LRRpredictor—A New LRR Motif Detection Method for Irregular Motifs of Plant NLR Proteins Using an Ensemble of Classifiers

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
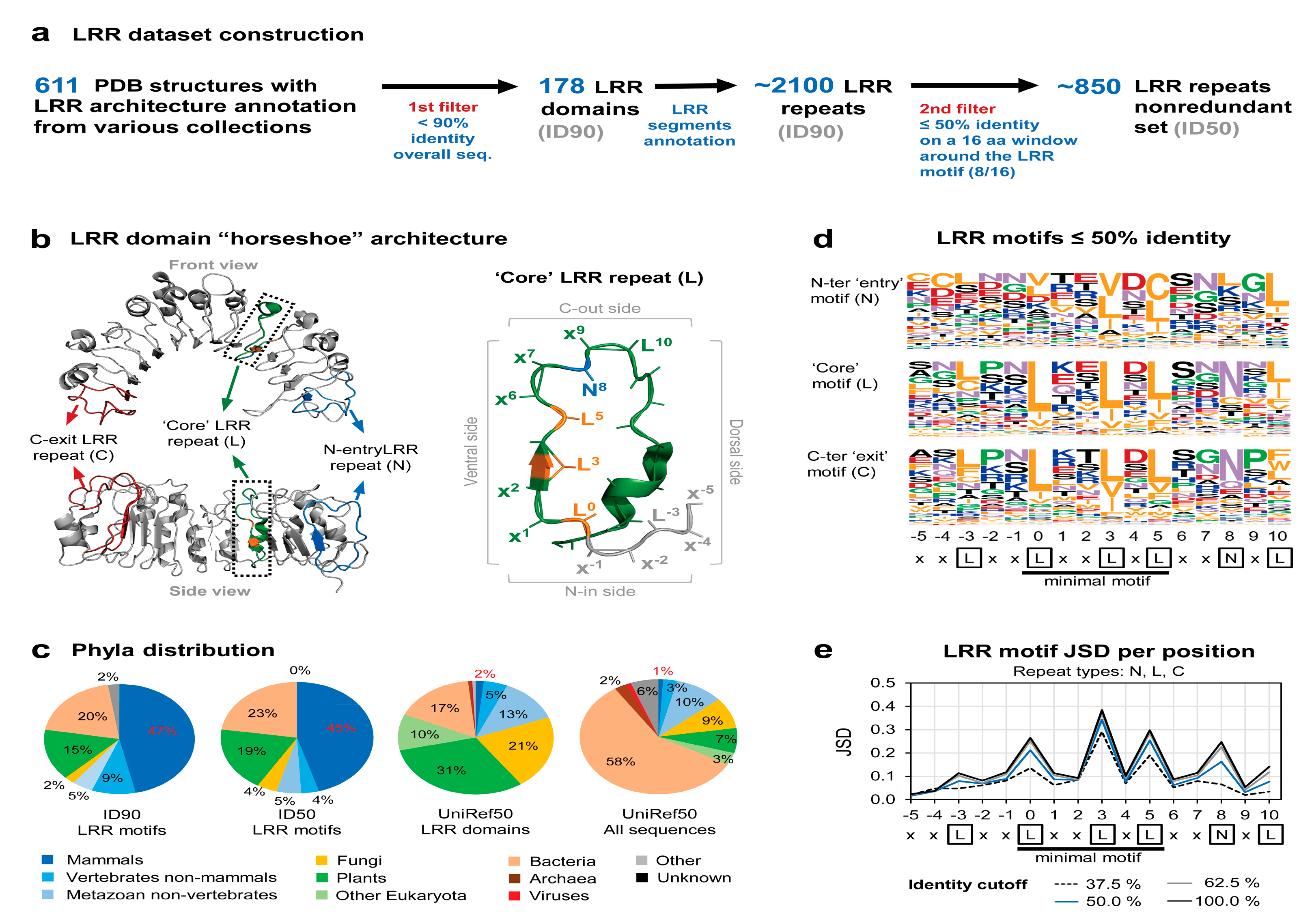
2.1. Assembly and Analysis of the LRR Structural Dataset
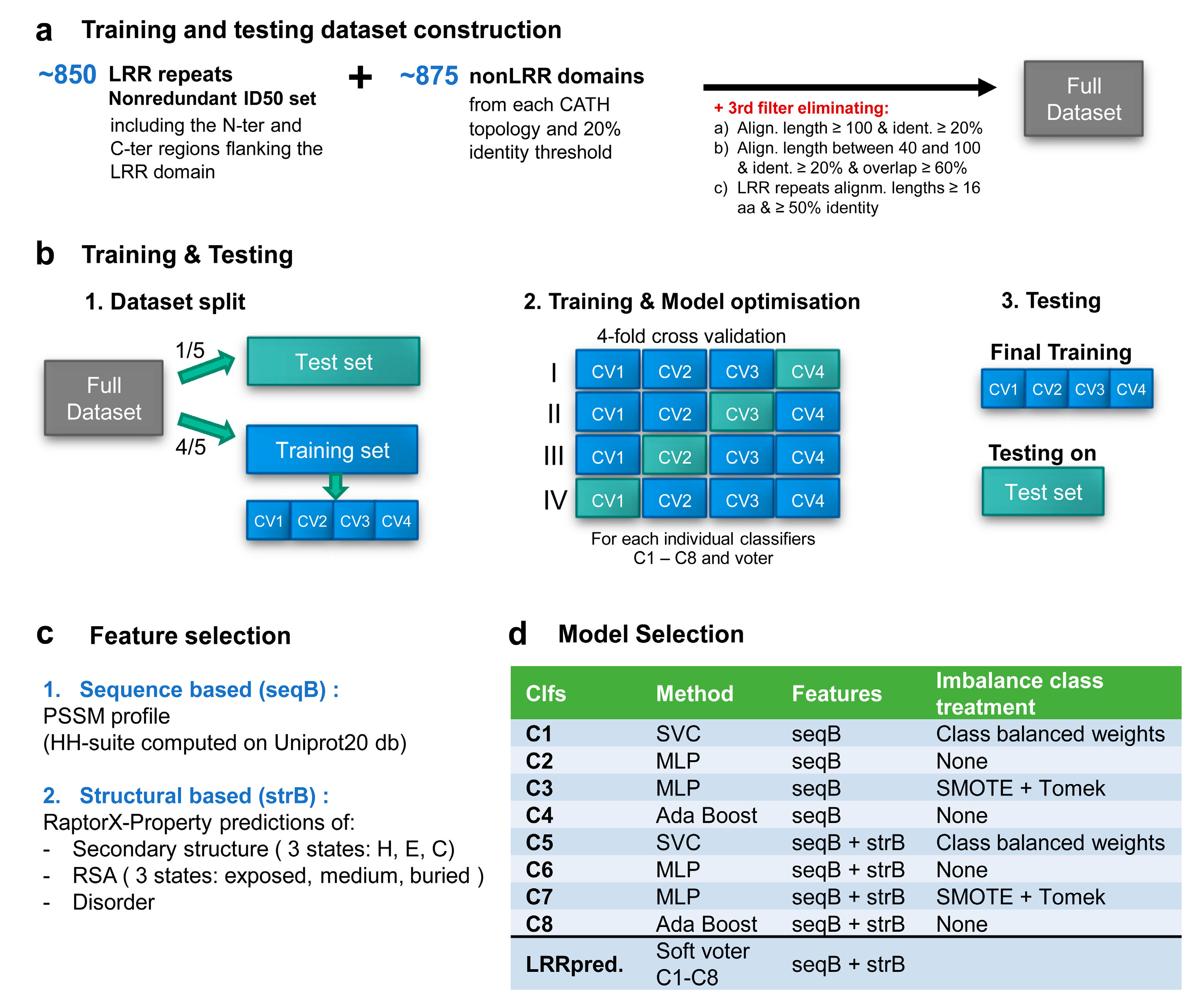
2.2. Training and Testing Datasets Construction
- the length of the alignment is over 100 and the identity is over 20%.
- length of the alignment is between 40 and 100 with an identity over 20% and the overlap with respect to both sequences is more than 60%.
- LRR repeats with alignments lengths ≥16 aa and ≥50% identical (equivalent of at most 8/16 aa constraint imposed initially on the motifs).
2.3. Feature Selection and Data Pre-Processing
2.4. Machine Learning Model Selection
2.5. Assembly of Protein Family Sets Containing LRR Domains
2.6. Assessment of LRR Motif Conservation Across Protein Groups
3. Results
3.1. Available LRR Domains in Structural Data
3.2. Development of the LRRpredictor Method
3.3. Assessment of LRRpredictor Performance
3.4. LRRpredictor Behavior on Protein Families Containing LRR Domains
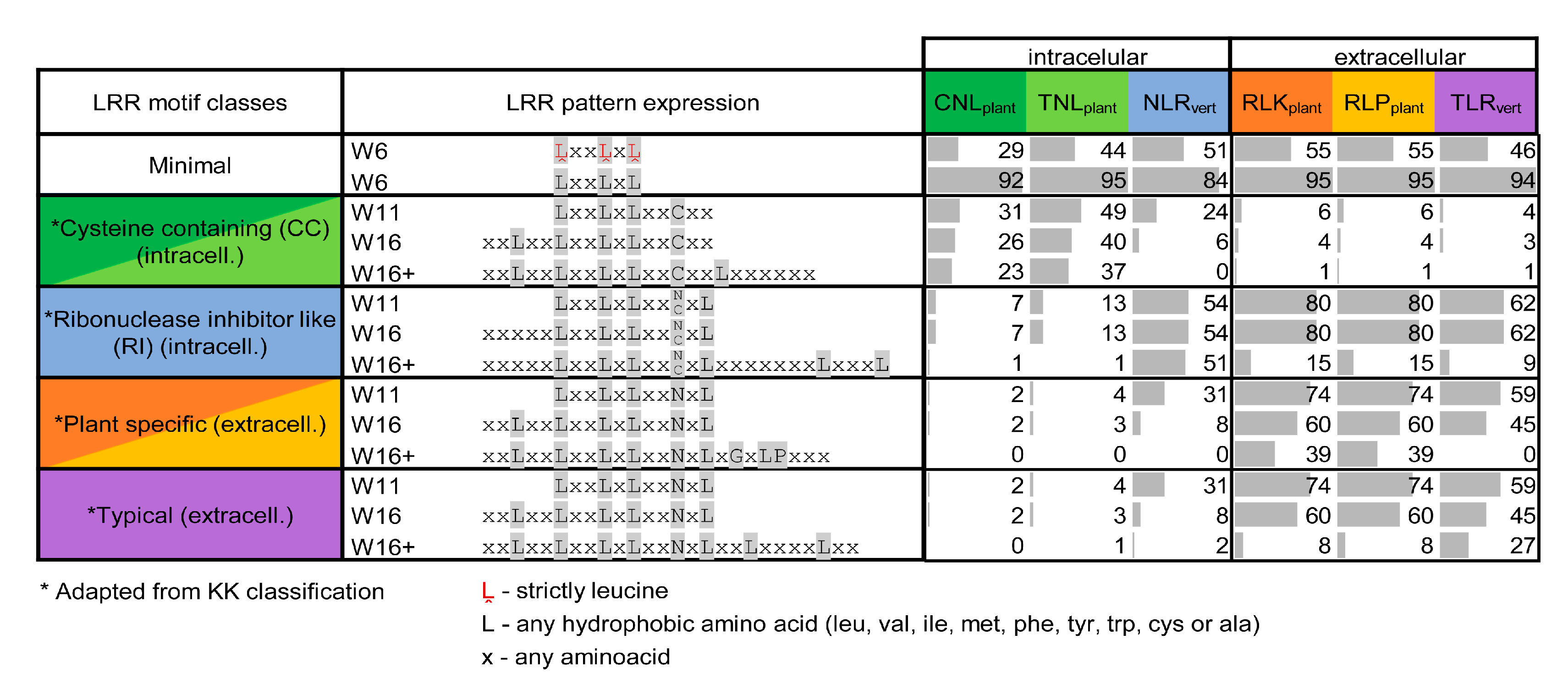
3.5. Predicted Repeats Consensus in Each Class
3.6. LRR Motifs Variability Across Classes
3.7. LRRpredictor Specificity Tested on Solenoid Architectures
4. Discussion
5. Conclusions
Supplementary Materials
Availability
Author Contributions
Funding
Conflicts of Interest
Appendix A



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Classifier | In-Sample | Out-Of Sample | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Precision | Recall | F1-Score | Precision | Recall | F1-Score | TN | FP | FN | TP | Recall on Core(L) Only | ||||||||
| - | Non-LRR Proteins | LRR Proteins | N-Entry (N) | Core (L) | C-Exit (C) | N-Entry (N) | Core (L) | C-Exit (C) | ||||||||||
| Cross validation | CV 1 | C1 | 0.897 | 0.998 | 0.945 | 0.930 | 0.930 | 0.930 | 33004 | 0 | 10 | 7 | 2 | 1 | 12 | 110 | 11 | 0.982 |
| C2 | 0.938 | 0.904 | 0.921 | 0.936 | 0.923 | 0.930 | 33005 | 0 | 9 | 8 | 2 | 1 | 11 | 110 | 11 | 0.982 | ||
| C3 | 0.877 | 0.887 | 0.882 | 0.904 | 0.923 | 0.914 | 33000 | 3 | 11 | 8 | 2 | 1 | 11 | 110 | 11 | 0.982 | ||
| C4 | 0.942 | 0.866 | 0.902 | 0.956 | 0.902 | 0.928 | 33008 | 0 | 6 | 9 | 3 | 2 | 10 | 109 | 10 | 0.973 | ||
| C5 | 0.894 | 0.925 | 0.910 | 0.924 | 0.937 | 0.931 | 33003 | 1 | 10 | 6 | 2 | 1 | 13 | 110 | 11 | 0.982 | ||
| C6 | 0.919 | 0.912 | 0.915 | 0.943 | 0.930 | 0.937 | 33006 | 0 | 8 | 7 | 2 | 1 | 12 | 110 | 11 | 0.982 | ||
| C7 | 0.976 | 1.000 | 0.988 | 0.938 | 0.951 | 0.944 | 33005 | 0 | 9 | 2 | 4 | 1 | 17 | 108 | 11 | 0.964 | ||
| C8 | 0.943 | 0.853 | 0.895 | 0.970 | 0.888 | 0.927 | 33010 | 0 | 4 | 10 | 3 | 3 | 9 | 109 | 9 | 0.973 | ||
| LRRpredictor | 0.930 | 0.918 | 0.924 | 0.950 | 0.923 | 0.936 | 33007 | 0 | 7 | 8 | 2 | 1 | 11 | 110 | 11 | 0.982 | ||
| CV 2 | C1 | 0.904 | 1.000 | 0.950 | 0.874 | 0.909 | 0.891 | 35819 | 0 | 23 | 9 | 4 | 3 | 12 | 135 | 12 | 0.971 | |
| C2 | 0.930 | 0.900 | 0.915 | 0.903 | 0.909 | 0.906 | 35825 | 0 | 17 | 8 | 5 | 3 | 13 | 134 | 12 | 0.964 | ||
| C3 | 0.901 | 0.890 | 0.895 | 0.892 | 0.897 | 0.895 | 35823 | 0 | 19 | 10 | 5 | 3 | 11 | 134 | 12 | 0.964 | ||
| C4 | 0.954 | 0.855 | 0.902 | 0.937 | 0.851 | 0.892 | 35832 | 0 | 10 | 13 | 8 | 5 | 8 | 131 | 10 | 0.942 | ||
| C5 | 0.904 | 0.937 | 0.920 | 0.914 | 0.914 | 0.914 | 35827 | 1 | 14 | 8 | 4 | 3 | 13 | 135 | 12 | 0.971 | ||
| C6 | 0.947 | 0.904 | 0.925 | 0.925 | 0.914 | 0.920 | 35829 | 0 | 13 | 8 | 4 | 3 | 13 | 135 | 12 | 0.971 | ||
| C7 | 0.974 | 1.000 | 0.987 | 0.898 | 0.909 | 0.903 | 35824 | 2 | 16 | 7 | 4 | 5 | 14 | 135 | 10 | 0.971 | ||
| C8 | 0.953 | 0.878 | 0.914 | 0.944 | 0.869 | 0.905 | 35833 | 0 | 9 | 10 | 9 | 4 | 11 | 130 | 11 | 0.935 | ||
| LRRpredictor | 0.941 | 0.908 | 0.924 | 0.924 | 0.897 | 0.910 | 35829 | 0 | 13 | 9 | 6 | 3 | 12 | 133 | 12 | 0.957 | ||
| CV 3 | C1 | 0.925 | 0.998 | 0.960 | 0.881 | 0.847 | 0.864 | 36028 | 0 | 18 | 7 | 10 | 7 | 12 | 115 | 6 | 0.920 | |
| C2 | 0.949 | 0.943 | 0.946 | 0.903 | 0.828 | 0.864 | 36032 | 0 | 14 | 8 | 12 | 7 | 11 | 113 | 6 | 0.904 | ||
| C3 | 0.922 | 0.904 | 0.913 | 0.879 | 0.834 | 0.856 | 36028 | 0 | 18 | 7 | 11 | 8 | 12 | 114 | 5 | 0.912 | ||
| C4 | 0.972 | 0.898 | 0.934 | 0.899 | 0.790 | 0.841 | 36032 | 0 | 14 | 7 | 18 | 8 | 12 | 107 | 5 | 0.856 | ||
| C5 | 0.932 | 0.945 | 0.938 | 0.887 | 0.847 | 0.866 | 36029 | 0 | 17 | 8 | 9 | 7 | 11 | 116 | 6 | 0.928 | ||
| C6 | 0.944 | 0.931 | 0.938 | 0.891 | 0.834 | 0.862 | 36030 | 0 | 16 | 9 | 10 | 7 | 10 | 115 | 6 | 0.920 | ||
| C7 | 0.979 | 1.000 | 0.989 | 0.861 | 0.866 | 0.864 | 36024 | 2 | 20 | 6 | 8 | 7 | 13 | 117 | 6 | 0.936 | ||
| C8 | 0.968 | 0.906 | 0.936 | 0.883 | 0.771 | 0.823 | 36030 | 0 | 16 | 10 | 18 | 8 | 9 | 107 | 5 | 0.856 | ||
| LRRpredictor | 0.950 | 0.939 | 0.945 | 0.893 | 0.854 | 0.873 | 36030 | 0 | 16 | 7 | 9 | 7 | 12 | 116 | 6 | 0.928 | ||
| CV 4 | C1 | 0.885 | 1.000 | 0.939 | 0.936 | 0.916 | 0.926 | 33343 | 0 | 12 | 8 | 6 | 2 | 14 | 147 | 13 | 0.961 | |
| C2 | 0.923 | 0.884 | 0.903 | 0.961 | 0.900 | 0.929 | 33348 | 0 | 7 | 9 | 7 | 3 | 13 | 146 | 12 | 0.954 | ||
| C3 | 0.818 | 0.901 | 0.858 | 0.851 | 0.932 | 0.890 | 33324 | 2 | 29 | 7 | 4 | 2 | 15 | 149 | 13 | 0.974 | ||
| C4 | 0.931 | 0.848 | 0.888 | 0.971 | 0.868 | 0.917 | 33350 | 0 | 5 | 12 | 10 | 3 | 10 | 143 | 12 | 0.935 | ||
| C5 | 0.907 | 0.920 | 0.913 | 0.906 | 0.916 | 0.911 | 33337 | 0 | 18 | 8 | 5 | 3 | 14 | 148 | 12 | 0.967 | ||
| C6 | 0.921 | 0.907 | 0.914 | 0.951 | 0.926 | 0.939 | 33346 | 0 | 9 | 7 | 5 | 2 | 15 | 148 | 13 | 0.967 | ||
| C7 | 0.983 | 1.000 | 0.992 | 0.922 | 0.926 | 0.924 | 33340 | 4 | 11 | 7 | 5 | 2 | 15 | 148 | 13 | 0.967 | ||
| C8 | 0.944 | 0.846 | 0.892 | 0.970 | 0.858 | 0.911 | 33350 | 0 | 5 | 15 | 9 | 3 | 7 | 144 | 12 | 0.941 | ||
| LRRpredictor | 0.923 | 0.912 | 0.917 | 0.967 | 0.916 | 0.941 | 33349 | 0 | 6 | 8 | 6 | 2 | 14 | 147 | 13 | 0.961 | ||
| Test | C1 | 0.900 | 0.997 | 0.946 | 0.874 | 0.880 | 0.877 | 35107 | 0 | 19 | 6 | 10 | 2 | 13 | 109 | 10 | 0.916 | |
| C2 | 0.962 | 0.956 | 0.959 | 0.941 | 0.847 | 0.891 | 35118 | 0 | 8 | 8 | 12 | 3 | 11 | 107 | 9 | 0.899 | ||
| C3 | 0.882 | 0.896 | 0.889 | 0.852 | 0.847 | 0.850 | 35104 | 1 | 21 | 8 | 13 | 2 | 11 | 106 | 10 | 0.891 | ||
| C4 | 0.940 | 0.874 | 0.906 | 0.907 | 0.840 | 0.872 | 35113 | 1 | 12 | 8 | 14 | 2 | 11 | 105 | 10 | 0.882 | ||
| C5 | 0.895 | 0.934 | 0.914 | 0.862 | 0.873 | 0.868 | 35105 | 1 | 20 | 7 | 10 | 2 | 12 | 109 | 10 | 0.916 | ||
| C6 | 0.940 | 0.901 | 0.920 | 0.928 | 0.860 | 0.893 | 35116 | 0 | 10 | 7 | 12 | 2 | 12 | 107 | 10 | 0.899 | ||
| C7 | 0.990 | 1.000 | 0.995 | 0.827 | 0.827 | 0.827 | 35100 | 4 | 22 | 7 | 16 | 3 | 12 | 103 | 9 | 0.866 | ||
| C8 | 0.942 | 0.874 | 0.906 | 0.920 | 0.840 | 0.878 | 35115 | 0 | 11 | 9 | 13 | 2 | 10 | 106 | 10 | 0.891 | ||
| LRRpredictor | 0.943 | 0.928 | 0.936 | 0.928 | 0.860 | 0.893 | 35116 | 0 | 10 | 7 | 12 | 2 | 12 | 107 | 10 | 0.899 | ||
References
- Enkhbayar, P.; Kamiya, M.; Osaki, M.; Matsumoto, T.; Matsushima, N. Structural Principles of Leucine-Rich Repeat (LRR) Proteins. Proteins Struct. Funct. Bioinform. 2004, 54, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.F.; Henk, A.; Mowery, P.; Holub, E.; Innes, R.W. A mutation within the leucine-rich repeat domain of the arabidopsis disease resistance gene RPS5 partially suppresses multiple bacterial and downy mildew resistance genes. Plant Cell 1998, 10, 1439–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; McAdams, S.A.; Bryan, G.T.; Hershey, H.P.; Valent, B. Direct interaction of resistance gene and avirulence gene products confers rice blast resistance. EMBO J. 2000, 19, 4004–4014. [Google Scholar] [CrossRef] [PubMed]
- Moghaddas, F.; Zeng, P.; Zhang, Y.; Schützle, H.; Brenner, S.; Hofmann, S.R.; Berner, R.; Zhao, Y.; Lu, B.; Chen, X.; et al. Autoinflammatory mutation in NLRC4 reveals a leucine-rich repeat (LRR)–LRR oligomerization interface. J. Allergy Clin. Immunol. 2018, 142, 1956–1967.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; Van Der Brug, M.; De Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, G.X.; Li, Z.; Zhou, Y.Z. The role of small leucine-rich proteoglycans in osteoarthritis pathogenesis. Osteoarthritis Cartilage. 2014, 22, 896–903. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M. PRELP, collagen, and a theory of Hutchinson-Gilford progeria. Ageing Res. Rev. 2003, 2, 95–105. [Google Scholar] [CrossRef]
- Sukarta, O.C.A.; Slootweg, E.J.; Goverse, A. Structure-informed insights for NLR functioning in plant immunity. Semin. Cell Dev. Biol. 2016, 56, 134–149. [Google Scholar] [CrossRef]
- Urbach, J.M.; Ausubel, F.M. The NBS-LRR architectures of plant R-proteins and metazoan NLRs evolved in independent events. Proc. Natl. Acad. Sci. USA 2017, 114, 1063–1068. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, N.; Miyashita, H.; Enkhbayar, P.; Kretsinger, R.H. Comparative Geometrical Analysis of Leucine-Rich Repeat Structures in the Nod-Like and Toll-Like Receptors in Vertebrate Innate Immunity. Biomolecules 2015, 5, 1955–1978. [Google Scholar] [CrossRef] [PubMed]
- Kajava, A.V.; Kobe, B. Assessment of the ability to model proteins with leucine-rich repeats in light of the latest structural information. Protein Sci. 2002, 11, 1082–1090. [Google Scholar] [CrossRef]
- Matsushima, N.; Tanaka, T.; Enkhbayar, P.; Mikami, T.; Taga, M.; Yamada, K.; Kuroki, Y. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genom. 2007, 8, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobe, B.; Kajava, A.V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 2001, 11, 725–732. [Google Scholar] [CrossRef]
- Ng, A.C.Y.; Eisenberg, J.M.; Heath, R.J.W.; Huett, A.; Robinson, C.M.; Nau, G.J.; Xavier, R.J. Human leucine-rich repeat proteins: A genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc. Natl. Acad. Sci. USA 2011, 108, 4631–4638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sela, H.; Spiridon, L.N.; Petrescu, A.J.; Akerman, M.; Mandel-Gutfreund, Y.; Nevo, E.; Loutre, C.; Keller, B.; Schulman, A.H.; Fahima, T. Ancient diversity of splicing motifs and protein surfaces in the wild emmer wheat (Triticum dicoccoides) LR10 coiled coil (CC) and leucine-rich repeat (LRR) domains. Mol. Plant Pathol. 2012, 13, 276–287. [Google Scholar] [CrossRef]
- Slootweg, E.J.; Spiridon, L.N.; Roosien, J.; Butterbach, P.; Pomp, R.; Westerhof, L.; Wilbers, R.; Bakker, E.; Bakker, J.; Petrescu, A.J.; et al. Structural determinants at the interface of the ARC2 and leucine-rich repeat domains control the activation of the plant immune receptors Rx1 and Gpa2. Plant Physiol. 2013, 162, 1510–1528. [Google Scholar] [CrossRef] [Green Version]
- Sela, H.; Spiridon, L.N.; Ashkenazi, H.; Bhullar, N.K.; Brunner, S.; Petrescu, A.-J.; Fahima, T.; Keller, B.; Jordan, T. Three-Dimensional Modeling and Diversity Analysis Reveals Distinct AVR Recognition Sites and Evolutionary Pathways in Wild and Domesticated Wheat Pm3 R Genes. Mol. Plant-Microbe Interact. 2014, 27, 835–845. [Google Scholar] [CrossRef]
- Rajaraman, J.; Douchkov, D.; Hensel, G.; Stefanato, F.L.; Gordon, A.; Ereful, N.; Caldararu, O.F.; Petrescu, A.J.; Kumlehn, J.; Boyd, L.A.; et al. An LRR/Malectin receptor-like kinase mediates resistance to non-adapted and adapted powdery mildew fungi in barley and wheat. Front. Plant Sci. 2016, 7, 1836. [Google Scholar] [CrossRef] [Green Version]
- Baudin, M.; Schreiber, K.J.; Martin, E.C.; Petrescu, A.J.; Lewis, J.D. Structure–function analysis of ZAR1 immune receptor reveals key molecular interactions for activity. Plant J 2020, 101, 352–370. [Google Scholar] [CrossRef]
- Wang, J.; Hu, M.; Wang, J.; Qi, J.; Han, Z.; Wang, G.; Qi, Y.; Wang, H.-W.; Zhou, J.-M.; Chai, J. Reconstitution and structure of a plant NLR resistosome conferring immunity. Science 2019, 364, eaav5870. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Hu, M.; Wu, S.; Qi, J.; Wang, G.; Han, Z.; Qi, Y.; Gao, N.; Wang, H.W.; et al. Ligand-triggered allosteric ADP release primes a plant NLR complex. Science 2019, 364, eaav5868. [Google Scholar] [CrossRef]
- Kajava, A.V.; Vassart, G.; Wodak, S.J. Modeling of the three-dimensional structure of proteins with the typical leucine-rich repeats. Structure 1995, 3, 867–877. [Google Scholar] [CrossRef] [Green Version]
- Helft, L.; Reddy, V.; Chen, X.; Koller, T.; Federici, L.; Fernández-Recio, J.; Gupta, R.; Bent, A. LRR conservation mapping to predict functional sites within protein leucine-rich repeat domains. PLoS ONE 2011, 6, e21614. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, W.; Zhang, T.; Gong, Z.; Zhao, H.; Han, G.Z. Out of Water: The Origin and Early Diversification of Plant R-Genes. Plant Physiol. 2018, 177, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Offord, V.; Werling, D. LRRfinder2.0: A webserver for the prediction of leucine-rich repeats. Innate Immun. 2013, 19, 398–402. [Google Scholar] [CrossRef] [Green Version]
- Bej, A.; Sahoo, B.R.; Swain, B.; Basu, M.; Jayasankar, P.; Samanta, M. LRRsearch: An asynchronous server-based application for the prediction of leucine-rich repeat motifs and an integrative database of NOD-like receptors. Comput. Biol. Med. 2014, 53, 164–170. [Google Scholar] [CrossRef]
- Dawson, N.L.; Lewis, T.E.; Das, S.; Lees, J.G.; Lee, D.; Ashford, P.; Orengo, C.A.; Sillitoe, I. CATH: An expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 2017, 45, D289–D295. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Dunbrack, R.L. PISCES: A protein sequence culling server. Bioinformatics 2003, 19, 1589–1591. [Google Scholar] [CrossRef] [Green Version]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, W.; Liu, S.; Xu, J. RaptorX-Property: A web server for protein structure property prediction. Nucleic Acids Res. 2016, 44, W430–W435. [Google Scholar] [CrossRef]
- Wang, S.; Sun, S.; Xu, J. AUC-maximized deep convolutional neural fields for protein sequence labeling. In Proceedings of the Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin, Germany, 2016; Volume 9852 LNAI, pp. 1–16. [Google Scholar]
- Wang, S.; Ma, J.; Xu, J. AUCpreD: Proteome-level protein disorder prediction by AUC-maximized deep convolutional neural fields. In Proceedings of the Bioinformatics; Oxford University Press: Oxford, UK, 2016; Volume 32, pp. i672–i679. [Google Scholar]
- Wang, S.; Peng, J.; Ma, J.; Xu, J. Protein Secondary Structure Prediction Using Deep Convolutional Neural Fields. Sci. Rep. 2016, 6, 18962. [Google Scholar] [CrossRef] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Pal, S.K.; Mitra, S. Multilayer Perceptron, Fuzzy Sets, and Classification. IEEE Trans. Neural Netw. 1992, 3, 683–697. [Google Scholar] [CrossRef]
- Rosenblatt, F. The perceptron: A probabilistic model for information storage and organization in the brain. Psychol. Rev. 1958, 65, 386–408. [Google Scholar] [CrossRef] [Green Version]
- Freund, Y.; Schapire, R.E. A Decision-Theoretic Generalization of On-Line Learning and an Application to Boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Bai, Y.; Garcia, E.A.; Li, S. ADASYN: Adaptive synthetic sampling approach for imbalanced learning. In Proceedings of the 2008 IEEE International Joint Conference on Neural Networks; IEEE: Piscataway, NJ, USA, 2008; pp. 1322–1328. [Google Scholar]
- Han, H.; Wang, W.-Y.; Mao, B.-H. Borderline-SMOTE: A New Over-Sampling Method in Imbalanced Data Sets Learning; Springer: Berlin/Heidelberg, Germany, 2005; Volume 3644. [Google Scholar]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Cooper, E.W.; Kamei, K. Borderline Over-sampling for Imbalanced Data Classification. In Proceedings: Fifth International Workshop on Computational Intelligence & Applications; IEEE: Piscataway, NJ, USA, 2009; Volume 2009, pp. 24–29. [Google Scholar]
- Batista, G.E.; Bazzan, A.L.C.; Monard, M.C. Balancing Training Data for Automated Annotation of Keywords: A Case Study. In WOB; UFRGS: Porto Alegre, Brazil, 2003; pp. 10–18. [Google Scholar]
- Batista, G.E.; Prati, R.C.; Monard, M.C. A study of the behavior of several methods for balancing machine learning training data. ACM SIGKDD Explor. Newsl. 2004, 6, 20–29. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Liu, D.C.; Nocedal, J. On the limited memory BFGS method for large scale optimization. Math. Program. 1989, 45, 503–528. [Google Scholar] [CrossRef] [Green Version]
- Kingma, D.P.; Ba, J.L. Adam: A method for stochastic optimization. In Proceedings of the 3rd International Conference on Learning Representations, ICLR 2015—Conference Track Proceedings; International Conference on Learning Representations; ICLR: San Diego, CA, USA, 2015. [Google Scholar]
- Robbins, H.; Monro, S. A Stochastic Approximation Method. Ann. Math. Stat. 1951, 22, 400–407. [Google Scholar] [CrossRef]
- Hahnioser, R.H.R.; Sarpeshkar, R.; Mahowald, M.A.; Douglas, R.J.; Seung, H.S. Digital selection and analogue amplification coexist in a cortex- inspired silicon circuit. Nature 2000, 405, 947–951. [Google Scholar] [CrossRef]
- Lemaitre, G.; Nogueira, F.; Aridas, C.K. Imbalanced-learn: A Python Toolbox to Tackle the Curse of Imbalanced Datasets in Machine Learning. J. Mach. Learn. Res. 2017, 18, 1–5. [Google Scholar]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef]
- Halperin, E.; Buhler, J.; Karp, R.; Krauthgamer, R.; Westover, B. Detecting protein sequence conservation via metric embeddings. Bioinformatics 2003, 19, 122–129. [Google Scholar] [CrossRef]
- Govindarajan, R.; Leela, B.C.; Nair, A.S. RBLOSUM performs better than CorBLOSUM with lesser error per query. BMC Res. Notes 2018, 11, 328. [Google Scholar] [CrossRef] [PubMed]
- Styczynski, M.P.; Jensen, K.L.; Rigoutsos, I.; Stephanopoulos, G. BLOSUM62 miscalculations improve search performance. Nat. Biotechnol. 2008, 26, 274–275. [Google Scholar] [CrossRef] [PubMed]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Kruskal, J.B. Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 1964, 29, 1–27. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L.L.C. The PyMOL Molecular Graphics System, Version 2.2.3. Available online: https://pymol.org/2/ (accessed on 10 November 2019).
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 99–104. [Google Scholar] [CrossRef]
- Kajava, A.V. Structural diversity of leucine-rich repeat proteins. J. Mol. Biol. 1998, 277, 519–527. [Google Scholar] [CrossRef]
- Borg, I.; Groenen, P.J.F. Modern Multidimensional Scaling, 2nd ed.; Chapter: MDS Models and Measures of Fit; Springer: Berlin, Germany, 2005; pp. 37–61. [Google Scholar]
- Ward, C.W.; Garrett, T.P.J. The relationship between the L1 and L2 domains of the insulin and epidermal growth factor receptors and leucine-rich repeat modules. BMC Bioinform. 2001, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, M.; Cournoyer, P.; Dinesh-Kumar, S.P. The leucine-rich repeat domain in plant innate immunity: A wealth of possibilities. Cell. Microbiol. 2009, 11, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plantĝ€ pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef]
- Ravensdale, M.; Nemri, A.; Thrall, P.H.; Ellis, J.G.; Dodds, P.N. Co-evolutionary interactions between host resistance and pathogen effector genes in flax rust disease. Mol. Plant Pathol. 2011, 12, 93–102. [Google Scholar] [CrossRef]
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef] [Green Version]
- Borrelli, G.M.; Mazzucotelli, E.; Marone, D.; Crosatti, C.; Michelotti, V.; Valè, G.; Mastrangelo, A.M. Regulation and evolution of NLR genes: A close interconnection for plant immunity. Int. J. Mol. Sci. 2018, 19, 1662. [Google Scholar] [CrossRef] [Green Version]
- Leister, R.T.; Dahlbeck, D.; Day, B.; Li, Y.; Chesnokova, O.; Staskawicz, B.J. Molecular genetic evidence for the role of SGT1 in the intramolecular complementation of Bs2 protein activity in Nicotiana benthamiana. Plant Cell 2005, 17, 1268–1278. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.D.; Lee, A.H.-Y.; Hassan, J.A.; Wan, J.; Hurley, B.; Jhingree, J.R.; Wang, P.W.; Lo, T.; Youn, J.-Y.; Guttman, D.S.; et al. The Arabidopsis ZED1 pseudokinase is required for ZAR1-mediated immunity induced by the Pseudomonas syringae type III effector HopZ1a. Proc. Natl. Acad. Sci. USA 2013, 110, 18722–18727. [Google Scholar] [CrossRef] [Green Version]
- Mestre, P.; Baulcombe, D.C. Elicitor-mediated oligomerization of the tobacco N disease resistance protein. Plant Cell 2006, 18, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Huh, S.U.; Cevik, V.; Ding, P.; Duxbury, Z.; Ma, Y.; Tomlinson, L.; Sarris, P.F.; Jones, J.D.G. Protein-protein interactions in the RPS4/RRS1 immune receptor complex. PLoS Pathog. 2017, 13, e1006376. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Yan, C.; Liu, P.; Huang, Z.; Ma, R.; Zhang, C.; Wang, R.; Zhang, Y.; Martinon, F.; Miao, D.; et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 2013, 341, 172–175. [Google Scholar] [CrossRef]
- Sun, Y.; Li, L.; Macho, A.P.; Han, Z.; Hu, Z.; Zipfel, C.; Zhou, J.-M.; Chai, J. Structural basis for flg22-induced activation of the Arabidopsis FLS2-BAK1 immune complex. Science 2013, 342, 624–628. [Google Scholar] [CrossRef]
- Choe, J.; Kelker, M.S.; Wilson, I.A. Crystal structure of human toll-like receptor 3. Science 2005, 22, 581–585. [Google Scholar] [CrossRef]
- Pickersgill, R.; Jenkins, J.; Harris, G.; Nasser, W.; Robert-Baudouy, J. The structure of Bacillus subtilis pectate lyase in complex with calcium. Nat. Struct. Biol. 1994, 1, 717–723. [Google Scholar] [CrossRef]
- Olivier, N.B.; Imperiali, B. Crystal structure and catalytic mechanism of PglD from Campylobacter jejuni. J. Biol. Chem. 2008, 283, 27937–27946. [Google Scholar] [CrossRef] [Green Version]
- Huber, A.H.; Nelson, W.J.; Weis, W.I. Three-dimensional structure of the armadillo repeat region of β-catenin. Cell 1997, 90, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Hamada-Nakahara, S.; Itoh, Y.; Takemura, K.; Shimada, A.; Ueda, Y.; Kitamata, M.; Matsuoka, R.; Hanawa-Suetsugu, K.; Senju, Y.; et al. TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P₂. Nat. Commun. 2014, 5, 4994. [Google Scholar] [CrossRef] [Green Version]








| Full Training & Testing Dataset (CV 1-4 and Test Sets) | ||||||
|---|---|---|---|---|---|---|
| NonLRR Proteins | LRR Proteins | |||||
| LRR-Like Pattern | Total Number | False Motifs | True Motifs | False Motifs | True Motifs | |
| ḼxxḼxḼ | (3Ḽ) | 296 | 114 | 0 | 27 | 155 |
| ḼxxḼxŁ/ḼxxŁxḼ/ŁxxḼxḼ | (2Ḽ) | 1,060 | 773 | 0 | 147 | 140 |
| ḼxxŁxŁ/ŁxxŁxŁ/ŁxxŁxḼ | (1Ḽ) | 7,239 | 5,875 | 0 | 1,192 | 172 |
| LxxLxL | 12,247 | 10,149 | 0 | 1,417 | 681 | |
| LxxLxLxxN | 811 | 438 | 0 | 76 | 297 | |
| LxxLxLxxC | 273 | 163 | 0 | 41 | 69 | |
| LxxLxLxx(N/C)xL | 618 | 269 | 0 | 39 | 310 | |
| Number of predicted positions (16 aa sliding windows): | 148,540 | 25,658 | ||||
| 815 LRR motifs | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, E.C.; Sukarta, O.C.A.; Spiridon, L.; Grigore, L.G.; Constantinescu, V.; Tacutu, R.; Goverse, A.; Petrescu, A.-J. LRRpredictor—A New LRR Motif Detection Method for Irregular Motifs of Plant NLR Proteins Using an Ensemble of Classifiers. Genes 2020, 11, 286. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030286
Martin EC, Sukarta OCA, Spiridon L, Grigore LG, Constantinescu V, Tacutu R, Goverse A, Petrescu A-J. LRRpredictor—A New LRR Motif Detection Method for Irregular Motifs of Plant NLR Proteins Using an Ensemble of Classifiers. Genes. 2020; 11(3):286. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030286
Chicago/Turabian StyleMartin, Eliza C., Octavina C. A. Sukarta, Laurentiu Spiridon, Laurentiu G. Grigore, Vlad Constantinescu, Robi Tacutu, Aska Goverse, and Andrei-Jose Petrescu. 2020. "LRRpredictor—A New LRR Motif Detection Method for Irregular Motifs of Plant NLR Proteins Using an Ensemble of Classifiers" Genes 11, no. 3: 286. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030286