High-Throughput Genetic Testing in ALS: The Challenging Path of Variant Classification Considering the ACMG Guidelines

 , ,
, ,
Abstract
:1. Introduction
2. Patients and Methods: A Brief Description of the Studies Performed in Our Patient Cohort
2.1. Patients
2.2. Methods
- The first version included the following genes: ANG, ATXN2, CHCHD10, CHMP2B, CHRNA4, DAO, DCTN1, EPHA4, EWSR1, FIG4, FUS, HNRNPA1, HNRNPA2B1, MATR3, OPTN, PFN1, SETX, SIGMAR1, SOD1, SQSTM1, TAF15, TARDBP, UBQLN2, VAPB, and VCP;
- In the second version C9orf72, GLE1, GRN, MAPT, NIPA1, SS18L1, TBK1, and TUBA4A were added;
- In the third version the following genes were further added: ALS2, ANXA11, ARPP21, CCNF, NEK1, and SPG11.
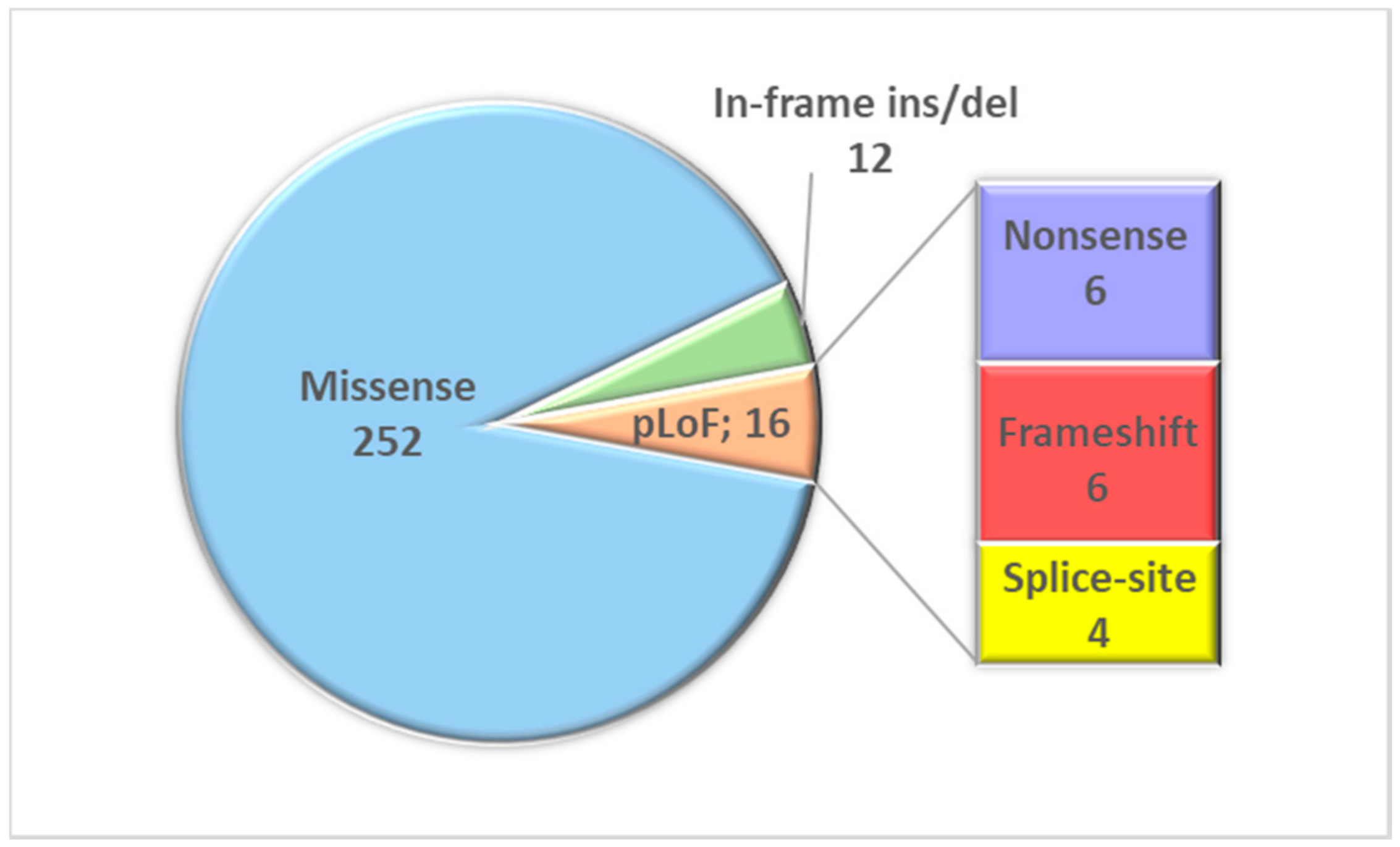
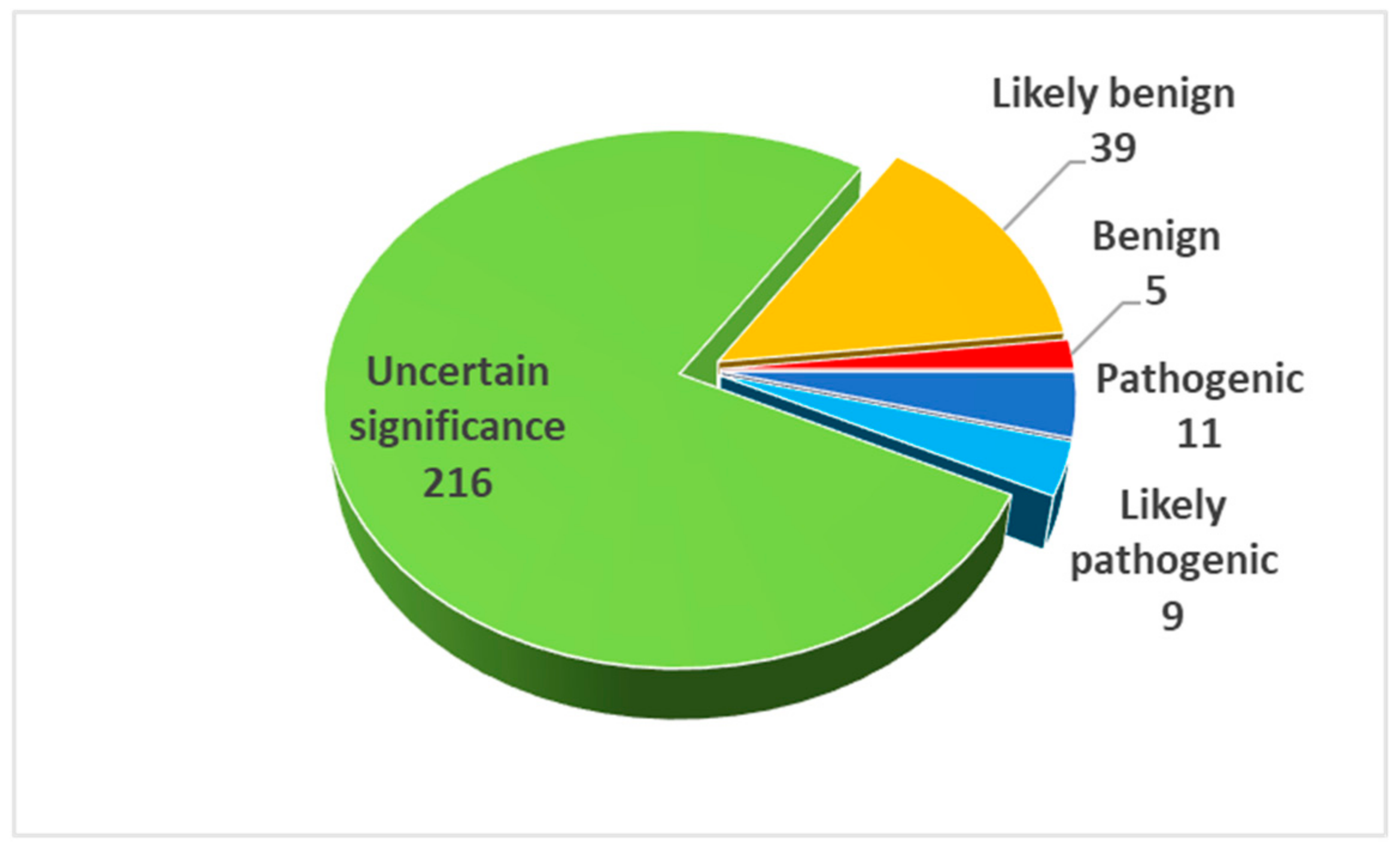
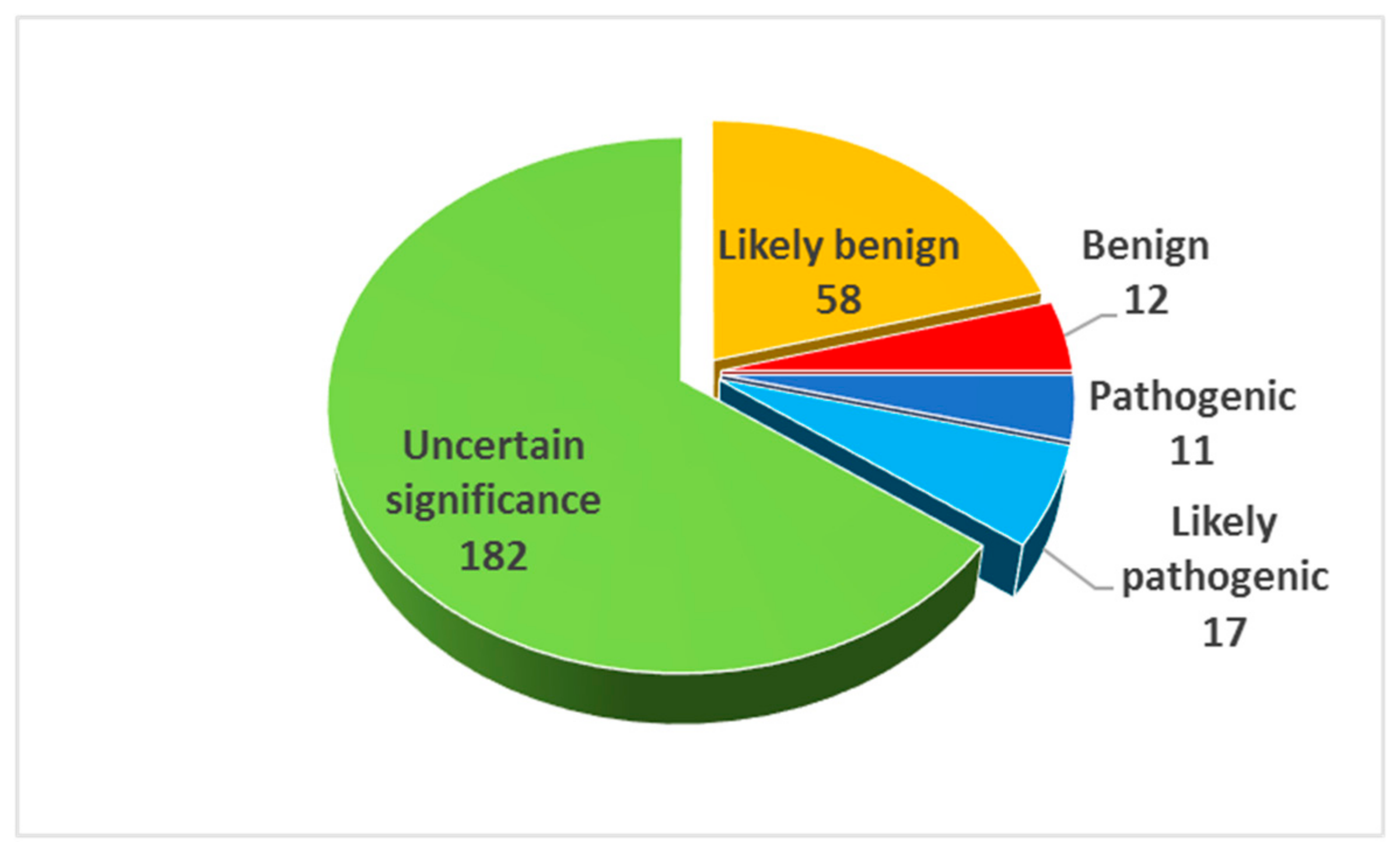
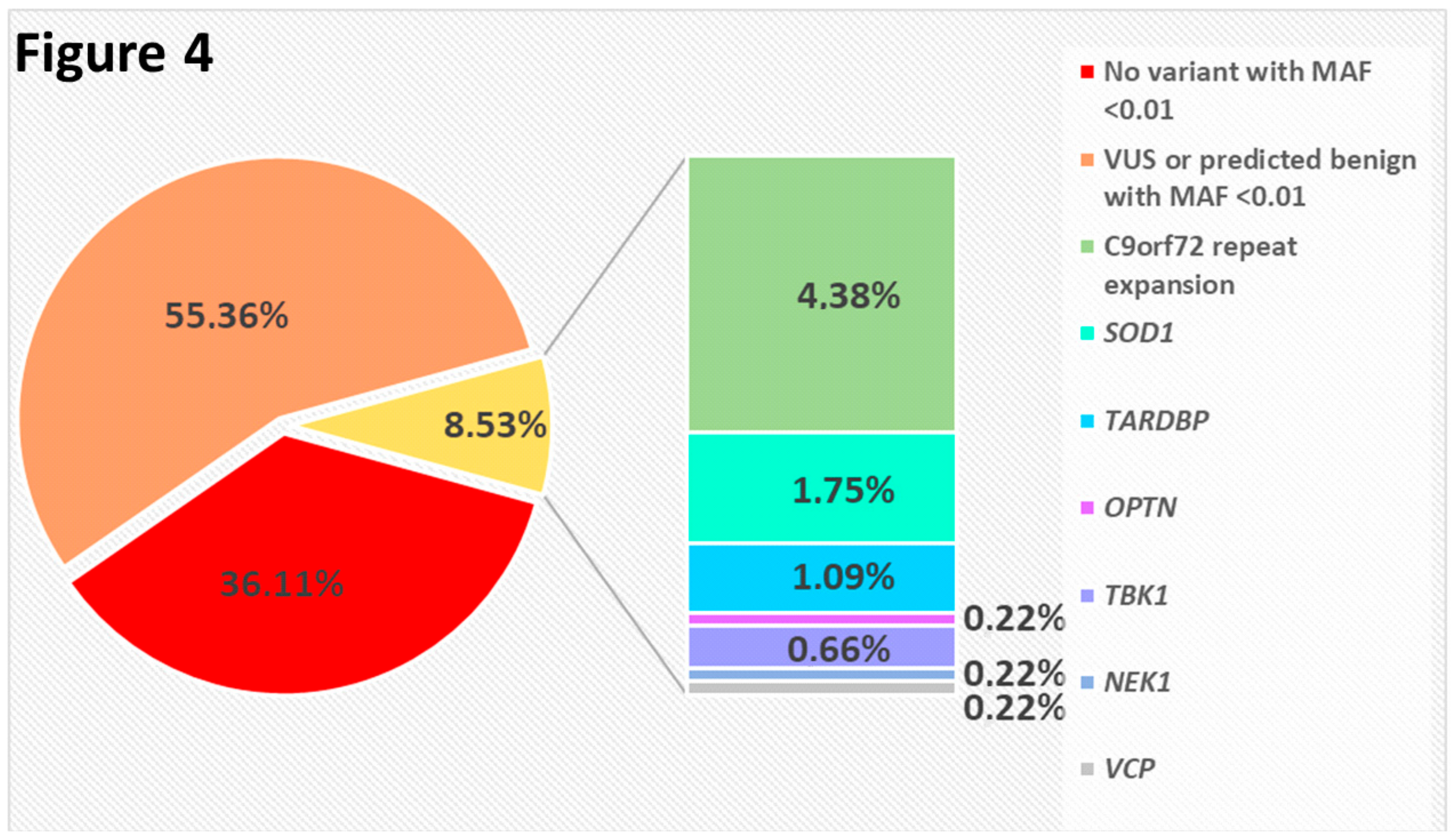
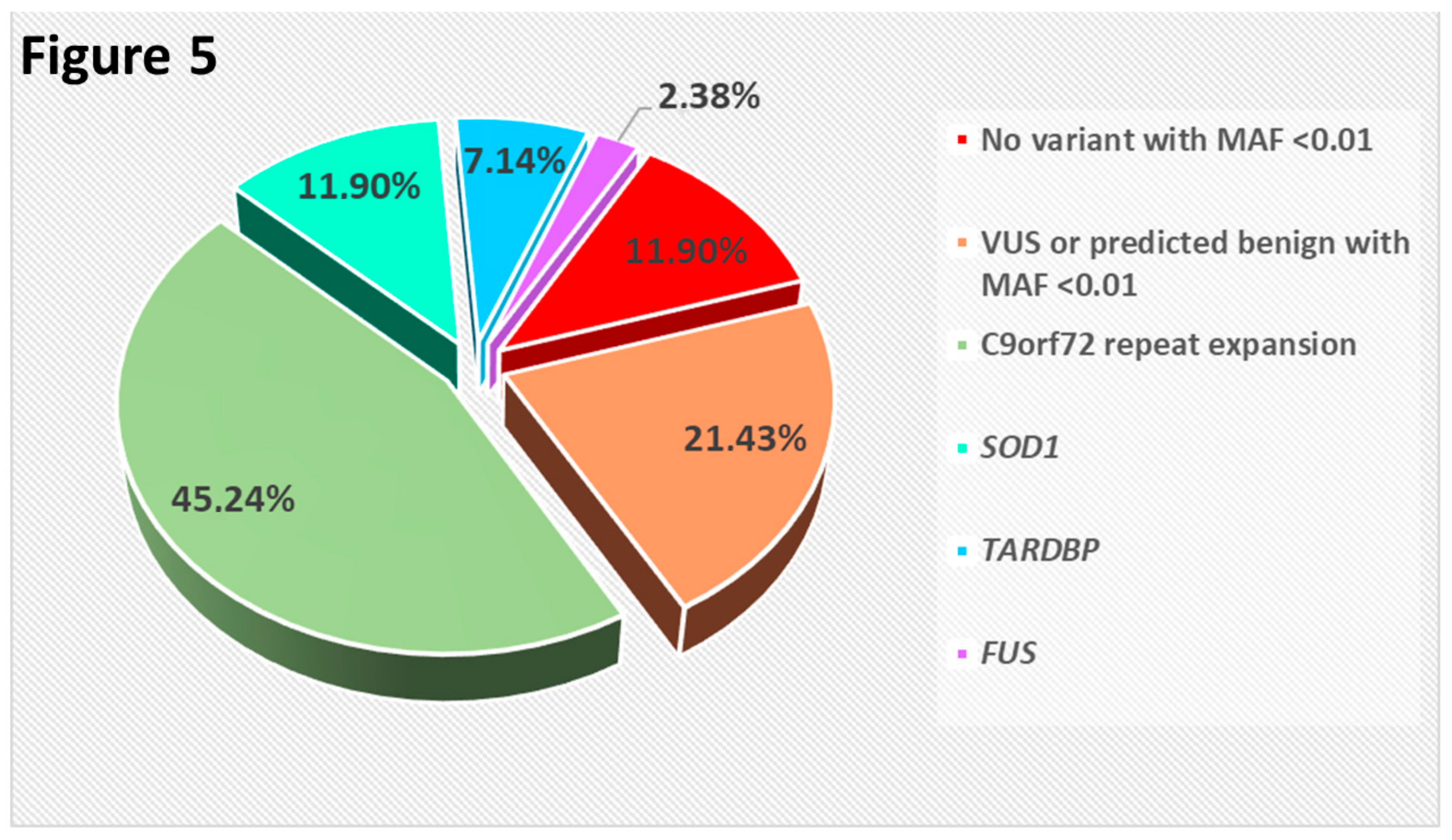
2.3. Result Summary
3. ALS Genes: What Are They?
- Pathogenic variants in certain genes can be identified only in a very small percentage of ALS patients and, for that reason, after papers initially proposed new candidates, additional confirmatory reports are still awaited. This holds even more true for the large number of genes recently identified, since time was insufficient to gather further independent evidences [17];
- Some genes were declared ALS-related as a result of large genome-wide association studies (GWAS). These studies contributed to identify some genes relevant for ALS, such as C9orf72 [14,18], KIF5A [19], and C21orf2 [20]. On the other hand, in remaining susceptibility loci, pathogenic variants have not been reported yet [21]. Nonetheless, the genotyping of the tagging SNPs identified (which are quite common in the general population) might play an important role in the near future in clinical trials and prognostic counseling [22];
- Another category of genes and loci efficiently detected by association studies are those acting as ALS phenotype modifiers, showing in a few cases an overlap with Mendelian genes and susceptibility genetic factors [23]. At present, the mutational screening of “modifier” genes would have little or no impact in a diagnostic setting;
- A final group of genes, often listed among those someway related to ALS, is constituted by genes coding for certain proteins, which play important roles in ALS pathogenesis, but in which no causative variant has been previously identified.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | ALS Locus | Chr | OMIM | Inherit | oe_Mis Upper | oe_Lof Upper | pLI | Other Associated Phenotypes | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| SOD1 | ALS1 | 21 | 147450 | AD,(AR) | 0.918 | 0.978 | 0.17 | - | [31] |
| ALS2 | ALS2 | 2 | 606352 | AR | 0.871 | 0.516 | <0.01 | Infantile onset ascending spastic paralysis | [32] |
| SETX | ALS4 | 9 | 608465 | AD | 1.054 | 0.296 | 0.95 | Spinocerebellar ataxia | [33] |
| SPG11 | ALS5 | 15 | 610844 | AR | 1.163 | 0.812 | <0.01 | Charcot–Marie–Tooth disease axonal type 2X, spastic paraplegia 11 (AR), juvenile amyotrophic lateral sclerosis | [34,35] |
| FUS | ALS6 | 16 | 137070 | AD,(AR) | 0.737 | 0.237 | 0.99 | Hereditary essential tremor | [36,37] |
| VAPB | ALS8 | 20 | 605704 | AD | 0.882 | 1.135 | 0.31 | Spinal muscular atrophy, late-onset | [38] |
| ANG | ALS9 | 14 | 105850 | AD | 1.172 | 1.893 | 0.28 | - | [39] |
| TARDBP | ALS10 | 1 | 605078 | AD | 0.385 | 0.275 | 0.98 | FTD | [40,41] |
| FIG4 | ALS11 | 6 | 609390 | AD | 0.831 | 1.24 | <0.01 | Charcot–Marie–Tooth disease, type 4J(AR); Yunis–Varon syndrome (AR) | [42] |
| OPTN | ALS12 | 10 | 602432 | AD,(AR) | 0.994 | 1.256 | <0.01 | Open angle glaucoma | [43] |
| VCP | ALS14 | 9 | 601023 | AD | 0.326 | 0.127 | 0.99 | FTD, inclusion body myopathy, Paget’s disease, Charcot–Marie–Tooth disease, type 2Y Charcot–Marie–Tooth disease, type 2Y | [44] |
| UBQLN2 | ALS15 | X | 300264 | XL | 0.824 | 0.448 | 0.84 | - | [45] |
| SIGMAR1 | ALS16 | 9 | 601978 | AR | 0.769 | 0.736 | 0.16 | Juvenile amyotrophic lateral sclerosis, distal hereditary motor neuropathies | [46] |
| CHMP2B | ALS17 | 3 | 609512 | AD | 1.072 | 1.216 | 0 | FTD | [47] |
| PFN1 | ALS18 | 17 | 176610 | AD | 0.54 | 0.686 | 0.73 | - | [48] |
| ERBB4 | ALS19 | 2 | 600543 | AD | 0.805 | 0.216 | 0.99 | - | [49] |
| HNRNPA1 | ALS20 | 12 | 164017 | AD | 0.461 | 0.364 | 0.93 | FTD, inclusion body myopathy, Paget’s disease | [50] |
| HNRNPA2B1 | 7 | 600124 | AD | 0.473 | 0.218 | 0.99 | FTD, inclusion body myopathy, Paget’s disease | [50] | |
| MATR3 | ALS21 | 5 | 164015 | AD | 0.704 | 0.079 | 1 | Distal myopathy with vocal cord and pharyngeal weakness | [51] |
| TUBA4A | ALS22 | 2 | 191110 | AD | 0.51 | 0.62 | 0.15 | - | [52] |
| ANXA11 | ALS23 | 10 | 602572 | AD | 1.143 | 0.98 | <0.01 | - | [53] |
| NEK1 | ALS24 | 4 | 604588 | AD | 0.948 | 0.864 | <0.01 | Short-rib thoracic dysplasia 6 with or without polydactyly (AR) | [54,55] |
| KIF5A | ALS25 | 12 | 602821 | AD | 0.652 | 0.252 | 0.99 | Spastic paraplegia 10 (AD), neonatal intractable myoclonus, | [19,56] |
| C9orf72 | FTDALS1 | 9 | 614260 | AD | 1.065 | 1.468 | <0.01 | FTD | [14,18] |
| CHCHD10 | FTDALS2 | 22 | 615903 | AD | 0.939 | 1.903 | <0.01 | Spinal muscular atrophy Jokela type, isolated mitochondrial myopathy, FTD | [57] |
| SQSTM1 | FTDALS3 | 5 | 601530 | AD | 1.359 | 0.745 | 0.01 | FTD, inclusion body myopathy, Paget’s disease, neurodegeneration with ataxia, dystonia, and gaze palsy, childhood-onset | [58] |
| TBK1 | FTDALS4 | 12 | 604834 | AD | 0.794 | 0.42 | 0.07 | - | [59,60] |
| CCNF | FTDALS5 | 16 | 600227 | AD | 0.979 | 0.419 | 0.12 | - | [61] |
| ATP13A2 | 1 | 610513 | AR | 0.913 | 0.584 | <0.01 | Kufor–Rakeb syndrome, spastic paraplegia 78 (AR) | [62] | |
| ATP7A | X | 300011 | AD | 0.883 | 0.216 | 0.99 | Menkes disease, occipital horn syndrome, spinal muscular atrophy (X-linked 3) | [63] | |
| C21orf2 | 21 | 603191 | AD | 1.205 | 1.35 | <0.01 | Retinal dystrophy with macular staphyloma, axial spondylometaphyseal dysplasia | [20] | |
| CACNA1A | 19 | 601011 | AD | 0.609 | 0.134 | 1 | Early infantile epileptic encephalopathy, episodic ataxia, migraine familial hemiplegic, spinocerebellar ataxia 6 | [64,65] | |
| CYLD | 16 | 605018 | AD | 0.612 | 0.204 | 0.99 | FTD, Familial cylindromatosis, Brooke–Spiegler syndrome, trichoepithelioma | [66] | |
| DAO | 12 | 124050 | AD | 1.102 | 1.697 | <0.01 | Schizophrenia | [67] | |
| DCTN1 | 2 | 601143 | AD | 0.967 | 0.364 | 0.08 | Perry syndrome (AD), neuronopathy, distal hereditary motor, type VIIB (AD) | [68] | |
| DNAJC7 | 17 | 601964 | AD | 0.621 | 0.28 | 0.99 | - | [69] | |
| ERLIN1 | 10 | 611604 | AR | 0.635 | 0.473 | 0.64 | Spastic paraplegia 62 AR | [70] | |
| ERLIN2 | 8 | 611605 | AR, AD | 0.709 | 0.833 | <0.01 | Spastic paraplegia 18 (AR) | [71] | |
| EWSR1 | 22 | 133450 | 0.718 | 0.278 | 0.99 | Ewing sarcoma | [72] | ||
| GARS | 7 | 600287 | AD | 0.911 | 0.402 | 0.3 | Distal hereditary motor neuronopathy | [73] | |
| GLE1 | 9 | 603371 | AD | 0.967 | 0.752 | <0.01 | Lethal congenital contracture syndrome 1 (AR); lethal arthrogryposis with anterior horn cell disease (AR) | [74] | |
| GLT8D1 | 3 | 618355 | AD | 0.993 | 1.134 | <0.01 | - | [75] | |
| GRN | 17 | 138945 | AD | 1.049 | 0.483 | 0.06 | FTD, neuronal ceroid lipofuscinosis, primary progressive aphasia | [76] | |
| MAPT | 17 | 157140 | AD | 0.873 | 0.596 | <0.01 | FTD, Pick’s disease, progressive supranuclear palsy | [77] | |
| NEFH | 22 | 162230 | 1.008 | 1.064 | <0.01 | Charcot–Marie–Tooth disease, axonal type 2CC | [78,79] | ||
| PRPH | 12 | 170710 | AD | 1.006 | 1.383 | <0.01 | - | [80] | |
| RAPGEF2 | 4 | 609530 | AD | 0.693 | 0.197 | 1 | - | [81] | |
| SPAST | 2 | 604277 | AD | 0.896 | 0.224 | 0.99 | Spastic paraplegia 4 (AD) | [82] | |
| SPG7 | 16 | 602783 | AD | 1.196 | 1.651 | <0.01 | Spastic paraplegia 7 (AR) | [83] | |
| SS18L1 | 20 | 606472 | AD | 0.818 | 0.306 | 0.98 | - | [84] | |
| TAF15 | 17 | 601574 | AD | 0.9 | 0.399 | 0.15 | Chondrosarcoma, extraskeletal myxoid | [85] | |
| TIA1 | 2 | 603518 | AD | 0.694 | 0.443 | 0.26 | Welander distal myopathy | [86] |
4. Applying the ACMG Standards and Guidelines for the Interpretation of ALS-Related Variants
4.1. The Role of Null Variants
4.2. The Role of Missense Variants
4.3. In-Frame Deletions and Insertions
4.4. De Novo Variants and Parental Testing
4.5. Variant Frequency in Cases and Control Populations
- The inheritance model to consider is “monoallelic” for most ALS genes;
- The “prevalence” value required should not correspond to ALS prevalence but rather to the cumulative lifetime risk of ALS (i.e., ~1/300) [130], which would more closely describe the fraction of the population carrying variants responsible for ALS;
- The allelic heterogeneity is likely very low and we can conservatively set it at 0.1, assuming that for each ALS-gene there should be at least 10 different pathogenic variants (the C9orf72 repeat expansion would be the only relevant exception, but we do not expect to find other sequence variants with such features in ALS);
- The allelic heterogeneity could also be set at 0.05, considering the amount of genes that have been claimed so far of being responsible for ALS and the fact that none of them appears to be responsible for more than 5% of cases (again, with the well-known and expectedly isolated exception of the C9orf72);
- The penetrance is even more difficult to evaluate, since a large difference among genes and variants may exist. A recent estimate of the C9orf72 repeat expansion penetrance shows that it might be nearly complete at 83 years [131]. Supposing that for other genes might be smaller at the same age, we could tentatively consider a value of 80%. Nonetheless, this value should be modelled to the age distribution of reference cohorts.
4.6. Familial Segregation
4.7. Functional Studies
4.8. Reputable Source
4.9. Further Considerations
- The definition of an allele frequency to be considered either in favor (PM2) or against (PP2) the pathogenic role of variants. Different thresholds should be established considering the different models of inheritance;
- The study of larger cohorts of patients and controls to allow for a more precise evaluation of the relative risk associated with as many different variants as possible (PS4);
- The collaborative efforts in sharing data about patients tested and variants identified from laboratories around the world. This would lead to a more frequent use of criteria that may have a strong impact on variant classification (i.e., PS1, PM1, PM5, PP5, and BP6);
- The development and the selection of computational tools that explore the possibility of a certain variant to have ALS-specific functional consequences (PP3 and BP4);
- The identification of causative variants in families with a relevant number of affected people. Though they are a rare finding, their relevance is invaluable for both the identification of new ALS-associated genes and the classification of further variants (PP1);
- The development of standardized functional assays for the assessment of the variant involvement in ALS pathogenesis.
5. The Complex Genetic Architecture of ALS and the Challenges in the Identification of Pathogenic Variants
6. Conclusions
- ALS diagnosis remains based on clinical, not genetic criteria. The improvement of the currently available variant classification systems might provide better foundations to use genetic results among the diagnostic criteria. This will acquire increasing importance if a presymptomatic diagnosis should become crucial in therapeutic interventions yet to come;
- Patient and family counseling is already a significant step in the process of care, yet consensus guidelines are missing, which could give indications of the results worth being communicated as useless or, even worse, a source of anxiety for patients and their relatives;
- Though the role of small-effect variants may appear of limited interest, their identification should not be neglected, since they could constitute a potential target for possible tailored therapeutic approaches in the context of the personalized medicine opportunity.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Chio, A.; Battistini, S.; Calvo, A.; Caponnetto, C.; Conforti, F.L.; Corbo, M.; Giannini, F.; Mandrioli, J.; Mora, G.; Sabatelli, M.; et al. Genetic counselling in ALS: Facts, uncertainties and clinical suggestions. J. Neurol. Neurosurg. Psychiatry 2014, 85, 478–485. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Li, H.-F.; Tan, G.-H.; Tao, Q.-Q.; Ni, W.; Cheng, X.-W.; Xiong, Z.-Q.; Wu, Z.-Y. Identify mutation in amyotrophic lateral sclerosis cases using HaloPlex target enrichment system. Neurobiol. Aging 2014, 35, 2881.e11–2881.e15. [Google Scholar] [CrossRef]
- Pensato, V.; Magri, S.; Dalla Bella, E.; Tannorella, P.; Bersano, E.; Sorarù, G.; Gatti, M.; Ticozzi, N.; Taroni, F.; Lauria, G.; et al. Sorting Rare ALS Genetic Variants by Targeted Re-Sequencing Panel in Italian Patients: OPTN, VCP, and SQSTM1 Variants Account for 3% of Rare Genetic Forms. J. Clin. Med. 2020, 9, 412. [Google Scholar] [CrossRef] [Green Version]
- Roggenbuck, J.; Palettas, M.; Vicini, L.; Patel, R.; Quick, A.; Kolb, S.J. Incidence of pathogenic, likely pathogenic, and uncertain ALS variants in a clinic cohort. Neurol. Genet. 2020, 6, e390. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Ludolph, A.; Drory, V.; Hardiman, O.; Nakano, I.; Ravits, J.; Robberecht, W.; Shefner, J. A revision of the El Escorial criteria-2015. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 291–292. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Lattante, S.; Conte, A.; Zollino, M.; Luigetti, M.; Del Grande, A.; Marangi, G.; Romano, A.; Marcaccio, A.; Meleo, E.; Bisogni, G.; et al. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology 2012, 79, 66–72. [Google Scholar] [CrossRef]
- Lattante, S.; Pomponi, M.G.; Conte, A.; Marangi, G.; Bisogni, G.; Patanella, A.K.; Meleo, E.; Lunetta, C.; Riva, N.; Mosca, L.; et al. ATXN1 intermediate-length polyglutamine expansions are associated with amyotrophic lateral sclerosis. Neurobiol. Aging 2018, 64, 157.e1–157.e5. [Google Scholar] [CrossRef] [Green Version]
- Project MinE ALS Sequencing Consortium. CHCHD10 variants in amyotrophic lateral sclerosis: Where is the evidence? Ann. Neurol. 2018, 84, 110–116. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e6. [Google Scholar] [CrossRef] [Green Version]
- van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Jiang, H.; Wang, F.; Li, S.; Wu, C.; Bao, J.; Zhu, Y.; Xu, Z.; Liu, B.; Ren, H.; et al. UNC13A variant rs12608932 is associated with increased risk of amyotrophic lateral sclerosis and reduced patient survival: A meta-analysis. Neurol. Sci. 2019, 40, 2293–2302. [Google Scholar] [CrossRef]
- Tan, H.H.G.; Westeneng, H.; Burgh, H.K.; Es, M.A.; Bakker, L.A.; Veenhuijzen, K.; Eijk, K.R.; Eijk, R.P.A.; Veldink, J.H.; Berg, L.H. The Distinct Traits of the UNC13A Polymorphism in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 88, 796–806. [Google Scholar] [CrossRef]
- Yanagi, K.S.; Wu, Z.; Amaya, J.; Chapkis, N.; Duffy, A.M.; Hajdarovic, K.H.; Held, A.; Mathur, A.D.; Russo, K.; Ryan, V.H.; et al. Meta-analysis of Genetic Modifiers Reveals Candidate Dysregulated Pathways in Amyotrophic Lateral Sclerosis. Neuroscience 2019, 396, A3–A20. [Google Scholar] [CrossRef]
- Sabatelli, M.; Marangi, G.; Conte, A.; Tasca, G.; Zollino, M.; Lattante, S. New ALS-Related Genes Expand the Spectrum Paradigm of Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 266–275. [Google Scholar] [CrossRef]
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Benatar, M.; Wuu, J.; Fernandez, C.; Weihl, C.C.; Katzen, H.; Steele, J.; Oskarsson, B.; Taylor, J.P. Motor neuron involvement in multisystem proteinopathy: Implications for ALS. Neurology 2013, 80, 1874–1880. [Google Scholar] [CrossRef] [Green Version]
- Rezaie, T. Adult-Onset Primary Open-Angle Glaucoma Caused by Mutations in Optineurin. Science 2002, 295, 1077–1079. [Google Scholar] [CrossRef]
- Thiel, C.; Kessler, K.; Giessl, A.; Dimmler, A.; Shalev, S.A.; von der Haar, S.; Zenker, M.; Zahnleiter, D.; Stöss, H.; Beinder, E.; et al. NEK1 Mutations Cause Short-Rib Polydactyly Syndrome Type Majewski. Am. J. Hum. Genet. 2011, 88, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Pecoraro, V.; Mandrioli, J.; Carone, C.; Chiò, A.; Traynor, B.J.; Trenti, T. The NGS technology for the identification of genes associated with the ALS. A systematic review. Eur. J. Clin. Investig. 2020, 50, e13228. [Google Scholar] [CrossRef]
- Van Hoecke, A.; Schoonaert, L.; Lemmens, R.; Timmers, M.; Staats, K.A.; Laird, A.S.; Peeters, E.; Philips, T.; Goris, A.; Dubois, B.; et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat. Med. 2012, 18, 1418–1422. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Hadano, S.; Yanagisawa, Y.; Skaug, J.; Fichter, K.; Nasir, J.; Martindale, D.; Koop, B.F.; Scherer, S.W.; Nicholson, D.W.; Rouleau, G.A.; et al. Cloning and Characterization of Three Novel Genes, ALS2CR1, ALS2CR2, and ALS2CR3, in the Juvenile Amyotrophic Lateral Sclerosis (ALS2) Critical Region at Chromosome 2q33–q34: Candidate Genes for ALS2. Genomics 2001, 71, 200–213. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA Helicase Gene Mutations in a Form of Juvenile Amyotrophic Lateral Sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef] [Green Version]
- Orlacchio, A.; Babalini, C.; Borreca, A.; Patrono, C.; Massa, R.; Basaran, S.; Munhoz, R.P.; Rogaeva, E.A.; St George-Hyslop, P.H.; Bernardi, G.; et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010, 133, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Daoud, H.; Zhou, S.; Noreau, A.; Sabbagh, M.; Belzil, V.; Dionne-Laporte, A.; Tranchant, C.; Dion, P.; Rouleau, G.A. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol. Aging 2012, 33, 839.e5–839.e9. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, T.J.; Bosco, D.A.; LeClerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.A.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.M.; Gillingwater, T.; Webb, J.; et al. A Mutation in the Vesicle-Trafficking Protein VAPB Causes Late-Onset Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Greenway, M.J.; Andersen, P.M.; Russ, C.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG mutations segregate with familial and “sporadic” amyotrophic lateral sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious Variants of FIG4, a Phosphoinositide Phosphatase, in Patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef]
- Parkinson, N.; Ince, P.G.; Smith, M.O.; Highley, R.; Skibinski, G.; Andersen, P.M.; Morrison, K.E.; Pall, H.S.; Hardiman, O.; Collinge, J.; et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006, 67, 1074–1077. [Google Scholar] [CrossRef]
- Wu, C.-H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fukuda, Y.; Yoshimura, J.; Toyoda, A.; Kurppa, K.; Moritoyo, H.; Belzil, V.V.; Dion, P.A.; Higasa, K.; Doi, K.; et al. ERBB4 Mutations that Disrupt the Neuregulin-ErbB4 Pathway Cause Amyotrophic Lateral Sclerosis Type 19. Am. J. Hum. Genet. 2013, 93, 900–905. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Ticozzi, N.; Fallini, C.; Gkazi, A.S.; Topp, S.; Kenna, K.P.; Scotter, E.L.; Kost, J.; Keagle, P.; Miller, J.W.; et al. Exome-wide Rare Variant Analysis Identifies TUBA4A Mutations Associated with Familial ALS. Neuron 2014, 84, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.N.; Topp, S.D.; Fallini, C.; Shibata, H.; Chen, H.-J.; Troakes, C.; King, A.; Ticozzi, N.; Kenna, K.P.; Soragia-Gkazi, A.; et al. Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaad9157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, D.; Müller, K.; Wieland, T.; Weydt, P.; Böhm, S.; Lulé, D.; Hübers, A.; Neuwirth, C.; Weber, M.; Borck, G.; et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 2016, 139, e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenna, K.P.; van Doormaal, P.T.C.; Dekker, A.M.; Ticozzi, N.; Kenna, B.J.; Diekstra, F.P.; van Rheenen, W.; van Eijk, K.R.; Jones, A.R.; Keagle, P.; et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Yilmaz, R.; Müller, K.; Grehl, T.; Petri, S.; Meyer, T.; Grosskreutz, J.; Weydt, P.; Ruf, W.; Neuwirth, C.; et al. Hot-spot KIF5A mutations cause familial ALS. Brain 2018, 141, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [Green Version]
- Fecto, F.; Yan, J.; Pavan Vemula, S.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68, 1440. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.L.; Topp, S.; Yang, S.; Smith, B.; Fifita, J.A.; Warraich, S.T.; Zhang, K.Y.; Farrawell, N.; Vance, C.; Hu, X.; et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 2016, 7, 11253. [Google Scholar] [CrossRef] [PubMed]
- Spataro, R.; Kousi, M.; Farhan, S.M.K.; Willer, J.R.; Ross, J.P.; Dion, P.A.; Rouleau, G.A.; Daly, M.J.; Neale, B.M.; La Bella, V.; et al. Mutations in ATP13A2 (PARK9) are associated with an amyotrophic lateral sclerosis-like phenotype, implicating this locus in further phenotypic expansion. Hum. Genom. 2019, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.; Hong, S.-A.; Kim, K.-K.; Baek, D.; Lee, D.; Londhe, A.M.; Lee, M.; Yu, J.; McEachin, Z.T.; Bassell, G.J.; et al. CRISPR-mediated gene correction links the ATP7A M1311V mutations with amyotrophic lateral sclerosis pathogenesis in one individual. Commun. Biol. 2020, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, D.; Müller, K.; Gastl, R.; Gorges, M.; Otto, M.; Pinkhardt, E.H.; Kassubek, J.; Weishaupt, J.H.; Ludolph, A.C. Analysis of CACNA1A CAG repeat lengths in patients with familial ALS. Neurobiol. Aging 2019, 74, 235.e5–235.e8. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Li, Y.R.; Chesi, A.; Hart, M.P.; Ramos, D.; Jethava, N.; Hosangadi, D.; Epstein, J.; Hodges, B.; Bonini, N.M.; et al. Evaluating the prevalence of polyglutamine repeat expansions in amyotrophic lateral sclerosis. Neurology 2011, 76, 2062–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson-Stone, C.; Hallupp, M.; Shahheydari, H.; Ragagnin, A.M.G.; Chatterton, Z.; Carew-Jones, F.; Shepherd, C.E.; Stefen, H.; Paric, E.; Fath, T.; et al. CYLD is a causative gene for frontotemporal dementia–amyotrophic lateral sclerosis. Brain 2020, 143, 783–799. [Google Scholar] [CrossRef]
- Mitchell, J.; Paul, P.; Chen, H.-J.; Morris, A.; Payling, M.; Falchi, M.; Habgood, J.; Panoutsou, S.; Winkler, S.; Tisato, V.; et al. Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc. Natl. Acad. Sci. USA 2010, 107, 7556–7561. [Google Scholar] [CrossRef] [Green Version]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.F.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef] [Green Version]
- Farhan, S.M.K.; Howrigan, D.P.; Abbott, L.E.; Klim, J.R.; Topp, S.D.; Byrnes, A.E.; Churchhouse, C.; Phatnani, H.; Smith, B.N.; Rampersaud, E.; et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nat. Neurosci. 2019, 22, 1966–1974. [Google Scholar] [CrossRef]
- Tunca, C.; Akçimen, F.; Coşkun, C.; Gündoğdu-Eken, A.; Kocoglu, C.; Çevik, B.; Bekircan-Kurt, C.E.; Tan, E.; Başak, A.N. ERLIN1 mutations cause teenage-onset slowly progressive ALS in a large Turkish pedigree. Eur. J. Hum. Genet. 2018, 26, 745–748. [Google Scholar] [CrossRef]
- Amador, M.-D.-M.; Muratet, F.; Teyssou, E.; Banneau, G.; Danel-Brunaud, V.; Allart, E.; Antoine, J.-C.; Camdessanché, J.-P.; Anheim, M.; Rudolf, G.; et al. Spastic paraplegia due to recessive or dominant mutations in ERLIN2 can convert to ALS. Neurol. Genet. 2019, 5, e374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couthouis, J.; Hart, M.P.; Erion, R.; King, O.D.; Diaz, Z.; Nakaya, T.; Ibrahim, F.; Kim, H.-J.; Mojsilovic-Petrovic, J.; Panossian, S.; et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 2899–2911. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Brulard, C.; Beltran, S.; Marouillat, S.; Bakkouche, S.E.; Andres, C.R.; Blasco, H.; Vourc’h, P. Typical bulbar ALS can be linked to GARS mutation. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 275–277. [Google Scholar] [CrossRef]
- Kaneb, H.M.; Folkmann, A.W.; Belzil, V.V.; Jao, L.-E.; Leblond, C.S.; Girard, S.L.; Daoud, H.; Noreau, A.; Rochefort, D.; Hince, P.; et al. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2015, 24, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Cooper-Knock, J.; Moll, T.; Ramesh, T.; Castelli, L.; Beer, A.; Robins, H.; Fox, I.; Niedermoser, I.; Van Damme, P.; Moisse, M.; et al. Mutations in the Glycosyltransferase Domain of GLT8D1 Are Associated with Familial Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 26, 2298–2306.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schymick, J.C.; Yang, Y.; Andersen, P.M.; Vonsattel, J.P.; Greenway, M.; Momeni, P.; Elder, J.; Chio, A.; Restagno, G.; Robberecht, W.; et al. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J. Neurol. Neurosurg. Psychiatry 2006, 78, 754–756. [Google Scholar] [CrossRef]
- Vance, C.; Al-Chalabi, A.; Ruddy, D.; Smith, B.N.; Hu, X.; Sreedharan, J.; Siddique, T.; Schelhaas, H.J.; Kusters, B.; Troost, D.; et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain 2006, 129, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Figlewicz, D.A.; Krizus, A.; Martinoli, M.G.; Meininger, V.; Dib, M.; Rouleau, G.A.; Julien, J.-P. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet. 1994, 3, 1757–1761. [Google Scholar] [CrossRef]
- Al-Chalabi, A. Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum. Mol. Genet. 1999, 8, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.L.; He, C.Z.; Kaufmann, P.; Chin, S.S.; Naini, A.; Liem, R.K.H.; Mitsumoto, H.; Hays, A.P. A Pathogenic Peripherin Gene Mutation in a Patient with Amyotrophic Lateral Sclerosis. Brain Pathol. 2006, 14, 290–296. [Google Scholar] [CrossRef]
- Heo, K.; Lim, S.M.; Nahm, M.; Kim, Y.-E.; Oh, K.-W.; Park, H.T.; Ki, C.-S.; Kim, S.H.; Lee, S. A De Novo RAPGEF2 Variant Identified in a Sporadic Amyotrophic Lateral Sclerosis Patient Impairs Microtubule Stability and Axonal Mitochondria Distribution. Exp. Neurobiol. 2018, 27, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Krüger, S.; Battke, F.; Sprecher, A.; Munz, M.; Synofzik, M.; Schöls, L.; Gasser, T.; Grehl, T.; Prudlo, J.; Biskup, S. Rare Variants in Neurodegeneration Associated Genes Revealed by Targeted Panel Sequencing in a German ALS Cohort. Front. Mol. Neurosci. 2016, 9, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmanovic, A.; Widjaja, M.; Förster, A.; Weder, J.; Wattjes, M.P.; Lange, I.; Sarikidi, A.; Auber, B.; Raab, P.; Christians, A.; et al. SPG7 mutations in amyotrophic lateral sclerosis: A genetic link to hereditary spastic paraplegia. J. Neurol. 2020, 267, 2732–2743. [Google Scholar] [CrossRef]
- Chesi, A.; Staahl, B.T.; Jovičić, A.; Couthouis, J.; Fasolino, M.; Raphael, A.R.; Yamazaki, T.; Elias, L.; Polak, M.; Kelly, C.; et al. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat. Neurosci. 2013, 16, 851–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ticozzi, N.; Vance, C.; LeClerc, A.L.; Keagle, P.; Glass, J.D.; McKenna-Yasek, D.; Sapp, P.C.; Silani, V.; Bosco, D.A.; Shaw, C.E.; et al. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 156, 285–290. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808–816.e9. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Tuo, M.; Li, P.; Zhou, C. Association between TBK1 mutations and risk of amyotrophic lateral sclerosis/frontotemporal dementia spectrum: A meta-analysis. Neurol. Sci. 2018, 39, 811–820. [Google Scholar] [CrossRef]
- de Majo, M.; Topp, S.D.; Smith, B.N.; Nishimura, A.L.; Chen, H.-J.; Gkazi, A.S.; Miller, J.; Wong, C.H.; Vance, C.; Baas, F.; et al. ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol. Aging 2018, 71, 266.e1–266.e10. [Google Scholar] [CrossRef]
- Kim, Y.-E.; Oh, K.-W.; Noh, M.-Y.; Nahm, M.; Park, J.; Lim, S.M.; Jang, J.-H.; Cho, E.-H.; Ki, C.-S.; Lee, S.; et al. Genetic and functional analysis of TBK1 variants in Korean patients with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2017, 50, 170.e1–170.e6. [Google Scholar] [CrossRef]
- Feng, S.; Che, C.; Feng, S.; Liu, C.; Li, L.; Li, Y.; Huang, H.; Zou, Z. Novel mutation in optineurin causing aggressive ALS+/−frontotemporal dementia. Ann. Clin. Transl. Neurol. 2019, 6, 2377–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, R.; Müller, K.; Brenner, D.; Volk, A.E.; Borck, G.; Hermann, A.; Meitinger, T.; Strom, T.M.; Danzer, K.M.; Ludolph, A.C.; et al. SQSTM1/p62 variants in 486 patients with familial ALS from Germany and Sweden. Neurobiol. Aging 2020, 87, 139.e9–139.e15. [Google Scholar] [CrossRef] [PubMed]
- Teyssou, E.; Takeda, T.; Lebon, V.; Boillée, S.; Doukouré, B.; Bataillon, G.; Sazdovitch, V.; Cazeneuve, C.; Meininger, V.; LeGuern, E.; et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: Genetics and neuropathology. Acta Neuropathol. 2013, 125, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Alonso, N.; Calero-Paniagua, I.; del Pino-Montes, J. Clinical and Genetic Advances in Paget’s Disease of Bone: A Review. Clin. Rev. Bone Miner. Metab. 2017, 15, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haack, T.B.; Ignatius, E.; Calvo-Garrido, J.; Iuso, A.; Isohanni, P.; Maffezzini, C.; Lönnqvist, T.; Suomalainen, A.; Gorza, M.; Kremer, L.S.; et al. Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am. J. Hum. Genet. 2016, 99, 735–743. [Google Scholar] [CrossRef]
- Duis, J.; Dean, S.; Applegate, C.; Harper, A.; Xiao, R.; He, W.; Dollar, J.D.; Sun, L.R.; Waberski, M.B.; Crawford, T.O.; et al. KIF5A mutations cause an infantile onset phenotype including severe myoclonus with evidence of mitochondrial dysfunction. Ann. Neurol. 2016, 80, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Rydzanicz, M.; Jagła, M.; Kosinska, J.; Tomasik, T.; Sobczak, A.; Pollak, A.; Herman-Sucharska, I.; Walczak, A.; Kwinta, P.; Płoski, R. KIF5A de novo mutation associated with myoclonic seizures and neonatal onset progressive leukoencephalopathy. Clin. Genet. 2017, 91, 769–773. [Google Scholar] [CrossRef]
- Huai, J.; Zhang, Z. Structural Properties and Interaction Partners of Familial ALS-Associated SOD1 Mutants. Front. Neurol. 2019, 10, 527. [Google Scholar] [CrossRef] [Green Version]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.A.; Fisher, E.M.C.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136, 2342–2358. [Google Scholar] [CrossRef]
- Park, J.H.; Elpers, C.; Reunert, J.; McCormick, M.L.; Mohr, J.; Biskup, S.; Schwartz, O.; Rust, S.; Grüneberg, M.; Seelhöfer, A.; et al. SOD1 deficiency: A novel syndrome distinct from amyotrophic lateral sclerosis. Brain 2019, 142, 2230–2237. [Google Scholar] [CrossRef]
- Roczniak-Ferguson, A.; Ferguson, S.M. Pleiotropic requirements for human TDP-43 in the regulation of cell and organelle homeostasis. Life Sci. Alliance 2019, 2, e201900358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daoud, H.; Valdmanis, P.N.; Kabashi, E.; Dion, P.; Dupre, N.; Camu, W.; Meininger, V.; Rouleau, G.A. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2008, 46, 112–114. [Google Scholar] [CrossRef] [PubMed]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS Mutations Associated with Amyotrophic Lateral Sclerosis: Summary and Update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef]
- An, H.; Rabesahala de Meritens, C.; Buchman, V.L.; Shelkovnikova, T.A. Frameshift peptides alter the properties of truncated FUS proteins in ALS-FUS. Mol. Brain 2020, 13, 77. [Google Scholar] [CrossRef]
- Naumann, M.; Peikert, K.; Günther, R.; Kooi, A.J.; Aronica, E.; Hübers, A.; Danel, V.; Corcia, P.; Pan-Montojo, F.; Cirak, S.; et al. Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 2384–2394. [Google Scholar] [CrossRef] [Green Version]
- Merner, N.D.; Girard, S.L.; Catoire, H.; Bourassa, C.V.; Belzil, V.V.; Rivière, J.-B.; Hince, P.; Levert, A.; Dionne-Laporte, A.; Spiegelman, D.; et al. Exome Sequencing Identifies FUS Mutations as a Cause of Essential Tremor. Am. J. Hum. Genet. 2012, 91, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef]
- Eilbeck, K.; Quinlan, A.; Yandell, M. Settling the score: Variant prioritization and Mendelian disease. Nat. Rev. Genet. 2017, 18, 599–612. [Google Scholar] [CrossRef]
- Chen, Y.; zheng, Z.-Z.; Huang, R.; Chen, K.; Song, W.; Zhao, B.; Chen, X.; Yang, Y.; Yuan, L.; Shang, H.-F. PFN1 mutations are rare in Han Chinese populations with amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, 1922.e1–1922.e5. [Google Scholar] [CrossRef]
- Hassan, M.S.; Shaalan, A.A.; Dessouky, M.I.; Abdelnaiem, A.E.; ElHefnawi, M. A review study: Computational techniques for expecting the impact of non-synonymous single nucleotide variants in human diseases. Gene 2019, 680, 20–33. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Conforti, F.L.; Spataro, R.; Sproviero, W.; Mazzei, R.; Cavalcanti, F.; Condino, F.; Simone, I.L.; Logroscino, G.; Patitucci, A.; Magariello, A.; et al. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology 2012, 79, 2315–2320. [Google Scholar] [CrossRef]
- Neuenschwander, A.G.; Thai, K.K.; Figueroa, K.P.; Pulst, S.M. Amyotrophic Lateral Sclerosis Risk for Spinocerebellar Ataxia Type 2 ATXN2 CAG Repeat Alleles. JAMA Neurol. 2014, 71, 1529. [Google Scholar] [CrossRef] [Green Version]
- van Blitterswijk, M.; Mullen, B.; Heckman, M.G.; Baker, M.C.; DeJesus-Hernandez, M.; Brown, P.H.; Murray, M.E.; Hsiung, G.-Y.R.; Stewart, H.; Karydas, A.M.; et al. Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol. Aging 2014, 35, 2421.e13–2421.e17. [Google Scholar] [CrossRef] [Green Version]
- Chio, A.; Calvo, A.; Moglia, C.; Canosa, A.; Brunetti, M.; Barberis, M.; Restagno, G.; Conte, A.; Bisogni, G.; Marangi, G.; et al. ATXN2 polyQ intermediate repeats are a modifier of ALS survival. Neurology 2015, 84, 251–258. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Kocerha, J.; Finch, N.; Crook, R.; Baker, M.; Desaro, P.; Johnston, A.; Rutherford, N.; Wojtas, A.; Kennelly, K.; et al. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum. Mutat. 2010, 31, E1377–E1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, A.; Moglia, C.; Canosa, A.; Brunetti, M.; Barberis, M.; Traynor, B.J.; Carrara, G.; Valentini, C.; Restagno, G.; Chiò, A. A de novo nonsense mutation of the FUS gene in an apparently familial amyotrophic lateral sclerosis case. Neurobiol. Aging 2014, 35, 1513.e7–1513.e11. [Google Scholar] [CrossRef] [Green Version]
- Conte, A.; Lattante, S.; Zollino, M.; Marangi, G.; Luigetti, M.; Del Grande, A.; Servidei, S.; Trombetta, F.; Sabatelli, M. P525L FUS mutation is consistently associated with a severe form of juvenile Amyotrophic Lateral Sclerosis. Neuromuscul. Disord. 2012, 22, 73–75. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Cui, L.-Y.; Sun, Q.; Li, X.-G.; Liu, M.-S.; Xu, Y.; Zhou, Y.; Yang, X.-Z. De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurobiol. Aging 2013, 34, 1312.e1–1312.e8. [Google Scholar] [CrossRef]
- Kim, Y.-E.; Oh, K.-W.; Kwon, M.-J.; Choi, W.-J.; Oh, S.; Ki, C.-S.; Kim, S.H. De novo FUS mutations in 2 Korean patients with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 1604.e17–1604.e19. [Google Scholar] [CrossRef]
- Hübers, A.; Just, W.; Rosenbohm, A.; Müller, K.; Marroquin, N.; Goebel, I.; Högel, J.; Thiele, H.; Altmüller, J.; Nürnberg, P.; et al. De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol. Aging 2015, 36, 3117.e1–3117.e6. [Google Scholar] [CrossRef]
- Leblond, C.S.; Webber, A.; Gan-Or, Z.; Moore, F.; Dagher, A.; Dion, P.A.; Rouleau, G.A. De novo FUS P525L mutation in Juvenile amyotrophic lateral sclerosis with dysphonia and diplopia. Neurol. Genet. 2016, 2, e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Wang, H.; Cai, Y.; Wen, H.; Wang, L.; Zhu, M.; Chen, Y.; Yu, Y.; Lu, X.; Zhou, M.; et al. FUS P525L mutation causing amyotrophic lateral sclerosis and movement disorders. Brain Behav. 2020, 10, e01625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Li, J.; Lu, H.; Liu, Y. A de novo c.1509dupA:p.R503fs mutation of FUS: Report of a girl with sporadic juvenile amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 5, 1–3. [Google Scholar] [CrossRef]
- Lim, S.M.; Kim, Y.-E.; Choi, W.J.; Oh, K.-W.; Noh, M.-Y.; Kwon, M.-S.; Nahm, M.; Kim, N.; Ki, C.-S.; Kim, S.H. CLEC4C p.K210del variant causes impaired cell surface transport in plasmacytoid dendritic cells of amyotrophic lateral sclerosis. Oncotarget 2016, 7, 24942–24949. [Google Scholar] [CrossRef] [PubMed]
- van Doormaal, P.T.C.; Ticozzi, N.; Weishaupt, J.H.; Kenna, K.; Diekstra, F.P.; Verde, F.; Andersen, P.M.; Dekker, A.M.; Tiloca, C.; Marroquin, N.; et al. The role of de novo mutations in the development of amyotrophic lateral sclerosis. Hum. Mutat. 2017, 38, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; van den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Whiffin, N.; Minikel, E.; Walsh, R.; O’Donnell-Luria, A.H.; Karczewski, K.; Ing, A.Y.; Barton, P.J.R.; Funke, B.; Cook, S.A.; MacArthur, D.; et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 2017, 19, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Johnston, C.A.; Stanton, B.R.; Turner, M.R.; Gray, R.; Blunt, A.H.-M.; Butt, D.; Ampong, M.-A.; Shaw, C.E.; Leigh, P.N.; Al-Chalabi, A. Amyotrophic lateral sclerosis in an urban setting. J. Neurol. 2006, 253, 1642–1643. [Google Scholar] [CrossRef] [Green Version]
- Murphy, N.A.; Arthur, K.C.; Tienari, P.J.; Houlden, H.; Chiò, A.; Traynor, B.J. Age-related penetrance of the C9orf72 repeat expansion. Sci. Rep. 2017, 7, 2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Chalabi, A. Recessive amyotrophic lateral sclerosis families with the D90A SOD1 mutation share a common founder: Evidence for a linked protective factor. Hum. Mol. Genet. 1998, 7, 2045–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Project MinE ALS Sequencing Consortium. Project MinE: Study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur. J. Hum. Genet. 2018, 26, 1537–1546. [Google Scholar] [CrossRef] [Green Version]
- Felbecker, A.; Camu, W.; Valdmanis, P.N.; Sperfeld, A.D.; Waibel, S.; Steinbach, P.; Rouleau, G.A.; Ludolph, A.C.; Andersen, P.M. Four familial ALS pedigrees discordant for two SOD1 mutations: Are all SOD1 mutations pathogenic? J. Neurol. Neurosurg. Psychiatry 2010, 81, 572–577. [Google Scholar] [CrossRef]
- Rezania, K.; Yan, J.; Dellefave, L.; Deng, H.; Siddique, N.; Pascuzzi, R.T.; Siddique, T.; Roos, R.P. A rare Cu/Zn superoxide dismutase mutation causing familial amyotrophic lateral sclerosis with variable age of onset, incomplete penetrance and a sensory neuropathy. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2003, 4, 162–166. [Google Scholar] [CrossRef]
- Nogales-Gadea, G.; Garcia-Arumi, E.; Andreu, A.L.; Cervera, C.; Gamez, J. A novel exon 5 mutation (N139H) in the SOD1 gene in a Spanish family associated with incomplete penetrance. J. Neurol. Sci. 2004, 219, 1–6. [Google Scholar] [CrossRef]
- Gamez, J.; Caponnetto, C.; Ferrera, L.; Syriani, E.; Marini, V.; Morales, M.; Bordo, D.; Pirro, C.; Garre, C.; Origone, P. I112M SOD1 mutation causes ALS with rapid progression and reduced penetrance in four Mediterranean families. Amyotroph. Lateral Scler. 2011, 12, 70–75. [Google Scholar] [CrossRef]
- Conforti, F.L.; Barone, R.; Fermo, S.L.; Giliberto, C.; Patti, F.; Gambardella, A.; Quattrone, A.; Zappia, M. Sporadic motor neuron disease in a familial novel SOD1 mutation: Incomplete penetrance or chance association? Amyotroph. Lateral Scler. 2011, 12, 220–222. [Google Scholar] [CrossRef]
- Uchida, A.; Sasaguri, H.; Kimura, N.; Tajiri, M.; Ohkubo, T.; Ono, F.; Sakaue, F.; Kanai, K.; Hirai, T.; Sano, T.; et al. Non-human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP-43. Brain 2012, 135, 833–846. [Google Scholar] [CrossRef]
- Crociara, P.; Chieppa, M.N.; Vallino Costassa, E.; Berrone, E.; Gallo, M.; Lo Faro, M.; Pintore, M.D.; Iulini, B.; D’Angelo, A.; Perona, G.; et al. Motor neuron degeneration, severe myopathy and TDP-43 increase in a transgenic pig model of SOD1-linked familiar ALS. Neurobiol. Dis. 2019, 124, 263–275. [Google Scholar] [CrossRef]
- Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Model. Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrice, J.; Gregory-Evans, C.; Shaw, C. Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural Regen. Res. 2018, 13, 2050. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, L.; Valenza, F.; Mosca, L.; Dal Mas, A.; Domi, T.; Romano, A.; Tarlarini, C.; Falzone, Y.M.; Tremolizzo, L.; Sorarù, G.; et al. TBK1 mutations in Italian patients with amyotrophic lateral sclerosis: Genetic and functional characterisation. J. Neurol. Neurosurg. Psychiatry 2017, 88, 869–875. [Google Scholar] [CrossRef] [Green Version]
- Lattante, S.; Doronzio, P.N.; Marangi, G.; Conte, A.; Bisogni, G.; Bernardo, D.; Russo, T.; Lamberti, D.; Patrizi, S.; Apollo, F.P.; et al. Coexistence of variants in TBK1 and in other ALS-related genes elucidates an oligogenic model of pathogenesis in sporadic ALS. Neurobiol. Aging 2019, 84, 239.e9–239.e14. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Bozzoni, V. Amyotrophic lateral sclerosis and environmental factors. Funct. Neurol. 2016, 31, 7–19. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Fang, F.; Hanby, M.F.; Leigh, P.N.; Shaw, C.E.; Ye, W.; Rijsdijk, F. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1324–1326. [Google Scholar] [CrossRef]
- Keller, M.F.; Ferrucci, L.; Singleton, A.B.; Tienari, P.J.; Laaksovirta, H.; Restagno, G.; Chiò, A.; Traynor, B.J.; Nalls, M.A. Genome-Wide Analysis of the Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2014, 71, 1123. [Google Scholar] [CrossRef] [Green Version]
- Fogh, I.; Ratti, A.; Gellera, C.; Lin, K.; Tiloca, C.; Moskvina, V.; Corrado, L.; Soraru, G.; Cereda, C.; Corti, S.; et al. A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 2220–2231. [Google Scholar] [CrossRef]
- Ryan, M.; Heverin, M.; McLaughlin, R.L.; Hardiman, O. Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2019, 76, 1367. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figley, M.D.; Thomas, A.; Gitler, A.D. Evaluating noncoding nucleotide repeat expansions in amyotrophic lateral sclerosis. Neurobiol. Aging 2014, 35, 936.e1–936.e4. [Google Scholar] [CrossRef] [Green Version]
- Paulson, H. Repeat expansion diseases. In The Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 147, pp. 105–123. [Google Scholar] [CrossRef]
- Yousefian-Jazi, A.; Sung, M.K.; Lee, T.; Hong, Y.-H.; Choi, J.K.; Choi, J. Functional fine-mapping of noncoding risk variants in amyotrophic lateral sclerosis utilizing convolutional neural network. Sci. Rep. 2020, 10, 12872. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.; Shatunov, A.; Sproviero, W.; Jones, A.R.; Shoai, M.; Hughes, D.; Al Khleifat, A.; Malaspina, A.; Morrison, K.E.; Shaw, P.J.; et al. A comprehensive analysis of rare genetic variation in amyotrophic lateral sclerosis in the UK. Brain 2017, 140, 1611–1618. [Google Scholar] [CrossRef]
- Theunissen, F.; Flynn, L.L.; Anderton, R.S.; Mastaglia, F.; Pytte, J.; Jiang, L.; Hodgetts, S.; Burns, D.K.; Saunders, A.; Fletcher, S.; et al. Structural Variants May Be a Source of Missing Heritability in sALS. Front. Neurosci. 2020, 14, 47. [Google Scholar] [CrossRef] [Green Version]
- van Blitterswijk, M.; van Es, M.A.; Hennekam, E.A.M.; Dooijes, D.; van Rheenen, W.; Medic, J.; Bourque, P.R.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 3776–3784. [Google Scholar] [CrossRef] [Green Version]
- Kenna, K.P.; McLaughlin, R.L.; Byrne, S.; Elamin, M.; Heverin, M.; Kenny, E.M.; Cormican, P.; Morris, D.W.; Donaghy, C.G.; Bradley, D.G.; et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J. Med. Genet. 2013, 50, 776–783. [Google Scholar] [CrossRef] [Green Version]
- Cooper-Knock, J.; Robins, H.; Niedermoser, I.; Wyles, M.; Heath, P.R.; Higginbottom, A.; Walsh, T.; Kazoka, M.; Ince, P.G.; Hautbergue, G.M.; et al. Targeted Genetic Screen in Amyotrophic Lateral Sclerosis Reveals Novel Genetic Variants with Synergistic Effect on Clinical Phenotype. Front. Mol. Neurosci. 2017, 10, 370. [Google Scholar] [CrossRef]
- Cady, J.; Allred, P.; Bali, T.; Pestronk, A.; Goate, A.; Miller, T.M.; Mitra, R.D.; Ravits, J.; Harms, M.B.; Baloh, R.H. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 2015, 77, 100–113. [Google Scholar] [CrossRef] [Green Version]
- Naruse, H.; Ishiura, H.; Mitsui, J.; Takahashi, Y.; Matsukawa, T.; Tanaka, M.; Doi, K.; Yoshimura, J.; Morishita, S.; Goto, J.; et al. Burden of rare variants in causative genes for amyotrophic lateral sclerosis (ALS) accelerates age at onset of ALS. J. Neurol. Neurosurg. Psychiatry 2019, 90, 537–542. [Google Scholar] [CrossRef]
- Pang, S.Y.-Y.; Hsu, J.S.; Teo, K.-C.; Li, Y.; Kung, M.H.W.; Cheah, K.S.E.; Chan, D.; Cheung, K.M.C.; Li, M.; Sham, P.-C.; et al. Burden of rare variants in ALS genes influences survival in familial and sporadic ALS. Neurobiol. Aging 2017, 58, 238.e9–238.e15. [Google Scholar] [CrossRef]
- Leija-Salazar, M.; Piette, C.; Proukakis, C. Review: Somatic mutations in neurodegeneration. Neuropathol. Appl. Neurobiol. 2018, 44, 267–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proukakis, C. Somatic mutations in neurodegeneration: An update. Neurobiol. Dis. 2020, 144, 105021. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Logroscino, G.; Hernan, M.A. Smoking and the risk of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1249–1252. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Richard, M.; Sandler, D.P.; Umbach, D.M.; Kamel, F. Head Injury and Amyotrophic Lateral Sclerosis. Am. J. Epidemiol. 2007, 166, 810–816. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Lee, M.-H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef]
- Oskarsson, B.; Horton, D.K.; Mitsumoto, H. Potential Environmental Factors in Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 877–888. [Google Scholar] [CrossRef] [Green Version]
- Al-Chalabi, A.; Calvo, A.; Chio, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.B.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014, 13, 1108–1113. [Google Scholar] [CrossRef]
- Chiò, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M.; et al. The multistep hypothesis of ALS revisited. Neurology 2018, 91, e635–e642. [Google Scholar] [CrossRef] [Green Version]
- Vucic, S.; Higashihara, M.; Sobue, G.; Atsuta, N.; Doi, Y.; Kuwabara, S.; Kim, S.H.; Kim, I.; Oh, K.-W.; Park, J.; et al. ALS is a multistep process in South Korean, Japanese, and Australian patients. Neurology 2020, 94, e1657–e1663. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.P.; Leblond, C.S.; Laurent, S.B.; Spiegelman, D.; Dionne-Laporte, A.; Camu, W.; Dupré, N.; Dion, P.A.; Rouleau, G.A. Oligogenicity, C9orf72 expansion, and variant severity in ALS. Neurogenetics 2020, 21, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar] [CrossRef] [Green Version]
- Kuuluvainen, L.; Kaivola, K.; Mönkäre, S.; Laaksovirta, H.; Jokela, M.; Udd, B.; Valori, M.; Pasanen, P.; Paetau, A.; Traynor, B.J.; et al. Oligogenic basis of sporadic ALS. Neurol. Genet. 2019, 5, e335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, E.P.; Henden, L.; Fifita, J.A.; Zhang, K.Y.; Grima, N.; Bauer, D.C.; Chan Moi Fat, S.; Twine, N.A.; Pamphlett, R.; Kiernan, M.C.; et al. Evidence for polygenic and oligogenic basis of Australian sporadic amyotrophic lateral sclerosis; jmedgenet-2020. J. Med. Genet. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Scarlino, S.; Domi, T.; Pozzi, L.; Romano, A.; Pipitone, G.B.; Falzone, Y.M.; Mosca, L.; Penco, S.; Lunetta, C.; Sansone, V.; et al. Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: Insights from a Next Generation Sequencing Study of an Italian ALS Cohort. Int. J. Mol. Sci. 2020, 21, 3346. [Google Scholar] [CrossRef] [PubMed]





| Code | Criteria | Level of Evidence | Relevance in ALS | Considerations for Their Application in ALS |
|---|---|---|---|---|
| PVS1 | Null variant (nonsense, frameshift, canonical splice sites, etc.) in a gene for which loss of function is a common mechanism of disease | Very strong | ++ | So far, applicable for variants in following genes (in ALS): ALS2, SPG11, TBK1, OPTN |
| PS1 | Same amino acid change as a previously established variant | Strong | ++ | Frequently applicable for genes more extensively investigated than others, e.g., SOD1, FUS, and TARDBP |
| PS2 | De novo variant in a patient with the disease and no family history | Strong | + | Parental testing rarely performed on routine basis. To be assessed mainly in early-onset cases (e.g., FUS-related ALS) |
| PS3 | Well-established functional studies supportive of a damaging effect on the gene or gene product | Strong | + | Functional studies already available for a large series of variants, but well-established tools are still missing for routine assessment of variant role |
| PS4 | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls | Strong | ++ | Data from large cohorts of patients and controls are already publicly available. Larger studies are still necessary to reach statistical significance for as many variants as possible |
| PM1 | Located in a mutational hotspot and/or critical and well-established functional domain without benign variation | Moderate | ++ | Easy to apply for variants in genes such as FUS, TARDBP, and KIF5A. Further studies needed for an extension of the list |
| PM2 | Absent from population controls | Moderate | + | To be considered in any case. Consensus is missing about allele frequency thresholds (i.e., variants responsible for ALS can be found in general population at low frequencies) |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant | Moderate | + | To be considered when variants in genes responsible for recessive forms of ALS are found |
| PM4 | Protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variant | Moderate | + | A small subset of variants found to date belong to this category, and their role is still not well-defined |
| PM5 | Novel missense at an amino acid residue where a different pathogenic missense change has been seen before | Moderate | ++ | Frequently applicable for genes more extensively investigated, e.g., SOD1, FUS, and TARDBP |
| PM6 | Assumed de novo, without confirmation of paternity and/or maternity | Moderate | + | Parental testing rarely performed on routine basis. To be assessed mainly in early-onset cases (e.g., FUS-related ALS) |
| PP1 | Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease | Supporting* | ++ | To be used for familial cases. Families with a larger number of affected members would allow for increased significance |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease | Supporting | ++ | Fulfilled for most ALS genes |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product | Supporting | ++ | Routinely used for variant evaluation in ALS |
| PP4 | Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology | Supporting | + | Applicable only in specific situations (i.e., large families with apparently Mendelian inheritance or suggestive phenotypes, such as juvenile forms) |
| PP5 | Reputable source reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation | Supporting* | ++ | Evidence strength depending on number and details of reports |
| BA1 | Allele frequency is >5% in general population | Stand-alone | ++ | To be considered for any variant |
| BS1 | Allele frequency is greater than expected for disorder | Strong | ++ | To be considered in any case, though a consensus is missing about allele frequency thresholds |
| BS2 | Observed in a healthy adult individual with full penetrance expected at an early age (the genotype must be consistent with the disease pattern of inheritance) | Strong | − | Not to be applied: In ALS, penetrance is not expected to be complete at an early age for any gene |
| BS3 | Well-established functional studies show no damaging effect on protein function or splicing | Strong | + | Functional studies already available for a large series of variants, but well-established tools are still missing for routine assessment of variant role |
| BS4 | Lack of segregation in affected members of a family | Strong | +/− | To be used in familial cases. Families with a larger number of affected members would allow for increased significance. However, reduced penetrance can explain the observation of healthy cases with the variant |
| BP1 | Missense variant in a gene for which primarily truncating variants are known to cause disease | Supporting | − | Not to be applied: Pathogenic missense variants are commonly found in ALS genes (apart from those in which a repeat expansion is the causative one) |
| BP2 | Observed in trans with a pathogenic variant for a fully penetrant dominant gene/disorder or observed in cis with a pathogenic variant in any inheritance pattern | Supporting | +/− | The co-occurrence of two or more pathogenic variants in ALS patients is not a rare event |
| BP3 | In-frame deletions/insertions in a repetitive region without a known function | Supporting | − | Not to be used, since repeat expansions are a common pathogenic mechanism in ALS (and may occur in introns) |
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product | Supporting | ++ | Routinely used for variant evaluation in ALS. However, there is the need for more specific tools |
| BP5 | Variant found in a case with an alternate molecular basis for disease | Supporting | + | Rarely useful |
| BP6 | Reputable source reports variant as benign, but the evidence is not available to the laboratory to perform an independent evaluation | Supporting | ++ | Evidence strength depending on number and details of reports |
| BP7 | A synonymous for which no splicing alteration is predicted AND the nucleotide is not highly conserved | Supporting | ++ | Routinely used for variant evaluation in ALS |
| Gene | Transcript | Exon | cDNA | Protein | Class | ACMG Criteria | Nr. |
|---|---|---|---|---|---|---|---|
| TARDBP | NM_007375 | 6 | c.881G > T | p.G294V | P | PM1, PM2, PM5, PP2, PP5(strong) | 1 f |
| TARDBP | NM_007375 | 6 | c.1127G > A | p.G376D | LP | PM1, PM2, PM5, PP2 | 1 f |
| TARDBP | NM_007375 | 6 | c.1144G > A | p.A382T | P | PM1, PP2, PP5 (very strong) | 6 f |
| OPTN | NM_021980 | 4 | c.451C > T | p.Q151* | P | PVS1 (very strong), PM2, PP3 | 1 |
| TBK1 | NM_013254 | 6 | c.684dupT | p.R229* | P | PVS1 (very strong), PM2, PP3 | 1 |
| TBK1 | NM_013254 | 13 | c.1445_1446delAT | p.Y482* | P | PVS1 (very strong), PM2, PP3 | 1 |
| TBK1 | NM_013254 | 19 | c.2040dupT | p.N681* | P | PVS1 (very strong), PM2, PP3 | 1 |
| FUS | NM_004960 | 14 | c.1540A > G | p.R514G | LP | PM1, PM2, PM5, PP2, PP3, PP5 | 1 f |
| SOD1 | NM_000454 | 1 | c.34G > T | p.D12Y | LP | PM1, PM2, PP2, PP3 | 3 f |
| SOD1 | NM_000454 | 3 | c.203T > C | p.L68P | LP | PM1, PM2, PM5, PP2, PP5 | 1 |
| SOD1 | NM_000454 | 3 | c.217G > A | p.G73S | P | PS1, PM1, PM2, PP2, PP3, PP5 (strong) | 1 |
| SOD1 | NM_000454 | 4 | c.255G > C | p.L85F | P | PS1, PM1 (strong), PM2, PM5, PP2, PP3 | 1 f |
| SOD1 | NM_000454 | 4 | c.256G > A | p.G86S | LP | PM1, PM2, PM5, PP2, PP3 | 1 |
| SOD1 | NM_000454 | 4 | c.272A > C | p.D91A | LP | PM1, PM2, PM5, PP2, PP5 | 2 # |
| SOD1 | NM_000454 | 4 | c.281G > A | p.G94D | LP | PM1, PM2, PM5, PP2, PP3, PP5 | 1 |
| SOD1 | NM_000454 | 4 | c.340A > T | p.I114F | LP | PM1, PM2, PM5, PP2, PP3, PP5 | 1 f |
| SOD1 | NM_000454 | 5 | c.435G > C | p.L145F | P | PS1, PM1 (strong), PM2, PM5, PP2, PP3, PP5 (strong) | 2 f |
| SOD1 | NM_000454 | 5 | c.449T > C | p.I150T | LP | PM1, PM2, PP2, PP3, PP5 | 1 f |
| NEK1 | NM_001199397 | 29 | c.2785_2786delGA | p.E929Nfs*12 | P | PVS1 (very strong), PM2, PP3 | 1 |
| VCP | NM_007126 | 5 | c.463C > T | p.R155C | P | PM1 (strong), PM2, PM5, PP2, PP3, PP5 (very strong) | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lattante, S.; Marangi, G.; Doronzio, P.N.; Conte, A.; Bisogni, G.; Zollino, M.; Sabatelli, M. High-Throughput Genetic Testing in ALS: The Challenging Path of Variant Classification Considering the ACMG Guidelines. Genes 2020, 11, 1123. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101123
Lattante S, Marangi G, Doronzio PN, Conte A, Bisogni G, Zollino M, Sabatelli M. High-Throughput Genetic Testing in ALS: The Challenging Path of Variant Classification Considering the ACMG Guidelines. Genes. 2020; 11(10):1123. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101123
Chicago/Turabian StyleLattante, Serena, Giuseppe Marangi, Paolo Niccolò Doronzio, Amelia Conte, Giulia Bisogni, Marcella Zollino, and Mario Sabatelli. 2020. "High-Throughput Genetic Testing in ALS: The Challenging Path of Variant Classification Considering the ACMG Guidelines" Genes 11, no. 10: 1123. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101123