An Improved Phenotype-Driven Tool for Rare Mendelian Variant Prioritization: Benchmarking Exomiser on Real Patient Whole-Exome Data

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Real Patient Whole-Exome Dataset with Known Molecular Diagnosis
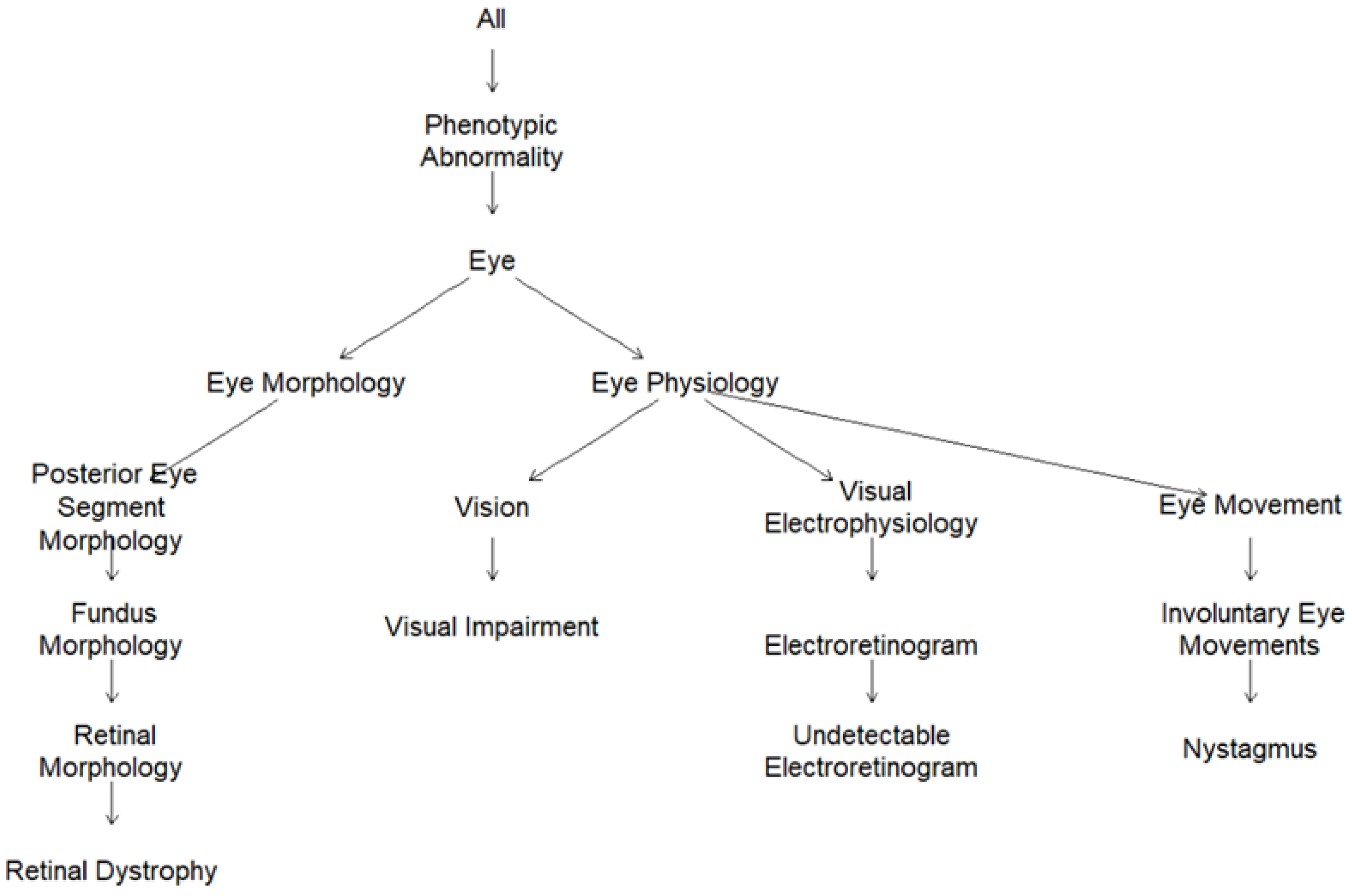
2.2. Human Phenotype Ontology (HPO)-Encoded Clinical Diagnoses
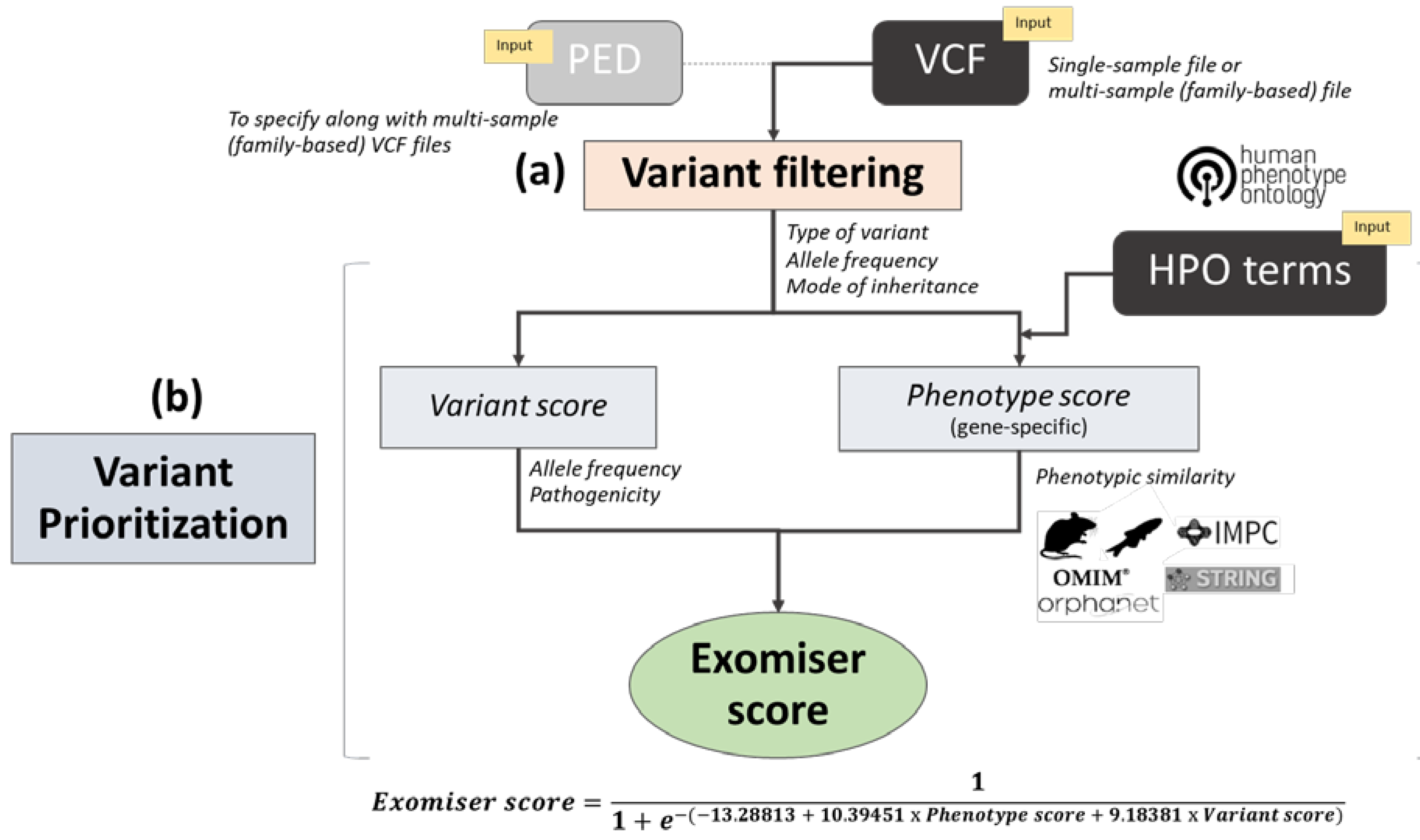
2.3. Exomiser Software
2.4. Software Analysis Settings
2.5. Software Performance Evaluation and Statistical Analysis
2.6. Whole-Exome Sequencing Data
3. Results
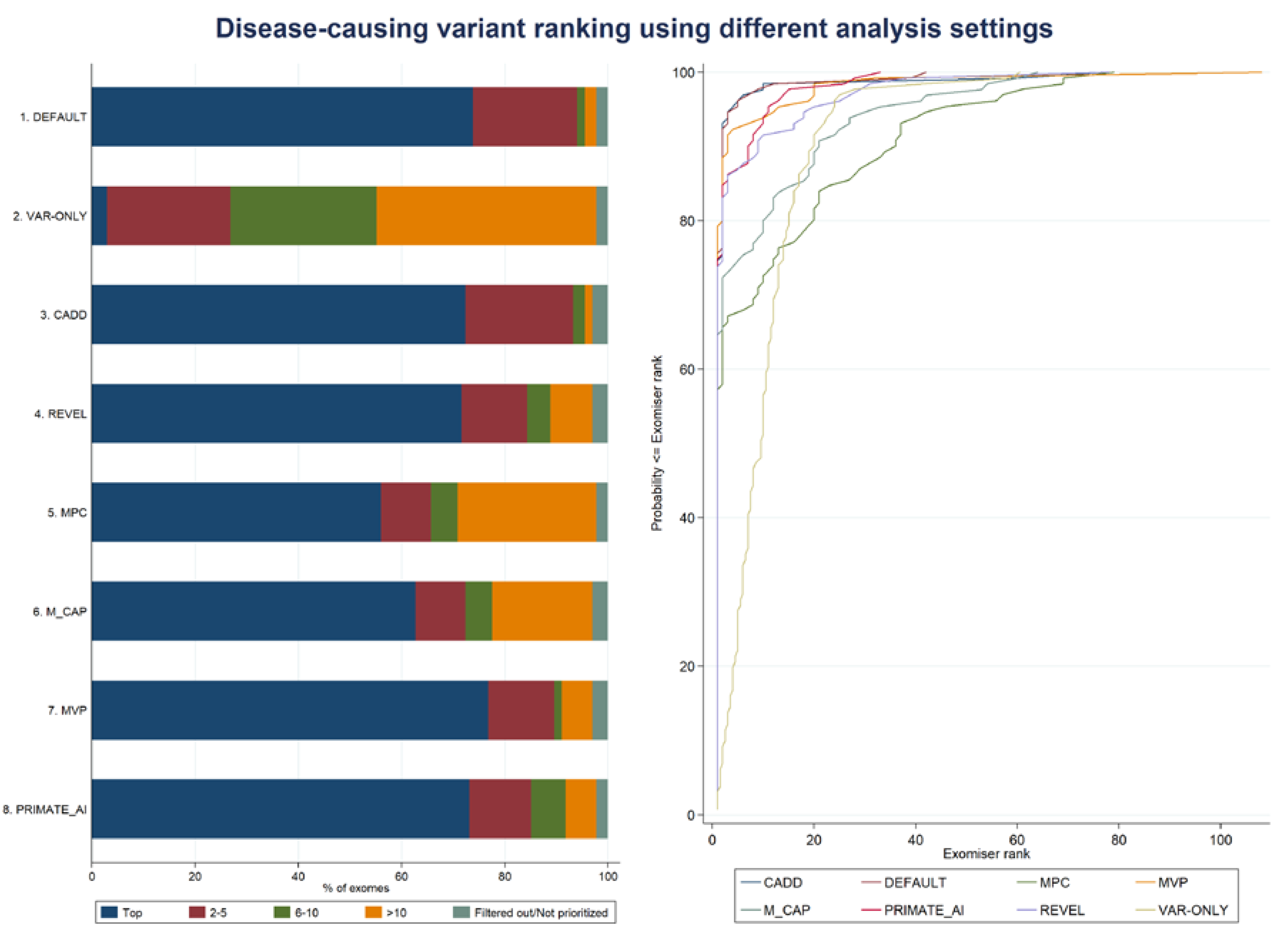
3.1. Exomiser Performance on the Inherited Retinal Disease (IRD) Patient Dataset
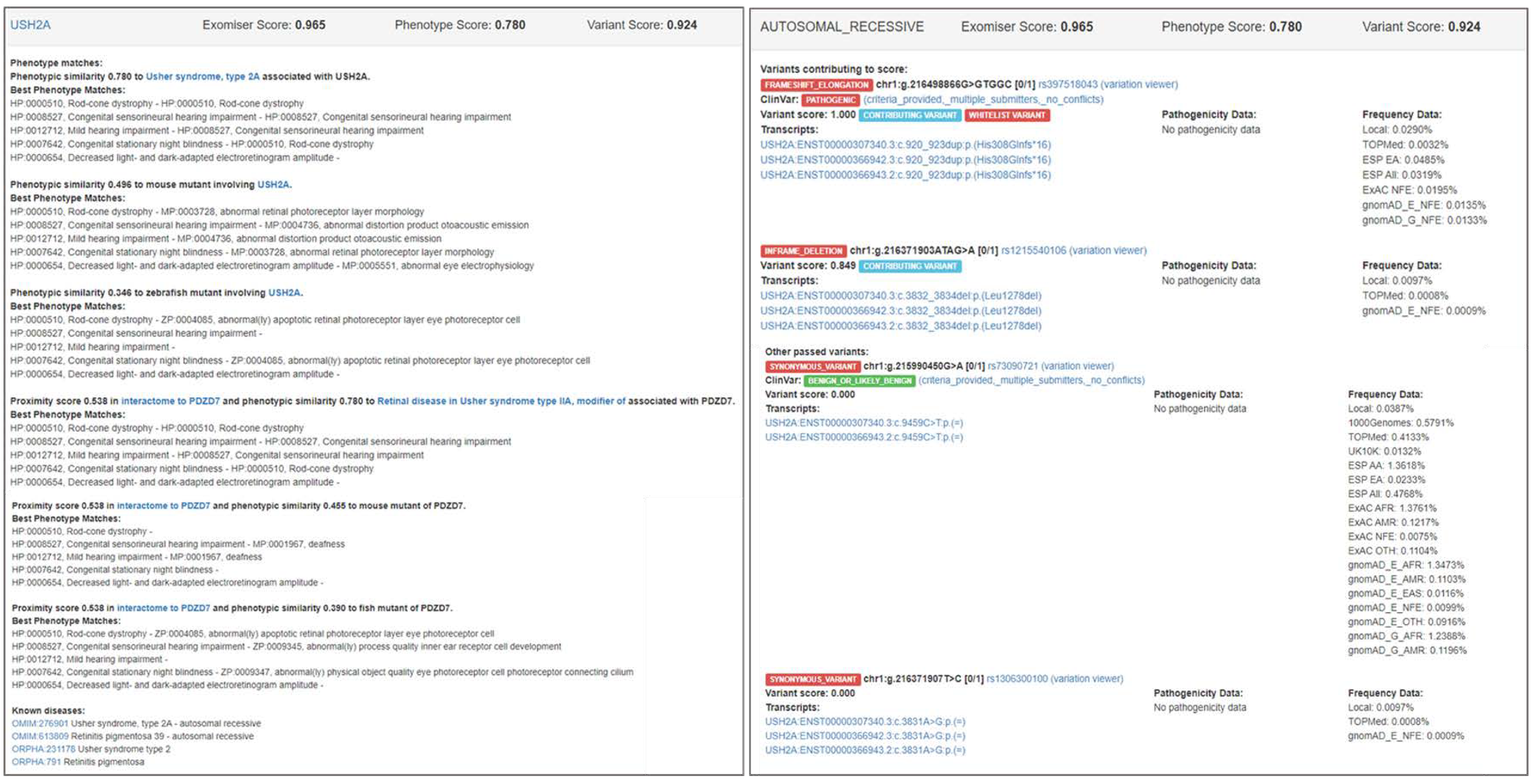
3.2. Examples of Exomiser Results on the IRD Patient Dataset
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; FitzPatrick, D.R.; Firth, H.V. Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 2018, 19, 327. [Google Scholar]
- Caspar, S.M.; Dubacher, N.; Kopps, A.M.; Meienberg, J.; Henggeler, C.; Matyas, G. Clinical sequencing: From raw data to diagnosis with lifetime value. Clin. Genet. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, A.; Caspar, S.M.; Meienberg, J.; Rohrbach, M.; Steinmann, B.; Matyas, G. Variant filtering, digenic variants, and other challenges in clinical sequencing: A lesson from fibrillinopathies. Clin. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Whiffin, N.; Minikel, E.; Walsh, R.; O’Donnell-Luria, A.H.; Karczewski, K.; Ing, A.Y.; Barton, P.J.; Funke, B.; Cook, S.A.; MacArthur, D.; et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 2017, 19, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Eilbeck, K.; Quinlan, A.; Yandell, M. Settling the score: Variant prioritization and Mendelian disease. Nat. Rev. Genet. 2017, 18, 599–612. [Google Scholar] [CrossRef]
- Li, J.C.; Zhao, T.T.; Zhang, Y.; Zhang, K.; Shi, L.S.; Chen, Y.; Wang, X.X.; Sun, Z.S. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018, 46, 7793–7804. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Gulko, B.; Hubisz, M.J.; Gronau, I.; Siepel, A. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat. Genet. 2015, 47, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, E118–U185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the Variant Effect Scoring tool. BMC Genom. 2013, 14, S3. [Google Scholar] [CrossRef] [Green Version]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP plus. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Siepel, A.; Pollard, K.S.; Haussler, D. New methods for detecting lineage-specific selection. Res. Comput. Mol. Biol. Proc. 2006, 3909, 190–205. [Google Scholar]
- Garber, M.; Guttman, M.; Clamp, M.; Zody, M.C.; Friedman, N.; Xie, X.H. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 2009, 25, I54–I62. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quang, D.; Chen, Y.F.; Xie, X.H. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Ionita-Laza, I.; McCallum, K.; Xu, B.; Buxbaum, J.D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016, 48, 214–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.S.; Hu, Y.M.; Sun, J.H.; Cheng, Y.W.; Cheung, K.H.; Zhao, H.Y. a statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data. Sci. Rep. 2015, 5, 10576. [Google Scholar] [CrossRef] [Green Version]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Dong, C.L.; Wei, P.; Jian, X.Q.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X.M. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Van der Velde, K.J.; de Boer, E.N.; van Diemen, C.C.; Sikkema-Raddatz, B.; Abbott, K.M.; Knopperts, A.; Franke, L.; Sijmons, R.H.; de Koning, T.J.; Wijmenga, C.; et al. GAVIN: Gene-Aware Variant INterpretation for medical sequencing. Genome Biol. 2017, 18, 6. [Google Scholar] [CrossRef] [Green Version]
- Samocha, K.E.; Kosmicki, J.A.; Karczewski, K.J.; O’Donnell-Luria, A.H.; Pierce-Hoffman, E.; MacArthur, D.G.; Neale, B.M.; Daly, M.J. Regional missense constraint improves variant deleteriousness prediction. bioRxiv 2017, 148353. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Chen, C.; Zhang, H.; Long, J.J.; Chung, W.K.; Guan, Y.; Shen, Y. MVP: Predicting pathogenicity of missense variants by deep learning. bioRxiv 2018, 259390. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, L.; Gao, H.; Padigepati, S.R.; McRae, J.F.; Li, Y.J.; Kosmicki, J.A.; Fritzilas, N.; Hakenberg, J.; Dutta, A.; Shon, J.; et al. Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet. 2018, 50, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Sifrim, A.; Popovic, D.; Tranchevent, L.C.; Ardeshirdavani, A.; Sakai, R.; Konings, P.; Vermeesch, J.R.; Aerts, J.; De Moor, B.; Moreau, Y. eXtasy: Variant prioritization by genomic data fusion. Nat. Methods 2013, 10, 1083–1084. [Google Scholar] [CrossRef] [Green Version]
- Aleman, A.; Garcia-Garcia, F.; Salavert, F.; Medina, I.; Dopazo, J. A web-based interactive framework to assist in the prioritization of disease candidate genes in whole-exome sequencing studies. Nucleic Acids Res. 2014, 42, W88–W93. [Google Scholar] [CrossRef] [Green Version]
- Javed, A.; Agrawal, S.; Ng, P.C. Phen-Gen: Combining phenotype and genotype to analyze rare disorders. Nat. Methods 2014, 11, 935–937. [Google Scholar] [CrossRef] [Green Version]
- Robinson, P.N.; Kohler, S.; Oellrich, A.; Wang, K.; Mungall, C.J.; Lewis, S.E.; Washington, N.; Bauer, S.; Seelow, D.; Krawitz, P.; et al. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res. 2014, 24, 340–348. [Google Scholar] [CrossRef] [Green Version]
- Singleton, M.V.; Guthery, S.L.; Voelkerding, K.V.; Chen, K.; Kennedy, B.; Margraf, R.L.; Durtschi, J.; Eilbeck, K.; Reese, M.G.; Jorde, L.B.; et al. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am. J. Hum. Genet. 2014, 94, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Trakadis, Y.J.; Buote, C.; Therriault, J.F.; Jacques, P.E.; Larochelle, H.; Levesque, S. PhenoVar: A phenotype-driven approach in clinical genomics for the diagnosis of polymalformative syndromes. BMC Med. Genom. 2014, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Zemojtel, T.; Kohler, S.; Mackenroth, L.; Jager, M.; Hecht, J.; Krawitz, P.; Graul-Neumann, L.; Doelken, S.; Ehmke, N.; Spielmann, M.; et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci. Transl. Med. 2014, 6, 252ra123. [Google Scholar] [CrossRef] [Green Version]
- Antanaviciute, A.; Watson, C.M.; Harrison, S.M.; Lascelles, C.; Crinnion, L.; Markham, A.F.; Bonthron, D.T.; Carr, I.M. OVA: Integrating molecular and physical phenotype data from multiple biomedical domain ontologies with variant filtering for enhanced variant prioritization. Bioinformatics 2015, 31, 3822–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Robinson, P.N.; Wang, K. Phenolyzer: Phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 2015, 12, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- James, R.A.; Campbell, I.M.; Chen, E.S.; Boone, P.M.; Rao, M.A.; Bainbridge, M.N.; Lupski, J.R.; Yang, Y.P.; Eng, C.M.; Posey, J.E.; et al. A visual and curatorial approach to clinical variant prioritization and disease gene discovery in genome-wide diagnostics. Genome Med. 2016, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoldi, L.; Forcato, C.; Vitulo, N.; Birolo, G.; De Pascale, F.; Feltrin, E.; Schiavon, R.; Anglani, F.; Negrisolo, S.; Zanetti, A.; et al. QueryOR: A comprehensive web platform for genetic variant analysis and prioritization. BMC Bioinform. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koile, D.; Cordoba, M.; Serro, M.D.; Kauffman, M.A.; Yankilevich, P. GenIO: A phenotype-genotype analysis web server for clinical genomics of rare diseases. BMC Bioinform. 2018, 19, 25. [Google Scholar] [CrossRef]
- Boudellioua, I.; Kulmanov, M.; Schofield, P.N.; Gkoutos, G.V.; Hoehndorf, R. DeepPVP: Phenotype-based prioritization of causative variants using deep learning. BMC Bioinform. 2019, 20, 65. [Google Scholar] [CrossRef]
- Hombach, D.; Schuelke, M.; Knierim, E.; Ehmke, N.; Schwarz, J.M.; Fischer-Zirnsak, B.; Seelow, D. MutationDistiller: User-driven identification of pathogenic DNA variants. Nucleic Acids Res. 2019, 47, W114–W120. [Google Scholar] [CrossRef]
- Jagadeesh, K.A.; Birgmeier, J.; Guturu, H.; Deisseroth, C.A.; Wenger, A.M.; Bernstein, J.A.; Bejerano, G. Phrank measures phenotype sets similarity to greatly improve Mendelian diagnostic disease prioritization. Genet. Med. 2019, 21, 464–470. [Google Scholar] [CrossRef]
- Li, Q.G.; Zhao, K.Y.; Bustamante, C.D.; Ma, X.; Wong, W.H. Xrare: A machine learning method jointly modeling phenotypes and genetic evidence for rare disease diagnosis. Genet. Med. 2019, 21, 2126–2134. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, F.; Wang, Y.; Qiu, Y.; Wu, Y.; Lu, Y.; Yang, L.; Qu, W.J.; Wang, H.; Zhou, W.; et al. PhenoPro: A novel toolkit for assisting in the diagnosis of Mendelian disease. Bioinformatics 2019, 35, 3559–3566. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Devkota, B.; Evans, P.; Zhao, X.N.; Baker, S.W.; Niazi, R.; Ca, K.J.; Gonzalez, M.A.; Jayaraman, P.; Conlin, L.K.; et al. Rapid and accurate interpretation of clinical exomes using Phenoxome: A computational phenotype-driven approach. Eur. J. Hum. Genet. 2019, 27, 612–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019, 47, D1018–D1027. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.; Doelken, S.C.; Mungall, C.J.; Bauer, S.; Firth, H.V.; Bailleul-Forestier, I.; Black, G.C.M.; Brown, D.L.; Brudno, M.; Campbell, J.; et al. The human phenotype ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014, 42, D966–D974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, S.; Vasilevsky, N.A.; Engelstad, M.; Foster, E.; McMurry, J.; Ayme, S.; Baynam, G.; Bello, S.M.; Boerkoel, C.F.; Boycott, K.M.; et al. The human phenotype ontology in 2017. Nucleic Acids Res. 2017, 45, D865–D876. [Google Scholar] [CrossRef]
- Robinson, P.N.; Kohler, S.; Bauer, S.; Seelow, D.; Horn, D.; Mundlos, S. The human phenotype ontology: A tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008, 83, 610–615. [Google Scholar] [CrossRef] [Green Version]
- Jager, M.; Wang, K.; Bauer, S.; Smedley, D.; Krawitz, P.; Robinson, P.N. Jannovar: A java library for exome annotation. Hum. Mutat. 2014, 35, 548–555. [Google Scholar] [CrossRef]
- Smedley, D.; Oellrich, A.; Kohler, S.; Ruef, B.; Westerfield, M.; Robinson, P.; Lewis, S.; Mungall, C.; Project, S.M.G. PhenoDigm: Analyzing curated annotations to associate animal models with human diseases. Database-J. Biol. Databases Curation 2013, 2013, bat025. [Google Scholar] [CrossRef] [Green Version]
- Bone, W.P.; Washington, N.L.; Buske, O.J.; Adams, D.R.; Davis, J.; Draper, D.; Flynn, E.D.; Girdea, M.; Godfrey, R.; Golas, G.; et al. Computational evaluation of exome sequence data using human and model organism phenotypes improves diagnostic efficiency. Genet. Med. 2016, 18, 608–617. [Google Scholar] [CrossRef] [Green Version]
- Smedley, D.; Jacobsen, J.O.B.; Jager, M.; Kohler, S.; Holtgrewe, M.; Schubach, M.; Siragusa, E.; Zemojtel, T.; Buske, O.J.; Washington, N.L.; et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc. 2015, 10, 2004–2015. [Google Scholar] [CrossRef] [Green Version]
- Smedley, D.; Schubach, M.; Jacobsen, J.O.B.; Kohler, S.; Zemojtel, T.; Spielmann, M.; Jager, M.; Hochheiser, H.; Washington, N.L.; McMurry, J.A.; et al. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in mendelian disease. Am. J. Hum. Genet. 2016, 99, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic basis of inherited retinal disease in a molecularly characterised cohort of over 3000 families from the United Kingdom. Ophthalmol 2020. [Google Scholar] [CrossRef]
- Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; Gabriel, S.B.; et al. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geihs, M.; Yan, Y.; Walter, K.; Huang, J.; Memari, Y.; Min, J.L.; Mead, D.; Hubbard, T.J.; Timpson, N.J.; Down, T.A.; et al. An interactive genome browser of association results from the UK10K cohorts project. Bioinformatics 2015, 31, 4029–4031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Landrum, M.J.; Kattman, B.L. ClinVar at five years: Delivering on the promise. Hum. Mutat. 2018, 39, 1623–1630. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef] [Green Version]
- Rath, A.; Olry, A.; Dhombres, F.; Brandt, M.M.; Urbero, B.; Ayme, S. Representation of rare diseases in health information systems: The orphanet approach to serve a wide range of end users. Hum. Mutat. 2012, 33, 803–808. [Google Scholar] [CrossRef]
- Smith, C.L.; Blake, J.A.; Kadin, J.A.; Richardson, J.E.; Bult, C.J.; Grp, M.G.D. Mouse Genome Database (MGD)-2018: Knowledgebase for the laboratory mouse. Nucleic Acids Res. 2018, 46, D836–D842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howe, D.G.; Bradford, Y.M.; Conlin, T.; Eagle, A.E.; Fashena, D.; Frazer, K.; Knight, J.; Mani, P.; Martin, R.; Moxon, S.A.T.; et al. ZFIN, the Zebrafish Model Organism Database: Increased support for mutants and transgenics. Nucleic Acids Res. 2013, 41, D854–D860. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Pontikos, N.; Yu, J.; Moghul, I.; Withington, L.; Blanco-Kelly, F.; Vulliamy, T.; Wong, T.L.E.; Murphy, C.; Cipriani, V.; Fiorentino, A.; et al. Phenopolis: An open platform for harmonization and analysis of genetic and phenotypic data. Bioinformatics 2017, 33, 2421–2423. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002, 12, 436–446. [Google Scholar] [CrossRef] [Green Version]
- Landis, J.R.; Koch, G.G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, D.; Richardson, S.; Turro, E. ontologyX: A suite of R packages for working with ontological data. Bioinformatics 2017, 33, 1104–1106. [Google Scholar] [CrossRef] [Green Version]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [Green Version]
- Ajmal, M.; Khan, M.I.; Neveling, K.; Khan, Y.M.; Azam, M.; Waheed, N.K.; Hamel, C.P.; Ben-Yosef, T.; De Baere, E.; Koenekoop, R.K.; et al. A missense mutation in the splicing factor gene DHX38 is associated with early-onset retinitis pigmentosa with macular coloboma. J. Med. Genet. 2014, 51, 444–448. [Google Scholar] [CrossRef]
- Latif, Z.; Chakchouk, I.; Schrauwen, I.; Lee, K.; Santos-Cortez, R.L.P.; Abbe, I.; Acharya, A.; Jarral, A.; Ali, I.; Ullah, E.; et al. Confirmation of the role of DHX38 in the etiology of early-onset retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4552–4557. [Google Scholar] [CrossRef] [Green Version]
- Girdea, M.; Dumitriu, S.; Fiume, M.; Bowdin, S.; Boycott, K.M.; Chenier, S.; Chitayat, D.; Faghfoury, H.; Meyn, M.S.; Ray, P.N.; et al. PhenoTips: Patient phenotyping software for clinical and research use. Hum. Mutat. 2013, 34, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Sobreira, N.; Hoover-Fong, J.; Sutton, V.R.; Boehm, C.; Schiettecatte, F.; Valle, D. PhenoDB: A new web-based tool for the collection, storage, and analysis of phenotypic features. Hum. Mutat. 2013, 34, 566–571. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Kury, F.S.P.; Li, Z.R.; Ta, C.; Wang, K.; Weng, C.H. Doc2Hpo: A web application for efficient and accurate HPO concept curation. Nucleic Acids Res. 2019, 47, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Son, J.H.; Xie, G.C.; Yuan, C.; Ena, L.; Li, Z.R.; Goldstein, A.; Huang, L.L.; Wang, L.W.; Shen, F.C.; Liu, H.F.; et al. Deep phenotyping on electronic health records facilitates genetic diagnosis by clinical exomes. Am. J. Hum. Genet. 2018, 103, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergouniotis, P.I.; Maxime, E.; Leroux, D.; Olry, A.; Thompson, R.; Rath, A.; Robinson, P.N.; Dollfus, H.; Ashworth, J.L.; Audo, I.; et al. An ontological foundation for ocular phenotypes and rare eye diseases. Orphanet J. Rare Dis. 2019, 14, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanciulli, M.; Petretto, E.; Aitman, T.J. Gene copy number variation and common human disease. Clin. Genet. 2010, 77, 201–213. [Google Scholar] [CrossRef]
- Baker, T.S.; Gambino, K.J.; Schriefer, L.; Lim, J.Y.; Steinberg, K.M.; Fajgenbaum, D.C.; Garcia-Sancho, A.M.; Byun, M. A novel FAS mutation with variable expressivity in a family with unicentric and idiopathic multicentric Castleman disease. Blood Adv. 2018, 2, 2959–2963. [Google Scholar] [CrossRef]
- Ji, J.; Shen, L.; Bootwalla, M.; Quindipan, C.; Tatarinova, T.; Maglinte, D.T.; Buckley, J.; Raca, G.; Saitta, S.C.; Biegel, J.A.; et al. A semiautomated whole-exome sequencing workflow leads to increased diagnostic yield and identification of novel candidate variants. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003756. [Google Scholar] [CrossRef] [Green Version]
- Martin-Sierra, C.; Gallego-Martinez, A.; Requena, T.; Frejo, L.; Batuecas-Caletrio, A.; Lopez-Escamez, J.A. Variable expressivity and genetic heterogeneity involving DPT and SEMA3D genes in autosomal dominant familial Meniere’s disease. Eur. J. Hum. Genet. 2017, 25, 200–207. [Google Scholar] [CrossRef]
- Martin-Sierra, C.; Requena, T.; Frejo, L.; Price, S.D.; Gallego-Martinez, A.; Batuecas-Caletrio, A.; Santos-Perez, S.; Soto-Varela, A.; Lysakowski, A.; Lopez-Escamez, J.A. A novel missense variant in PRKCB segregates low-frequency hearing loss in an autosomal dominant family with Meniere’s disease. Hum. Mol. Genet. 2016, 25, 3407–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Requena, T.; Cabrera, S.; Martin-Sierra, C.; Price, S.D.; Lysakowski, A.; Lopez-Escamez, J.A. Identification of two novel mutations in FAM136A and DTNA genes in autosomal-dominant familial Meniere’s disease. Hum. Mol. Genet. 2015, 24, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Toro, C.; Hori, R.T.; Malicdan, M.C.V.; Tifft, C.J.; Goldstein, A.; Gahl, W.A.; Adams, D.R.; Harper, F.; Wolfe, L.A.; Xiao, J.F.; et al. A recurrent de novo missense mutation in UBTF causes developmental neuroregression. Hum. Mol. Genet. 2018, 27, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Valkanas, E.; Schaffer, K.; Dunham, C.; Maduro, V.; du Souich, C.; Rupps, R.; Adams, D.R.; Baradaran-Heravi, A.; Flynn, E.; Malicdan, M.C.; et al. Phenotypic evolution of UNC80 loss of function. Am. J. Med. Genet. Part A 2016, 170, 3106–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.D.; Metikala, S.; Wheeler, P.G.; Sherpa, M.D.; Houten, S.M.; Horb, M.E.; Schadt, E.E. Heterozygous pathogenic variant in DACT1 causes an autosomal-dominant syndrome with features overlapping townes-brocks syndrome. Hum. Mutat. 2017, 38, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, C.; Scott, R.H.; Thomas, E.; Jones, L.; Murugaesu, N.; Pretty, F.B.; Halai, D.; Baple, E.; Craig, C.; Hamblin, A.; et al. The 100 000 Genomes Project: Bringing whole genome sequencing to the NHS. BMJ Br. Med. J. 2018, 361, k1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Diagnosis a | N | % |
|---|---|---|
| Retinitis pigmentosa (RP) | 36 | 26.9 |
| Leber congenital amaurosis (LCA) | 25 | 18.7 |
| Macular dystrophy (MD) | 16 | 11.9 |
| Cone-rod dystrophy (CRD) | 14 | 10.4 |
| Early onset retinal dystrophy (EORD) | 9 | 6.7 |
| Usher syndrome type II (USH2) | 8 | 6.0 |
| Achromatopsia (ACHM) | 6 | 4.5 |
| Congenital stationary night blindness (CSNB) | 5 | 3.7 |
| Retinal dystrophy (RD) | 3 | 2.2 |
| Usher syndrome type I (USH1) | 2 | 1.5 |
| Stargardt disease (STGD) | 2 | 1.5 |
| Occult macular dystrophy (OCMD) | 1 | 0.7 |
| Benign fleck retina (BFR) | 1 | 0.7 |
| Coloboma (COLOB) | 1 | 0.7 |
| Familial exudative vitreoretinopathy (FEVR) | 1 | 0.7 |
| Foveal hypoplasia (FH) | 1 | 0.7 |
| Myopia and deafness (Stickler syndrome) (STICKL) | 1 | 0.7 |
| Ocular albinism (OALB) | 1 | 0.7 |
| Optic atrophy (OATR) | 1 | 0.7 |
| Total | 134 | 100.0 |
| Genotype | N | % |
|---|---|---|
| Homozygote | 72 | 53.7 |
| Compound heterozygote | 39 | 29.1 |
| Heterozygote | 13 | 9.7 |
| Hemizygote a | 10 | 7.5 |
| Total | 134 | 100.0 |
| Analysis YML File | |
|---|---|
| 1. DEFAULT analysis: genomeAssembly: hg19 vcf: path-to-VCF-file hpoIds: [comma-separated-list-of-HPO-terms] inheritanceModes: { AUTOSOMAL_DOMINANT: 0.1, AUTOSOMAL_RECESSIVE_HOM_ALT: 0.5, AUTOSOMAL_RECESSIVE_COMP_HET: 2.0, X_DOMINANT: 0.1, X_RECESSIVE_HOM_ALT: 0.5, X_RECESSIVE_COMP_HET: 2.0, } analysisMode: PASS_ONLY frequencySources: [LOCAL, THOUSAND_GENOMES, TOPMED, UK10K, ESP_AFRICAN_AMERICAN, ESP_EUROPEAN_AMERICAN, ESP_ALL, EXAC_AFRICAN_INC_AFRICAN_AMERICAN, EXAC_AMERICAN, EXAC_SOUTH_ASIAN, EXAC_EAST_ASIAN, EXAC_FINNISH, EXAC_NON_FINNISH_EUROPEAN, EXAC_OTHER, GNOMAD_E_AFR, GNOMAD_E_AMR, GNOMAD_E_EAS, GNOMAD_E_FIN, GNOMAD_E_NFE, GNOMAD_E_OTH, GNOMAD_E_SAS, GNOMAD_G_AFR, GNOMAD_G_AMR, GNOMAD_G_EAS, GNOMAD_G_FIN, GNOMAD_G_NFE, GNOMAD_G_OTH, GNOMAD_G_SAS] pathogenicitySources: [POLYPHEN, MUTATION_TASTER, SIFT] steps: [ qualityFilter: {minQuality: 30.0} variantEffectFilter: { remove: [FIVE_PRIME_UTR_EXON_VARIANT, FIVE_PRIME_UTR_INTRON_VARIANT, THREE_PRIME_UTR_EXON_VARIANT, THREE_PRIME_UTR_INTRON_VARIANT, NON_CODING_TRANSCRIPT_EXON_VARIANT, UPSTREAM_GENE_VARIANT, INTERGENIC_VARIANT, REGULATORY_REGION_VARIANT, CODING_TRANSCRIPT_INTRON_VARIANT, NON_CODING_TRANSCRIPT_INTRON_VARIANT, DOWNSTREAM_GENE_VARIANT] }, frequencyFilter: {maxFrequency: 2.0}, pathogenicityFilter: {keepNonPathogenic: true}, inheritanceFilter: {}, omimPrioritiser: {}, hiPhivePrioritiser: {} ] | 2. VAR-ONLY As per DEFAULT, but without omimPrioritiser: {} and hiPhivePrioritiser: {} |
| 3. CADD As per DEFAULT, but with pathogenicitySources: [CADD] | |
| 4. REVEL As per DEFAULT, but with pathogenicitySources: [REVEL] | |
| 5. MPC As per DEFAULT, but with pathogenicitySources: [MPC] | |
| 6. M_CAP As per DEFAULT, but with pathogenicitySources: [M_CAP] | |
| 7. MVP As per DEFAULT, but with pathogenicitySources: [MVP] | |
| 8. PRIMATE-AI As per DEFAULT, but with pathogenicitySources: [PRIMATE-AI] | |
| Analysis Setting | Variants Filtered out | Variants not Prioritized a | Mean Rank (SD) | Median Rank | Min Rank | Max Rank | Top Ranked, % (N = 134) |
|---|---|---|---|---|---|---|---|
| 1. DEFAULT | 2 | 1 (gene rank: 1) | 2.1 (5.0) | 1 | 1 | 42 | 73.9 |
| 2. VAR-ONLY | 2 | 1 (gene rank: 30) | 10.8 (9.0) | 9.5 | 1 | 60.5 | 3.0 |
| 3. CADD | 2 | 2 (gene ranks: 1, 2) | 2.5 (8.4) | 1 | 1 | 77 | 72.4 |
| 4. REVEL | 2 | 2 (gene ranks: 2, 9) | 3.9 (9.2) | 1 | 1 | 78 | 71.6 |
| 5. MPC | 2 | 1 (gene rank: 2) | 10.1 (16.6) | 1 | 1 | 79 | 56.0 |
| 6. M_CAP | 2 | 2 (gene ranks: 2, 18) | 6.8 (12.5) | 1 | 1 | 64 | 62.7 |
| 7. MVP | 2 | 2 (gene ranks: 2, 4) | 3.1 (10.3) | 1 | 1 | 108 | 76.9 |
| 8. PRIMATE-AI | 2 | 1 (gene rank: 2) | 2.7 (5.0) | 1 | 1 | 33 | 73.1 |
| DEFAULT vs. VAR-ONLY | Top | 2–5 | 6–10 | >10 | Filtered out/Not Prioritized | Total | DEFAULT vs. M_CAP | Top | 2–5 | 6–10 | >10 | Filtered out/Not Prioritized | Total | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Top | 4 | 28 | 28 | 39 | 99 | Agreement, % | Top | 74 | 3 | 4 | 18 | 99 | Agreement, % | ||
| 2–5 | 3 | 9 | 15 | 27 | 7.6 | 2–5 | 10 | 9 | 1 | 6 | 1 | 27 | 64.6 | ||
| 6–10 | 1 | 1 | 2 | Cohen’s kappa | 6–10 | 1 | 1 | 2 | Cohen’s kappa | ||||||
| >10 | 1 | 2 | 3 | −0.013 (“poor”) | >10 | 2 | 1 | 3 | 0.27 (“fair”) | ||||||
| Filtered out/Not prioritized | 3 | 3 | Stuart–Maxwell P | Filtered out/Not prioritized | 3 | 3 | Stuart–Maxwell P | ||||||||
| Total | 4 | 32 | 38 | 57 | 3 | 134 | 3.4 × 10−22 | Total | 84 | 13 | 7 | 26 | 4 | 134 | 8.5 × 10−6 |
| DEFAULT vs. CADD | Top | 2–5 | 6–10 | >10 | Filtered out/Not prioritized | Total | DEFAULT vs. MVP | Top | 2–5 | 6–10 | >10 | Filtered out/Not prioritized | Total | ||
| Top | 93 | 5 | 1 | 99 | Agreement, % | Top | 93 | 1 | 1 | 4 | 99 | Agreement, % | |||
| 2–5 | 2 | 23 | 1 | 1 | 27 | 92.3 | 2–5 | 10 | 15 | 1 | 1 | 27 | 84.6 | ||
| 6–10 | 1 | 1 | 2 | Cohen’s kappa | 6–10 | 1 | 1 | 2 | Cohen’s kappa | ||||||
| >10 | 1 | 2 | 3 | 0.80 (“substantial”) | >10 | 1 | 2 | 3 | 0.58 (“moderate”) | ||||||
| Filtered out/Not prioritized | 1 | 2 | 3 | Stuart–Maxwell P | Filtered out/Not prioritized | 3 | 3 | Stuart–Maxwell P | |||||||
| Total | 97 | 28 | 3 | 2 | 4 | 134 | 0.818 | Total | 103 | 17 | 2 | 8 | 4 | 134 | 0.040 |
| DEFAULT vs. REVEL | Top | 2–5 | 6–10 | >10 | Filtered out/Not prioritized | Total | DEFAULT vs. PRIMATE_AI | Top | 2–5 | 6–10 | >10 | Filtered out/Not prioritized | Total | ||
| Top | 85 | 4 | 3 | 7 | 99 | Agreement, % | Top | 88 | 3 | 5 | 3 | 99 | Agreement, % | ||
| 2–5 | 11 | 12 | 1 | 2 | 1 | 27 | 75.4 | 2–5 | 10 | 12 | 1 | 3 | 1 | 27 | 79.2 |
| 6–10 | 1 | 1 | 2 | Cohen’s kappa | 6–10 | 1 | 1 | 2 | Cohen’s kappa | ||||||
| >10 | 2 | 1 | 3 | 0.40 (“fair”) | >10 | 1 | 2 | 3 | 0.48 (“moderate”) | ||||||
| Filtered out/Not prioritized | 3 | 3 | Stuart–Maxwell P | Filtered out/Not prioritized | 1 | 2 | 3 | Stuart–Maxwell P | |||||||
| Total | 96 | 17 | 6 | 11 | 4 | 134 | 0.011 | Total | 98 | 16 | 9 | 8 | 3 | 134 | 0.011 |
| DEFAULT vs. MPC | Top | 2–5 | 6–10 | >10 | Filtered out/Not prioritized | Total | |||||||||
| Top | 66 | 3 | 5 | 25 | 99 | Agreement, % | |||||||||
| 2–5 | 9 | 9 | 1 | 7 | 1 | 27 | 59.2 | ||||||||
| 6–10 | 1 | 1 | 2 | Cohen’s kappa | |||||||||||
| >10 | 1 | 2 | 3 | 0.24 (“fair”) | |||||||||||
| Filtered out/Not prioritized | 1 | 2 | 3 | Stuart–Maxwell P | |||||||||||
| Total | 75 | 13 | 7 | 36 | 3 | 134 | 1.0 × 10−7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cipriani, V.; Pontikos, N.; Arno, G.; Sergouniotis, P.I.; Lenassi, E.; Thawong, P.; Danis, D.; Michaelides, M.; Webster, A.R.; Moore, A.T.; et al. An Improved Phenotype-Driven Tool for Rare Mendelian Variant Prioritization: Benchmarking Exomiser on Real Patient Whole-Exome Data. Genes 2020, 11, 460. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11040460
Cipriani V, Pontikos N, Arno G, Sergouniotis PI, Lenassi E, Thawong P, Danis D, Michaelides M, Webster AR, Moore AT, et al. An Improved Phenotype-Driven Tool for Rare Mendelian Variant Prioritization: Benchmarking Exomiser on Real Patient Whole-Exome Data. Genes. 2020; 11(4):460. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11040460
Chicago/Turabian StyleCipriani, Valentina, Nikolas Pontikos, Gavin Arno, Panagiotis I. Sergouniotis, Eva Lenassi, Penpitcha Thawong, Daniel Danis, Michel Michaelides, Andrew R. Webster, Anthony T. Moore, and et al. 2020. "An Improved Phenotype-Driven Tool for Rare Mendelian Variant Prioritization: Benchmarking Exomiser on Real Patient Whole-Exome Data" Genes 11, no. 4: 460. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11040460