tagged proteins was transformed in the cells and colocalization of all three foci (red, blue, yellow) was monitored.

Figure 7.
Assays to test non-homologous repair. (A) A plasmid-based assay to study non-homologous end joining. A λ DNA fragment is flanked by three unique restriction sites on each side. The plasmid is cut with different left and right restriction enzymes then transformed as a linear fragment into cells. Propagation of the transformed fragment in cells is only possible upon re-ligation of linear fragment ends. Thus, transformation frequency can be used as an indicator of repair efficiency. PCR across the junction (black arrows represent primers) followed by sequencing can determine the ligation patterns. (B) An assay similar to (A) except that the plasmid is cut with only one enzyme (PvuII). After relegation, the junction is analyzed for reconstitution of the PvuII restriction site or deletion and insertion of various nucleotides. (C) In this assay, PCR primers (black arrows) can be designed with various sequence overhangs (red lines). Linear fragments are amplified and transformed in yeast. Repair is analyzed as in (A). (D) The HO restriction site marked with natMX was cloned at the arg1+ locus on Ch.I. Continuous expression of HO endonuclease will kill all cells that do not repair the break or repair it correctly because it reconstitutes the restriction site and are vulnerable to re-cutting. Incorrect repair destroys the restriction site and allows cells to live. The authors used both Sanger sequencing and Illumina next generation sequencing to analyze the junction. (E) A transposon-based assay to study genome integration and excision. A plasmid encoding the transposase is co-transformed with another plasmid with transposon integration sequences (Hermes) flanking a KanMX cassette. De-repression of the transposase allows integration of the KanMX cassette randomly in the genome. Following integration, re-expression of the transposase will cause excision of the transposon. Analysis of the junction sequences can determine the form of repair. (F) An assay to test the role of telomeres in preventing chromosome end fusions. Two head to head telomeric repeats were cloned in the second intron of the his3+ gene. Each repeat also contains 80 bp of subtelomeric sequences. This telomere repeat interruption does not disrupt transcription of the his3+ gene. Consequently, the plasmid is his+leu+. The plasmid is linearized (destroying transcription of his3+) between the telomeric repeats and introduced into cells. Because each end is capped by telomeres, the fragment can be maintained as a mini-chromosome, which is leu+his-. Deletion of telomerase causes telomere attrition and fusion of the ends which reconstitutes the his3+ gene. Such fusions can be selected for on media lacking both histidine and leucine. Analysis of the fusion junction can determine the precision of repair.
Figure 7.
Assays to test non-homologous repair. (A) A plasmid-based assay to study non-homologous end joining. A λ DNA fragment is flanked by three unique restriction sites on each side. The plasmid is cut with different left and right restriction enzymes then transformed as a linear fragment into cells. Propagation of the transformed fragment in cells is only possible upon re-ligation of linear fragment ends. Thus, transformation frequency can be used as an indicator of repair efficiency. PCR across the junction (black arrows represent primers) followed by sequencing can determine the ligation patterns. (B) An assay similar to (A) except that the plasmid is cut with only one enzyme (PvuII). After relegation, the junction is analyzed for reconstitution of the PvuII restriction site or deletion and insertion of various nucleotides. (C) In this assay, PCR primers (black arrows) can be designed with various sequence overhangs (red lines). Linear fragments are amplified and transformed in yeast. Repair is analyzed as in (A). (D) The HO restriction site marked with natMX was cloned at the arg1+ locus on Ch.I. Continuous expression of HO endonuclease will kill all cells that do not repair the break or repair it correctly because it reconstitutes the restriction site and are vulnerable to re-cutting. Incorrect repair destroys the restriction site and allows cells to live. The authors used both Sanger sequencing and Illumina next generation sequencing to analyze the junction. (E) A transposon-based assay to study genome integration and excision. A plasmid encoding the transposase is co-transformed with another plasmid with transposon integration sequences (Hermes) flanking a KanMX cassette. De-repression of the transposase allows integration of the KanMX cassette randomly in the genome. Following integration, re-expression of the transposase will cause excision of the transposon. Analysis of the junction sequences can determine the form of repair. (F) An assay to test the role of telomeres in preventing chromosome end fusions. Two head to head telomeric repeats were cloned in the second intron of the his3+ gene. Each repeat also contains 80 bp of subtelomeric sequences. This telomere repeat interruption does not disrupt transcription of the his3+ gene. Consequently, the plasmid is his+leu+. The plasmid is linearized (destroying transcription of his3+) between the telomeric repeats and introduced into cells. Because each end is capped by telomeres, the fragment can be maintained as a mini-chromosome, which is leu+his-. Deletion of telomerase causes telomere attrition and fusion of the ends which reconstitutes the his3+ gene. Such fusions can be selected for on media lacking both histidine and leucine. Analysis of the fusion junction can determine the precision of repair.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hylton, H.M.; Lucas, B.E.; Petreaca, R.C. Schizosaccharomyces pombe Assays to Study Mitotic Recombination Outcomes. Genes 2020, 11, 79. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010079
AMA Style
Hylton HM, Lucas BE, Petreaca RC. Schizosaccharomyces pombe Assays to Study Mitotic Recombination Outcomes. Genes. 2020; 11(1):79. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010079
Chicago/Turabian StyleHylton, Hannah M., Bailey E. Lucas, and Ruben C. Petreaca. 2020. "Schizosaccharomyces pombe Assays to Study Mitotic Recombination Outcomes" Genes 11, no. 1: 79. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010079
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.