Differences in Placental Imprinted Gene Expression across Preeclamptic and Non-Preeclamptic Pregnancies


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
Statistical Analysis
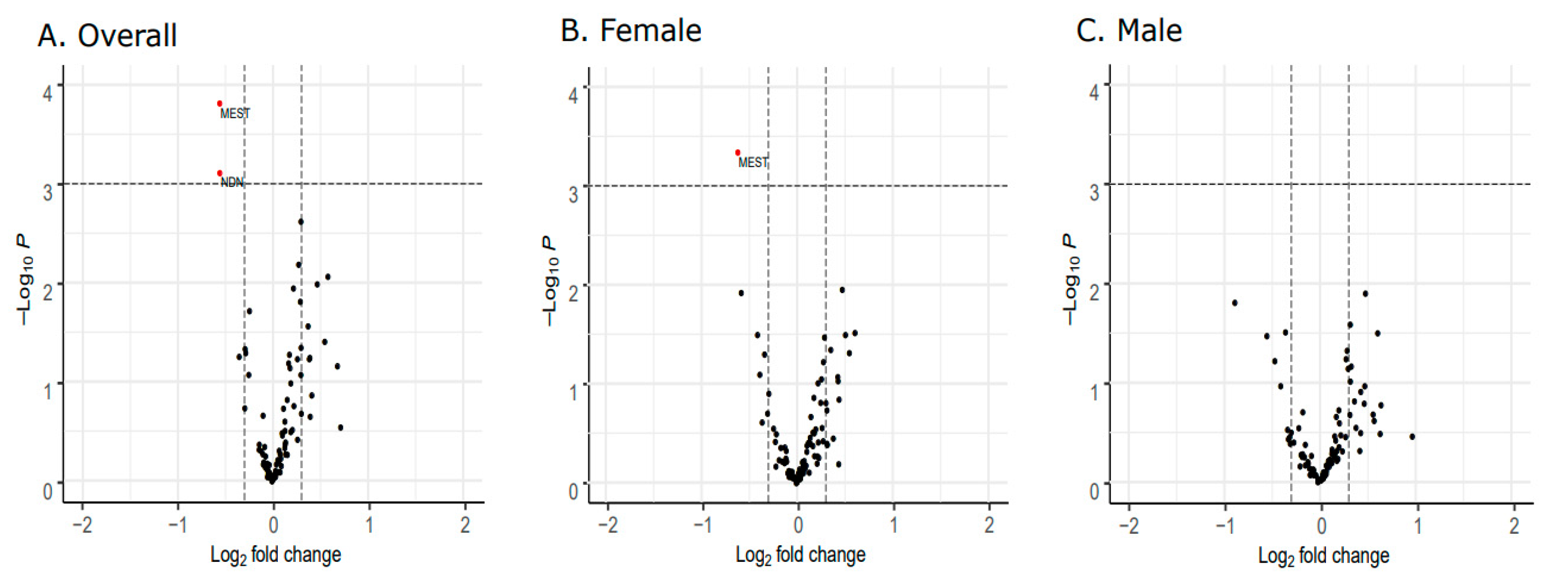
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Roberts, J.M.; Bell, M.J. If we know so much about preeclampsia, why haven’t we cured the disease? J. Reprod. Immunol. 2013, 99, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Abalos, E.; Cuesta, C.; Grosso, A.L.; Chou, D.; Say, L. Global and regional estimates of preeclampsia and eclampsia: A systematic review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 170, 1–7. [Google Scholar] [CrossRef]
- American College of Obstetricians. Task Force on Hypertension in Pregnancy. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar] [CrossRef]
- Roberts, J.M.; Mascalzoni, D.; Ness, R.B.; Poston, L. Collaboration to understand complex diseases: Preeclampsia and adverse pregnancy outcomes. Hypertension 2016, 67, 681–687. [Google Scholar] [CrossRef]
- Romundstad, P.R.; Magnussen, E.B.; Smith, G.D.; Vatten, L.J. Hypertension in Pregnancy and Later Cardiovascular Risk: Common antecedents? Circulation 2010, 122, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Baecke, M.; Spaanderman, M.E.A.; Van Der Werf, S.P. Cognitive function after pre-eclampsia: An explorative study. J. Psychosom. Obstet. Gynecol. 2009, 30, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Postma, I.; Bouma, A.; De Groot, J.C.; Aukes, A.M.; Aarnoudse, J.G.; Zeeman, G.G. Cerebral white matter lesions, subjective cognitive failures, and objective neurocognitive functioning: A follow-up study in women after hypertensive disorders of pregnancy. J. Clin. Exp. Neuropsychol. 2016, 38, 585–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, E.F.; Lazdam, M.; Lewandowski, A.J.; Worton, S.A.; Kelly, B.; Kenworthy, Y.; Adwani, S.; Wilkinson, A.R.; McCormick, K.; Sargent, I.; et al. Cardiovascular Risk Factors in Children and Young Adults Born to Preeclamptic Pregnancies: A Systematic Review. Pediatrics 2012, 129, e1552–e1561. [Google Scholar] [CrossRef] [PubMed]
- Kajantie, E.; Eriksson, J.G.; Osmond, C.; Thornburg, K.; Barker, D.J. Pre-Eclampsia Is Associated With Increased Risk of Stroke in the Adult Offspring the helsinki birth cohort study. Stroke 2009, 40, 1176–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuovinen, S.; Räikkönen, K.; Pesonen, A.-K.; Lahti-Pulkkinen, M.; Heinonen, K.; Wahlbeck, K.; Kajantie, E.; Osmond, C.; Barker, D.J.; Eriksson, J.G. Hypertensive disorders in pregnancy and risk of severe mental disorders in the offspring in adulthood: The Helsinki Birth Cohort Study. J. Psychiatr. Res. 2012, 46, 303–310. [Google Scholar] [CrossRef]
- Walker, C.K.; Krakowiak, P.; Baker, A.; Hansen, R.; Ozonoff, S.; Hertz-Picciotto, I. Preeclampsia, placental insufficiency, and autism spectrum disorder or developmental delay. JAMA Pediatr. 2015, 169, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Fowden, A.L.; Thornburg, K.L. Placental Origins of Chronic Disease. Physiol. Rev. 2016, 96, 1509–1565. [Google Scholar] [CrossRef] [PubMed]
- Ness, R.B.; Sibai, B.M. Shared and disparate components of the pathophysiologies of fetal growth restriction and preeclampsia. Am. J. Obstet. Gynecol. 2006, 195, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Mongraw-Chaffin, M.L.; Cirillo, P.M.; Cohn, B.A. Preeclampsia and Cardiovascular Disease Death: Prospective evidence from the child health and development studies cohort. Hypertension 2010, 56, 166–171. [Google Scholar] [CrossRef] [Green Version]
- Schalekamp-Timmermans, S.; Arends, L.R.; Alsaker, E.; Chappell, L.; Hansson, S.; Harsem, N.K.; Jälmby, M.; Jeyabalan, A.; Laivuori, H.; Lawlor, D.A.; et al. Fetal sex-specific differences in gestational age at delivery in pre-eclampsia: A meta-analysis. Int. J. Epidemiol. 2017, 46, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Lain, K.Y.; Roberts, J.M. Contemporary concepts of the pathogenesis and management of preeclampsia. JAMA 2002, 287, 3183–3186. [Google Scholar] [CrossRef]
- Roberts, J.M.; Escudero, C. The placenta in preeclampsia. Pregnancy Hypertens. 2012, 2, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-eclampsia: Pathophysiology and clinical implications. BMJ 2019, 366, l2381. [Google Scholar] [CrossRef] [Green Version]
- Kajantie, E.; Thornburg, K.L.; Eriksson, J.G.; Osmond, L.; Barker, D.J.P.; Osmond, C. In preeclampsia, the placenta grows slowly along its minor axis. Int. J. Dev. Biol. 2010, 54, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Salomon, C.; Guanzon, D.; Scholz-Romero, K.; Longo, S.; Correa, P.; Illanes, S.E.; Rice, G. Placental exosomes as early biomarker of preeclampsia—Potential role of exosomal microRNAs across gestation. J. Clin. Endocrinol. Metab. 2017, 102, 3182–3194. [Google Scholar] [CrossRef]
- Levine, L.; Habertheuer, A.; Ram, C.; Korutla, L.; Schwartz, N.; Hu, R.W.; Reddy, S.; Freas, A.; Zielinski, P.D.; Harmon, J.; et al. Syncytiotrophoblast extracellular microvesicle profiles in maternal circulation for noninvasive diagnosis of preeclampsia. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Ferguson-Smith, A.C. Mammalian Genomic Imprinting. Cold Spring Harb. Perspect. Biol. 2011, 3, a002592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson-Smith, A.C.; Surani, M.A. Imprinting and the Epigenetic Asymmetry Between Parental Genomes. Science 2001, 293, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Coan, P.; Burton, G.; Ferguson-Smith, A.C. Imprinted genes in the placenta—A review. Placenta 2005, 26 (Suppl. A), S10–S20. [Google Scholar] [CrossRef] [PubMed]
- Tycko, B. Imprinted genes in placental growth and obstetric disorders. Cytogenet. Genome Res. 2006, 113, 271–278. [Google Scholar] [CrossRef]
- Zadora, J.; Singh, M.; Herse, F.; Przybyl, L.; Haase, N.; Golic, M.; Yung, H.W.; Huppertz, B.; Cartwright, J.E.; Whitley, G.; et al. Disturbed Placental Imprinting in Preeclampsia Leads to Altered Expression of DLX5, a Human-Specific Early Trophoblast Marker. Circulation 2017, 136, 1824–1839. [Google Scholar] [CrossRef]
- Christians, J.K.; Leavey, K.; Cox, B.J. Associations between imprinted gene expression in the placenta, human fetal growth and preeclampsia. Biol. Lett. 2017, 13, 20170643. [Google Scholar] [CrossRef]
- Kanayama, N.; Takahashi, K.; Matsuura, T.; Sugimura, M.; Kobayashi, T.; Moniwa, N.; Tomita, M.; Nakayama, K. Deficiency in p57Kip2 expression induces preeclampsia-like symptoms in mice. Mol. Hum. Reprod. 2002, 8, 1129–1135. [Google Scholar] [CrossRef]
- Baumgartel, K.L.; Terhorst, L.; Conley, Y.P.; Roberts, J.M. Psychometric evaluation of the Epworth sleepiness scale in an obstetric population. Sleep Med. 2012, 14, 116–121. [Google Scholar] [CrossRef] [Green Version]
- Sween, L.K.; Althouse, A.D.; Roberts, J.M. Early-pregnancy percent body fat in relation to preeclampsia risk in obese women. Am. J. Obstet. Gynecol. 2015, 212, 84.e1. [Google Scholar] [CrossRef]
- Roccella, E.J. Report of the National High Blood Pressure Education Program Working Group on High Blood Pressure in Pregnancy. Am. J. Obstet. Gynecol. 2000, 183, s1–s22. [Google Scholar] [CrossRef]
- Carter, R.C.; Chen, J.; Li, Q.; Deyssenroth, M.; Dodge, N.C.; Wainwright, H.C.; Molteno, C.D.; Meintjes, E.M.; Jacobson, J.L.; Jacobson, S.W. Alcohol-Related Alterations in Placental Imprinted Gene Expression in Humans Mediate Effects of Prenatal Alcohol Exposure on Postnatal Growth. Alcohol. Clin. Exp. Res. 2018, 42, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. 2018. Available online: https://www.r-project.org/ (accessed on 29 September 2020).
- Mayer, W.; Hemberger, M.; Frank, H.G.; Grümmer, R.; Winterhager, E.; Kaufmann, P.; Fundele, R. Expression of the Imprinted Genes MEST/Mest in Human and Murine Placenta Suggests a Role in Angiogenesis. Dev. Dyn. 2000, 217. [Google Scholar] [CrossRef]
- Peng, W.; Chen, Y.; Luo, X.; Shan, N.; Lan, X.; Olson, D.; Zhang, H.; Ding, Y.-B.; Qi, H.-B. DNA methylation-associated repression of MEST/PEG1 expression contributes to the invasion of extravillous trophoblast cells. Placenta 2016, 46, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Kuwajima, T.; Nishimura, I.; Yoshikawa, K. Necdin Promotes GABAergic Neuron Differentiation in Cooperation with Dlx Homeodomain Proteins. J. Neurosci. 2006, 26, 5383–5392. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Kamei, Y.; Ezaki, O. Mest/Peg1 imprinted gene enlarges adipocytes and is a marker of adipocyte size. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E117–E124. [Google Scholar] [CrossRef] [Green Version]
- Nikonova, L.; Koza, R.A.; Mendoza, T.; Chao, P.-M.; Curley, J.P.; Kozak, L.P. Mesoderm-specific transcript is associated with fat mass expansion in response to a positive energy balance. FASEB J. 2008, 22, 3925–3937. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, K.; Hasegawa, K.; Ohkumo, T.; Miyoshi, H.; Tseng, Y.-H.; Yoshikawa, K. Necdin Controls Proliferation of White Adipocyte Progenitor Cells. PLoS ONE 2012, 7, e30948. [Google Scholar] [CrossRef] [Green Version]
- Pagliardini, S.; Ren, J.; Wevrick, R.; Greer, J.J. Developmental Abnormalities of Neuronal Structure and Function in Prenatal Mice Lacking the Prader-Willi Syndrome Gene Necdin. Am. J. Pathol. 2005, 167, 175–191. [Google Scholar] [CrossRef] [Green Version]
- Muscatelli, F.; Abrous, D.N.; Massacrier, A.; Boccaccio, I.; Le Moal, M.; Cau, P.; Cremer, H. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum. Mol. Genet. 2000, 9, 3101–3110. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Kohda, T.; Miyoshi, N.; Kuroiwa, Y.; Aisaka, K.; Tsutsumi, O.; Kaneko-Ishino, T.; Ishino, F. Human PEG1/MEST, an imprinted gene on chromosome 7. Hum. Mol. Genet. 1997, 6, 781–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelissen, E.C.; Dumoulin, J.C.; Daunay, A.; Evers, J.L.; Tost, J.; Van Montfoort, A. Placentas from pregnancies conceived by IVF/ICSI have a reduced DNA methylation level at the H19 and MEST differentially methylated regions. Hum. Reprod. 2013, 28, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Baines, K.J.; Rampersaud, A.M.; Hillier, D.M.; Jeyarajah, M.J.; Grafham, G.K.; Eastabrook, G.; Lacefield, J.C.; Renaud, S.J. Antiviral Inflammation during Early Pregnancy Reduces Placental and Fetal Growth Trajectories. J. Immunol. 2019, 204, 694–706. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, N.; Pliushch, G.; Schneider, E.; Dittrich, M.; Müller, T.; Korenkov, M.; Aretz, M.; Zechner, U.; Lehnen, H.; Haaf, T. Metabolic Programming of MEST DNA Methylation by Intrauterine Exposure to Gestational Diabetes Mellitus. Diabetes 2013, 62, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wan, L.; Weng, X.; Xie, J.; Zhang, A.; Liu, Y.; Dong, M. Alteration in methylation level at differential methylated regions of MEST and DLK1 in fetus of preeclampsia. Hypertens. Pregnancy 2017, 37, 1–8. [Google Scholar] [CrossRef] [PubMed]
- A Kappil, M.; Green, B.B.; A Armstrong, D.; Sharp, A.J.; Lambertini, L.; Marsit, C.J.; Chen, J. Placental expression profile of imprinted genes impacts birth weight. Epigenetics 2015, 10, 842–849. [Google Scholar] [CrossRef] [Green Version]
- McMINN, J.; Wei, M.; Sadovsky, Y.; Thaker, H.; Tycko, B. Imprinting of PEG1/MEST Isoform 2 in Human Placenta. Placenta 2006, 27, 119–126. [Google Scholar] [CrossRef]
- Deyssenroth, M.A.; Marsit, C.J.; Chen, J.; Lambertini, L. In-depth characterization of the placental imprintome reveals novel differentially methylated regions across birth weight categories. Epigenetics 2019, 15, 47–60. [Google Scholar] [CrossRef]
- Lefebvre, L.; Viville, S.; Barton, S.C.; Ishino, F.; Keverne, E.B.; Surani, M.A. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat. Genet. 1998, 20, 163–169. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Early Onset Cases (n = 24) | Preterm Controls (n = 19) | p-Value | Late Onset Cases (n = 25) | Term Controls (n = 31) | p-Value * |
|---|---|---|---|---|---|---|
| Mean (SD) | Mean (SD) | Mean (SD) | Mean (SD) | |||
| Gestational age (weeks) | 32.4 (1.5) | 34.6 (3.1) | <0.01 | 39.0 (1.4) | 39.2 (0.9) | 0.39 |
| Birthweight (grams) | 1645.5 (508.6) | 2351.8 (651.1) | <0.01 | 3099.1 (547.9) | 3312.2 (405.0) | 0.10 |
| Maternal age (years) | 27.0 (6.8) | 26.9 (5.6) | 0.96 | 26.9 (5.9) | 25.3 (4.8) | 0.27 |
| N (%) | N (%) | N (%) | N (%) | |||
| Delivery method: | ||||||
| Caesarean | 14 (58.3) | 7 (36.8) | 0.27 | 8 (32.0) | 6 (19.4) | 0.44 |
| Vaginal | 10 (41.7) | 12 (63.2) | 17 (68.0) | 25 (80.6) | ||
| Infant sex: | ||||||
| Female | 11 (45.8) | 9 (47.4) | 1.00 | 9 (36.0) | 12 (38.7) | 1.00 |
| Male | 13 (54.2) | 10 (52.6) | 16 (64.0) | 19 (61.3) | ||
| Race/ethnicity: | ||||||
| White | 18 (75.0) | 6 (31.6) | 0.01 | 19 (76.0) | 25 (80.6) | 0.93 |
| Non-white | 6 (25.0) | 13 (68.4) | 6 (24.0) | 6 (19.4) | ||
| Parity: | ||||||
| Nulliparous | 18 (75.0) | 12 (63.2) | 0.61 | 22 (88.0) | 26 (83.9) | 0.96 |
| Parous | 6 (25.0) | 7 (36.8) | 3 (12.0) | 5 (16.1) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deyssenroth, M.A.; Li, Q.; Escudero, C.; Myatt, L.; Chen, J.; Roberts, J.M. Differences in Placental Imprinted Gene Expression across Preeclamptic and Non-Preeclamptic Pregnancies. Genes 2020, 11, 1146. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101146
Deyssenroth MA, Li Q, Escudero C, Myatt L, Chen J, Roberts JM. Differences in Placental Imprinted Gene Expression across Preeclamptic and Non-Preeclamptic Pregnancies. Genes. 2020; 11(10):1146. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101146
Chicago/Turabian StyleDeyssenroth, Maya A., Qian Li, Carlos Escudero, Leslie Myatt, Jia Chen, and James M. Roberts. 2020. "Differences in Placental Imprinted Gene Expression across Preeclamptic and Non-Preeclamptic Pregnancies" Genes 11, no. 10: 1146. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101146