A Novel Aminoacyl-tRNA Synthetase Appended Domain Can Supply the Core Synthetase with Its Amino Acid Substrate

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cloning, Mutagenesis, and Expression
2.3. Western Blot Analysis
2.4. In Vitro tRNA Synthesis
2.5. tRNA Radiolabeling and Aminoacylation
2.6. Liquid Chromatography–Mass Spectrometry Detection of Transamination
3. Results
3.1. The MpMetRS metS Gene
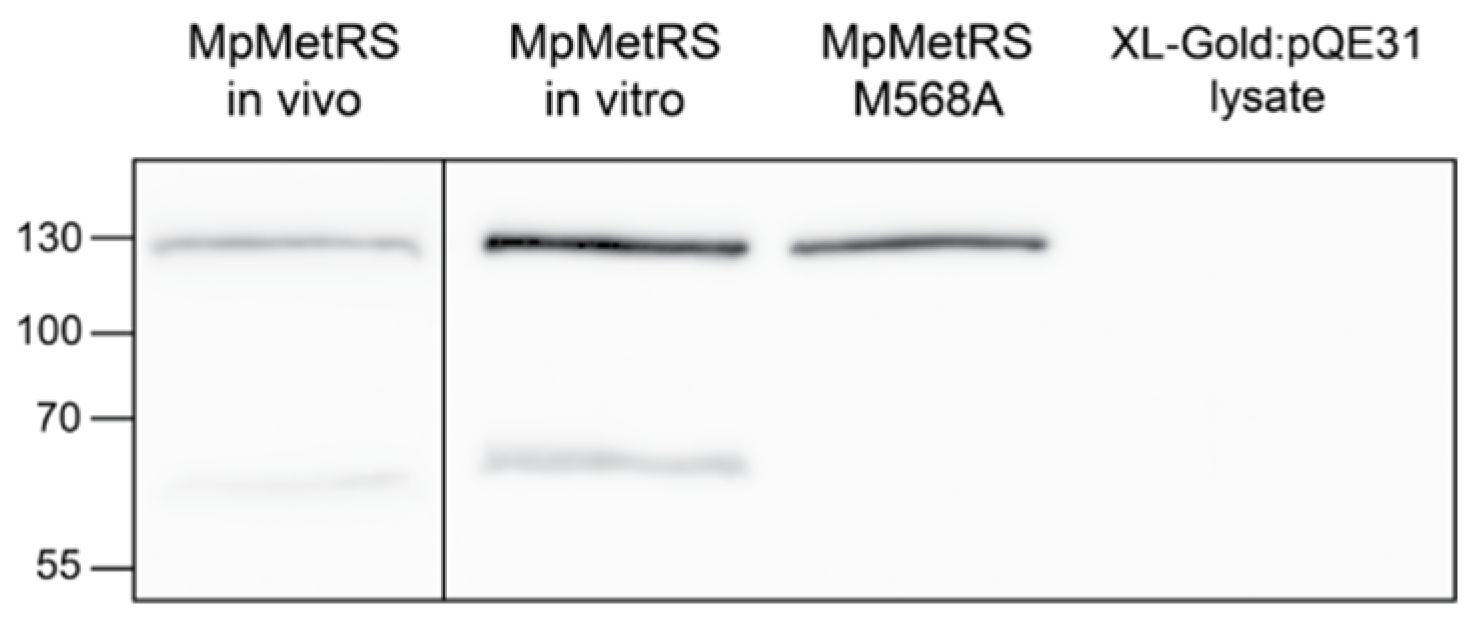
3.2. Evidence for Full-Length MpMetRS In Vivo
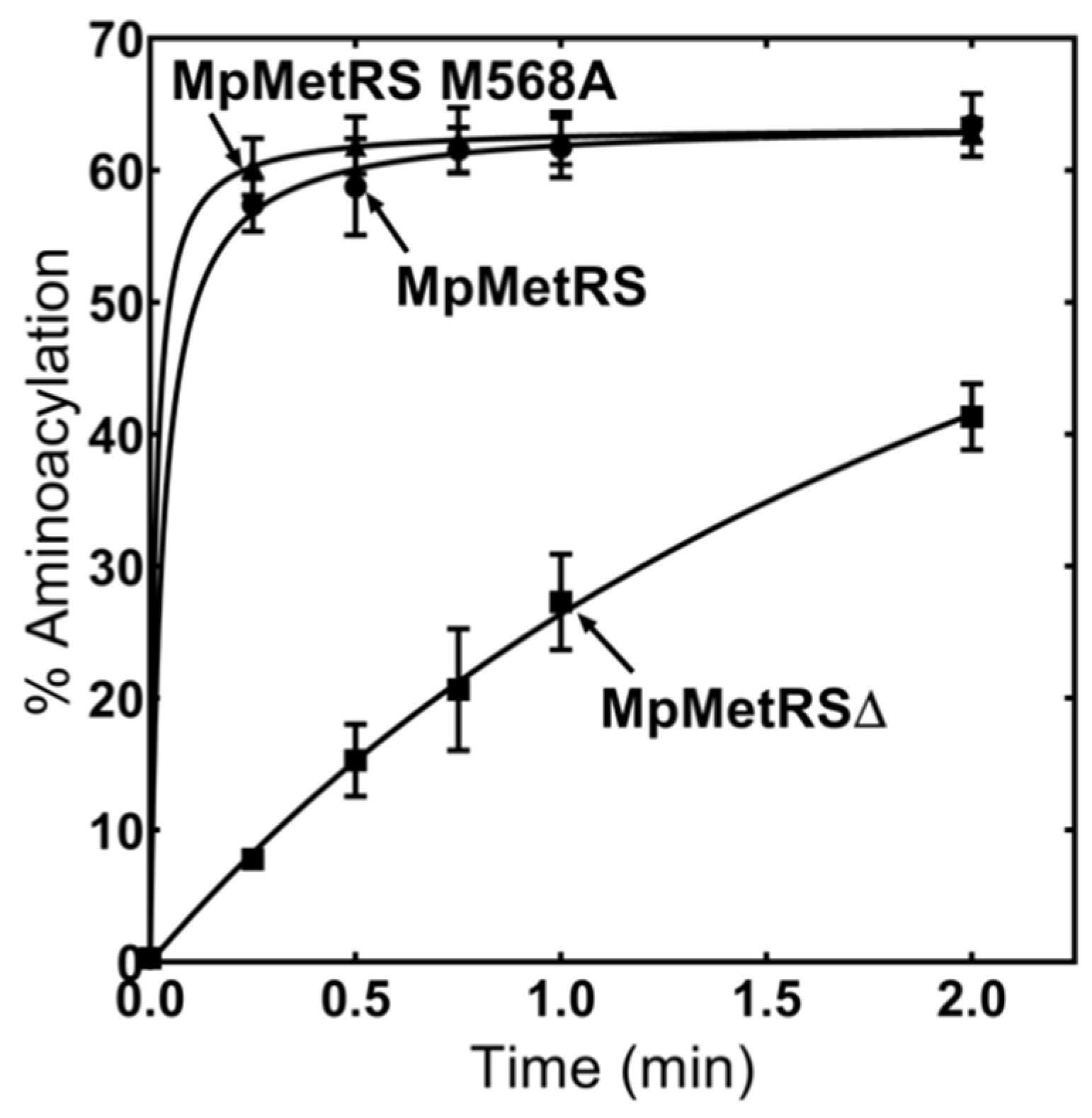
3.3. MpMetRS Is a Bifunctional Enzyme
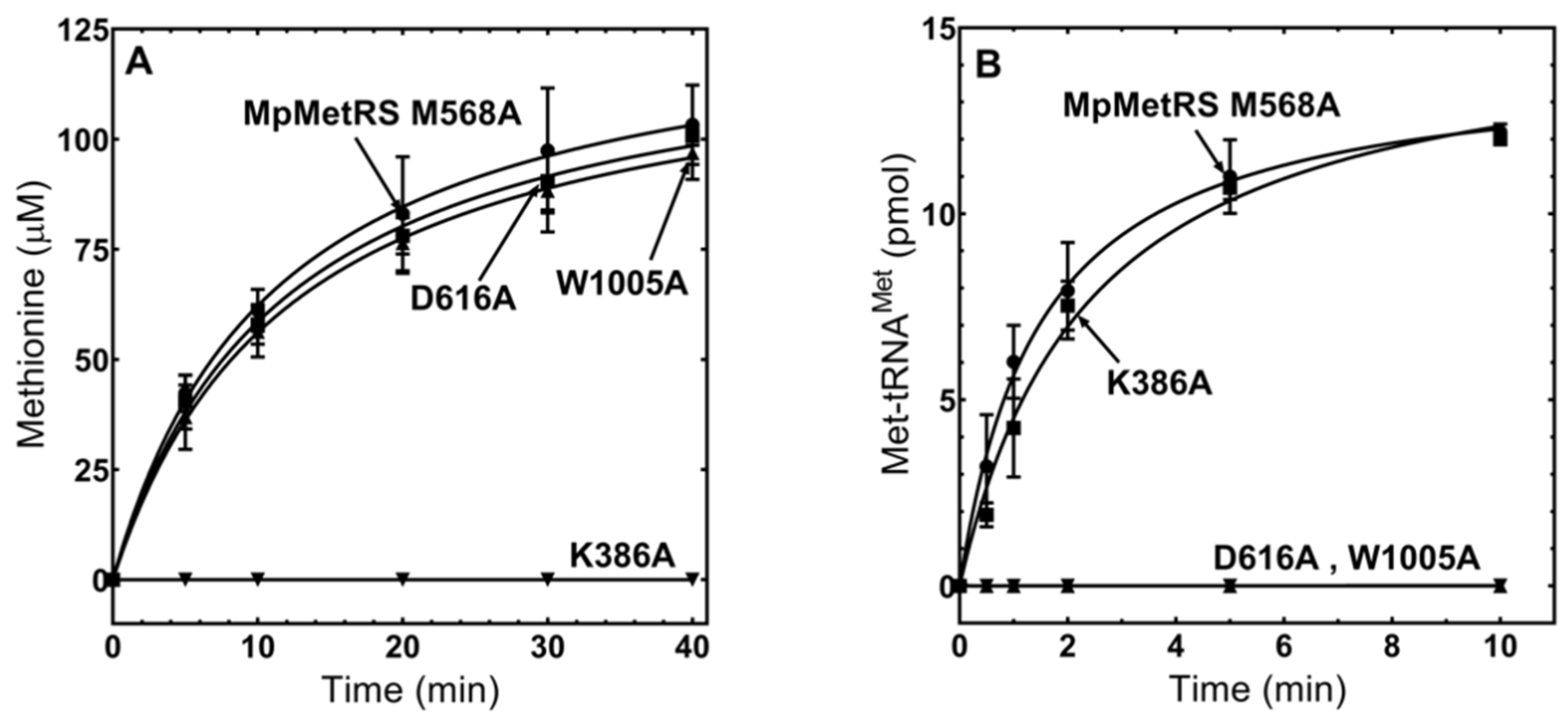
3.4. The Domains of MpMetRS Are Functionally Independent
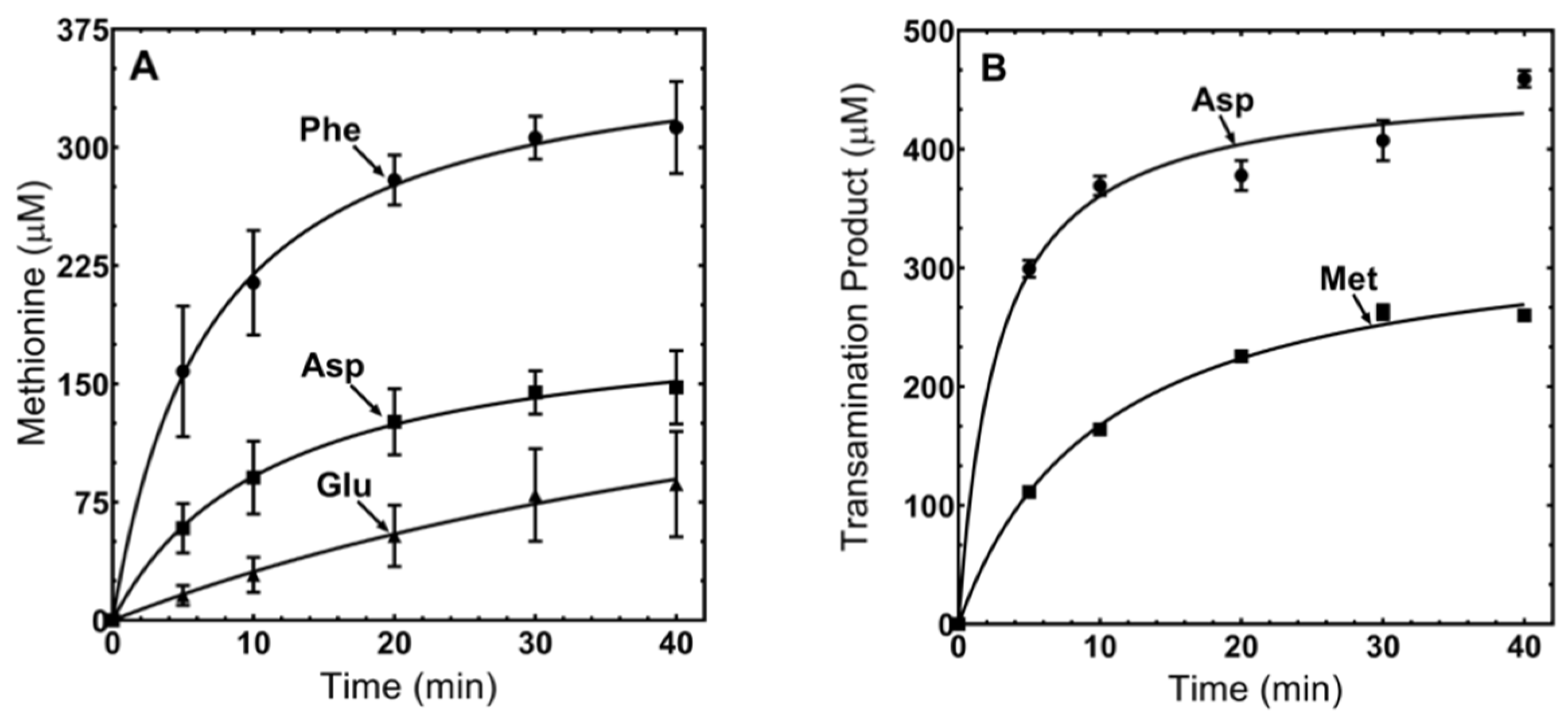
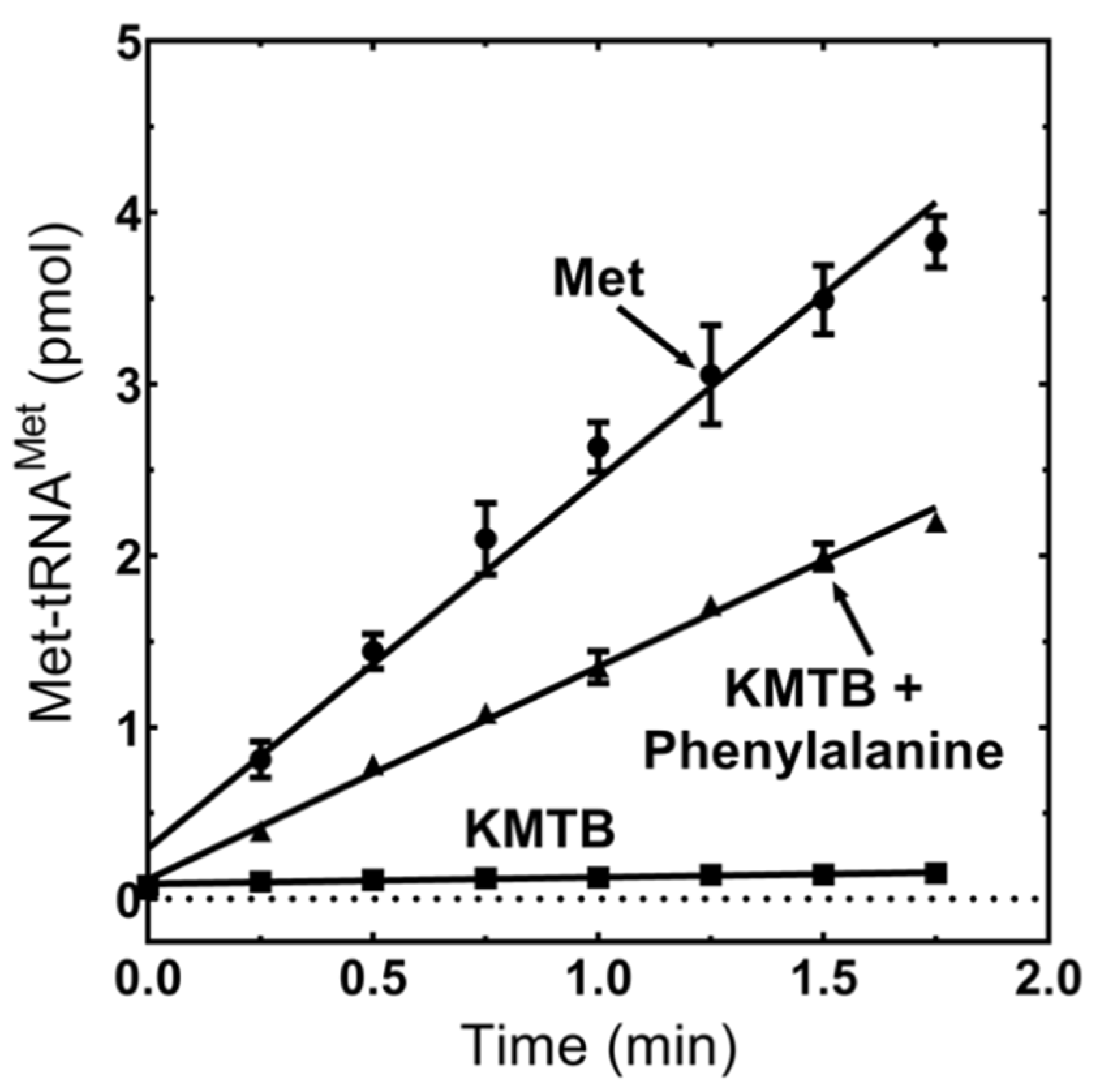
3.5. MpMetRS Aminotransferase Domain Can Supply tRNAMet with Methionine
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perona, J.J.; Hadd, A. Structural Diversity and Protein Engineering of the Aminoacyl-tRNA Synthetases. Biochemistry 2012, 51, 8705–8729. [Google Scholar] [CrossRef] [PubMed]
- Eriani, G.; Delarue, M.; Poch, O.; Gangloff, J.; Moras, D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 1990, 347, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Cusack, S.; Berthet-Colominas, C.; Hartlein, M.; Nassar, N. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 Å. Nature 1990, 347, 7. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.W.; Schimmel, P. Domain-domain communication in aminoacyl-tRNA synthetases. Prog. Nucleic Acid Res. Mol. Biol. 2001, 69, 317–349. [Google Scholar] [PubMed]
- Cvetesic, N.; Gruic-Sovulj, I. Synthetic and editing reactions of aminoacyl-tRNA synthetases using cognate and non-cognate amino acid substrates. Methods 2017, 113, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Schimmel, P. Essential nontranslational functions of tRNA synthetases. Nat. Chem. Biol. 2013, 9, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crepin, T.; Schmitt, E.; Blanquet, S.; Mechulam, Y. Three-dimensional structure of methionyl-tRNA synthetase from Pyrococcus abyssi. Biochemistry 2004, 43, 2635–2644. [Google Scholar] [CrossRef] [PubMed]
- Cassio, D.; Waller, J.-P. Modification of methionyl-tRNA synthetase by proteolytic cleavage and properties of the trypsin-modified enzyme. Eur. J. Biochem. 1971, 20, 283–300. [Google Scholar] [CrossRef]
- Lo, S.; Hayes, M.; Kotani, H.; Pierce, P.; Wear, D.; Newton, P.; Tully, J.; Shih, J. Adhesion onto and invasion into mammalian cells by Mycoplasma Penetrans: A newly isolated mycoplasma from patients with AIDS. Mod. Pathol. J. U. S. Can. Acad. Pathol. 1993, 6, 276–280. [Google Scholar]
- Sasaki, Y. The complete genomic sequence of Mycoplasma penetrans, an intracellular bacterial pathogen in humans. Nucleic Acids Res. 2002, 30, 5293–5300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Mark Roe, S.; Hou, Y.; Bartlam, M.; Rao, Z.; Pearl, L.H.; Danpure, C.J. Crystal structure of alanine: Glyoxylate aminotransferase and the relationship between genotype and enzymatic phenotype in primary hyperoxaluria Type 1. J. Mol. Biol. 2003, 331, 643–652. [Google Scholar] [CrossRef]
- Jones, T.E.; Brown, C.L.; Geslain, R.; Alexander, R.W.; Ribas de Pouplana, L. An operational RNA code for faithful assignment of AUG triplets to methionine. Mol. Cell 2008, 29, 401–407. [Google Scholar] [CrossRef]
- Jones, T.E.; Ribas de Pouplana, L.; Alexander, R.W. Evidence for late resolution of the AUX codon box in evolution. J. Biol. Chem. 2013, 288, 19625–19632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backlund, P.S.; Chang, C.P.; Smith, R.A. Identification of 2-keto-4-methylthiobutyrate as an intermediate compound in methionine synthesis from 5′-methylthioadenosine. J. Biol. Chem. 1982, 257, 4196–4202. [Google Scholar]
- Berger, L.C.; Wilson, J.; Wood, P.; Berger, B.J. Methionine regeneration and aspartate aminotransferase in parasitic protozoa. J. Bacteriol. 2001, 183, 4421–4434. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. 4. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Curnow, A.W.; Hong, K.W.; Yuan, R.; Kim, S.I.; Martins, O.; Winkler, W.; Henkin, T.M.; Söll, D. Glu-tRNAGln amidotransferase: A novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proc. Natl. Acad. Sci. USA 1997, 94, 11819–11826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherlin, L.D.; Bullock, T.L.; Nissan, T.A.; Perona, J.J.; Lariviere, F.J.; Uhlenbeck, O.C.; Scaringe, S.A. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA 2001, 7, 1671–1678. [Google Scholar]
- Ledoux, S.; Uhlenbeck, O.C. [3′-32P]-labeling tRNA with nucleotidyltransferase for assaying aminoacylation and peptide bond formation. Methods 2008, 44, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Wolfson, A.D.; Pleiss, J.A.; Uhlenbeck, O.C. A new assay for tRNA aminoacylation kinetics. RNA 1998, 4, 1019–1023. [Google Scholar] [CrossRef] [Green Version]
- Won Han, G.; Schwarzenbacher, R.; Page, R.; Jaroszewski, L.; Abdubek, P.; Ambing, E.; Biorac, T.; Canaves, J.M.; Chiu, H.-J.; Dai, X.; et al. Crystal structure of an alanine-glyoxylate aminotransferase from Anabaena sp. at 1.70 Å resolution reveals a noncovalently linked PLP cofactor. Proteins Struct. Funct. Bioinform. 2005, 58, 971–975. [Google Scholar] [CrossRef]
- Wada, H.; Esmond, S. The enzymatic oxidation of pyridoxine and pyridoxamine phosphates. J. Biol. Chem. 1961, 236, 8. [Google Scholar]
- Notarnicola, S.M.; McIntosh, M.A.; Wise, K.S. Multiple translational products from a Mycoplasma hyorhinis gene expressed in Escherichia coli. J. Bacteriol. 1990, 172, 2986–2995. [Google Scholar] [CrossRef] [Green Version]
- Loberg, M.A.; Hurtig, J.E.; Graff, A.H.; Allan, K.M.; Buchan, J.A.; Spencer, M.K.; Kelly, J.E.; Clodfelter, J.E.; Morano, K.A.; Lowther, W.T.; et al. Aromatic residues at the dimer−dimer interface in the peroxiredoxin Tsa1 facilitate decamer formation and biological function. Chem. Res. Toxicol. 2019, 32, 474–483. [Google Scholar] [CrossRef]
- Mauney, C.H.; Rogers, L.C.; Harris, R.S.; Daniel, L.W.; Devarie-Baez, N.O.; Wu, H.; Furdui, C.M.; Poole, L.B.; Perrino, F.W.; Hollis, T. The SAMHD1 dNTP triphosphohydrolase is controlled by a redox switch. Antioxid. Redox Signal. 2017, 27, 1317–1331. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, G.; Pelka, H.; Schulman, L.H.; Brunie, S. Activation of methionine by Escherichia coli methionyl-tRNA synthetase. Biochemistry 1991, 30, 9569–9575. [Google Scholar] [CrossRef]
- Ghosh, G.; Pelka, H.; Schulman, L.H. Identification of the tRNA anticodon recognition site of Escherichia coli methionyl-tRNA synthetase. Biochemistry 1990, 29, 2220–2225. [Google Scholar] [CrossRef]
- Sayer, C.; Bommer, M.; Isupov, M.; Ward, J.; Littlechild, J. Crystal structure and substrate specificity of the thermophilic serine: Pyruvate aminotransferase from Sulfolobus solfataricus. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Cui, Y.; Li, T.; Li, R.; Guo, L.; Li, D.; Wu, B. Biochemical and structural characterization of a highly active branched-chain amino acid aminotransferase from Pseudomonas sp. for efficient biosynthesis of chiral amino acids. Appl. Microbiol. Biotechnol. 2019, 103, 8051–8062. [Google Scholar] [CrossRef] [PubMed]
- Mironov, G.G.; St-Jacques, A.D.; Mungham, A.; Eason, M.G.; Chica, R.A.; Berezovski, M.V. Bioanalysis for biocatalysis: Multiplexed capillary electrophoresis–mass spectrometry assay for aminotransferase substrate discovery and specificity profiling. J. Am. Chem. Soc. 2013, 135, 13728–13736. [Google Scholar] [CrossRef]
- Onuffer, J.J.; Ton, B.T.; Klement, I. The use of natural and unnatural amino acid substrates to define the substrate specificity differences of Escherichia coli aspartate and tyrosine aminotransferases. Protein Sci. 1995, 4, 1743–1749. [Google Scholar] [CrossRef] [Green Version]
- Berger, B.J.; Dai, W.W.; Wang, H.; Stark, R.E.; Cerami, A. Aromatic amino acid transamination and methionine recycling in trypanosomatids. Proc. Natl. Acad. Sci. USA 1996, 93, 4126–4130. [Google Scholar] [CrossRef] [Green Version]
- Mechulam, Y.; Schmitt, E.; Maveyraud, L.; Zelwer, C.; Nureki, O.; Yokoyama, S.; Konno, M.; Blanquet, S. Crystal structure of Escherichia coli methionyl-tRNA synthetase highlights species-specific features. J. Mol. Biol. 1999, 294, 1287–1297. [Google Scholar] [CrossRef]
- Bera, A.K.; Smith, J.L.; Zalkin, H. Dual role for the glutamine phosphoribosylpyrophosphate amidotransferase ammonia channel: Interdomain signaling and intermediate channeling. J. Biol. Chem. 2000, 275, 7975–7979. [Google Scholar] [CrossRef] [Green Version]
- Geck, M.K.; Kirsch, J.F. A Novel, definitive test for substrate channeling illustrated with the aspartate aminotransferase malate dehydrogenase system. Biochemistry 1999, 38, 8032–8037. [Google Scholar] [CrossRef]
- Anderson, K.S. Understanding the molecular mechanism of substrate channeling and domain communication in protozoal bifunctional TS-DHFR. Protein Eng. Des. Sel. PEDS 2017, 30, 253–261. [Google Scholar] [CrossRef]
- Sharma, N.; Ahalawat, N.; Sandhu, P.; Strauss, E.; Mondal, J.; Anand, R. Role of allosteric switches and adaptor domains in long-distance cross-talk and transient tunnel formation. Sci. Adv. 2020, 6, eaay7919. [Google Scholar] [CrossRef] [Green Version]
- Bizarro, C.V.; Schuck, D.C. Purine and pyrimidine nucleotide metabolism in Mollicutes. Genet. Mol. Biol. 2007, 30, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Pollack, J.D. The necessity of combining genomic and enzymatic data to infer metabolic function and pathways in the smallest bacteria: Amino acid, purine and pyrimidine metabolism in mollicutes. Front. Biosci. 2002, 7, d1762–d1781. [Google Scholar] [PubMed] [Green Version]
- Rice, K.; Batul, K.; Whiteside, J.; Kelso, J.; Papinski, M.; Schmidt, E.; Pratasouskaya, A.; Wang, D.; Sullivan, R.; Bartlett, C.; et al. The predominance of nucleotidyl activation in bacterial phosphonate biosynthesis. Nat. Commun. 2019, 10, 3698. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AARS Domain Kinetics | Substrate | KM (μM) | kcat (s−1) | kcat/KM (M−1 s−1) |
| M568A:tRNAMet | 2.7 ± 0.7 | 6.2 ± 1.5 | 2.30 × 10−6 | |
| MpMetRS∆:tRNAMet | 2.3 | 0.5 | 0.22 × 10−6 | |
| ADT Domain Kinetics | Substrate | KM (mM) | kcat (s−1) | kcat/KM (M−1 s−1) |
| KMTB | 4.1 ± 0.3 | 6.9 ± 0.7 | 1.7 × 10−3 | |
| Phenylalanine | 0.5 ± 0.1 | 5.4 ± 0.4 | 10.9 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muraski, M.; Nilsson, E.; Weekley, B.; Sharma, S.B.; Alexander, R.W. A Novel Aminoacyl-tRNA Synthetase Appended Domain Can Supply the Core Synthetase with Its Amino Acid Substrate. Genes 2020, 11, 1320. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111320
Muraski M, Nilsson E, Weekley B, Sharma SB, Alexander RW. A Novel Aminoacyl-tRNA Synthetase Appended Domain Can Supply the Core Synthetase with Its Amino Acid Substrate. Genes. 2020; 11(11):1320. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111320
Chicago/Turabian StyleMuraski, Marc, Emil Nilsson, Benjamin Weekley, Sandhya Bharti Sharma, and Rebecca W. Alexander. 2020. "A Novel Aminoacyl-tRNA Synthetase Appended Domain Can Supply the Core Synthetase with Its Amino Acid Substrate" Genes 11, no. 11: 1320. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111320