Cognitive Decline in Alzheimer’s Disease: Limited Clinical Utility for GWAS or Polygenic Risk Scores in a Clinical Trial Setting

 ,
,
Abstract
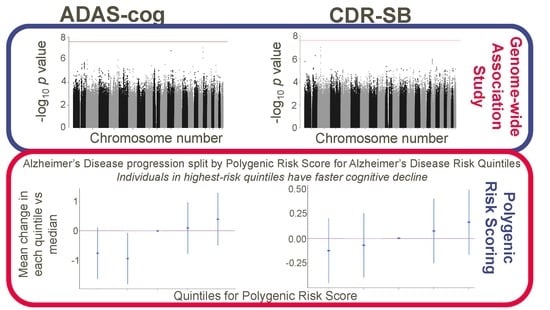
:
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Phenotype Data
2.3. Regression of Genotypes by Phenotypic Data
2.4. Construction of Polygenic Risk Scores
2.5. Genome-Wide Complex Trait Analysis
3. Results
3.1. Subjects
3.2. Association Analysis Results
3.3. Polygenic Risk Scores
3.4. GCTA Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Guerreiro, R.; Wojtas, A.; Brás, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2012, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.; Hooli, B.; Divito, J.; Ionita, I.; et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele ϵ4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Risacher, S.L.; Nho, K.; Kim, S.; Shen, L.; McDonald, B.C.; Yoder, K.K.; Hutchins, G.D.; West, J.D.; Tallman, E.F.; et al. GWAS of longitudinal amyloid accumulation on 18F-florbetapir PET in Alzheimer’s disease implicates microglial activation gene IL1RAP. Brain 2015, 138, 3076–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del-Aguila, J.L.; Fernandez, M.V.; Schindler, S.; Ibanez, L.; Deming, Y.; Ma, S.; Saef, B.; Black, K.; Budde, J.; Norton, J.; et al. Assessment of the Genetic Architecture of Alzheimer’s Disease Risk in Rate of Memory Decline. J. Alzheimer’s Dis. 2018, 62, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.; Sawchak, S.; Chiang, C.; Davies, J.; Donovan, C.; Saunders, A.M.; Irizarry, M.; Jeter, B.; Zvartau-Hind, M.; Van Dyck, C.H.; et al. Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer’s disease: Two phase 3 studies. Curr. Alzheimer Res. 2011, 8, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Euesden, J.; Lewis, C.M.; O’Reilly, P.F. PRSice: Polygenic Risk Score software. Bioinformatics 2014, 31, 1466–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
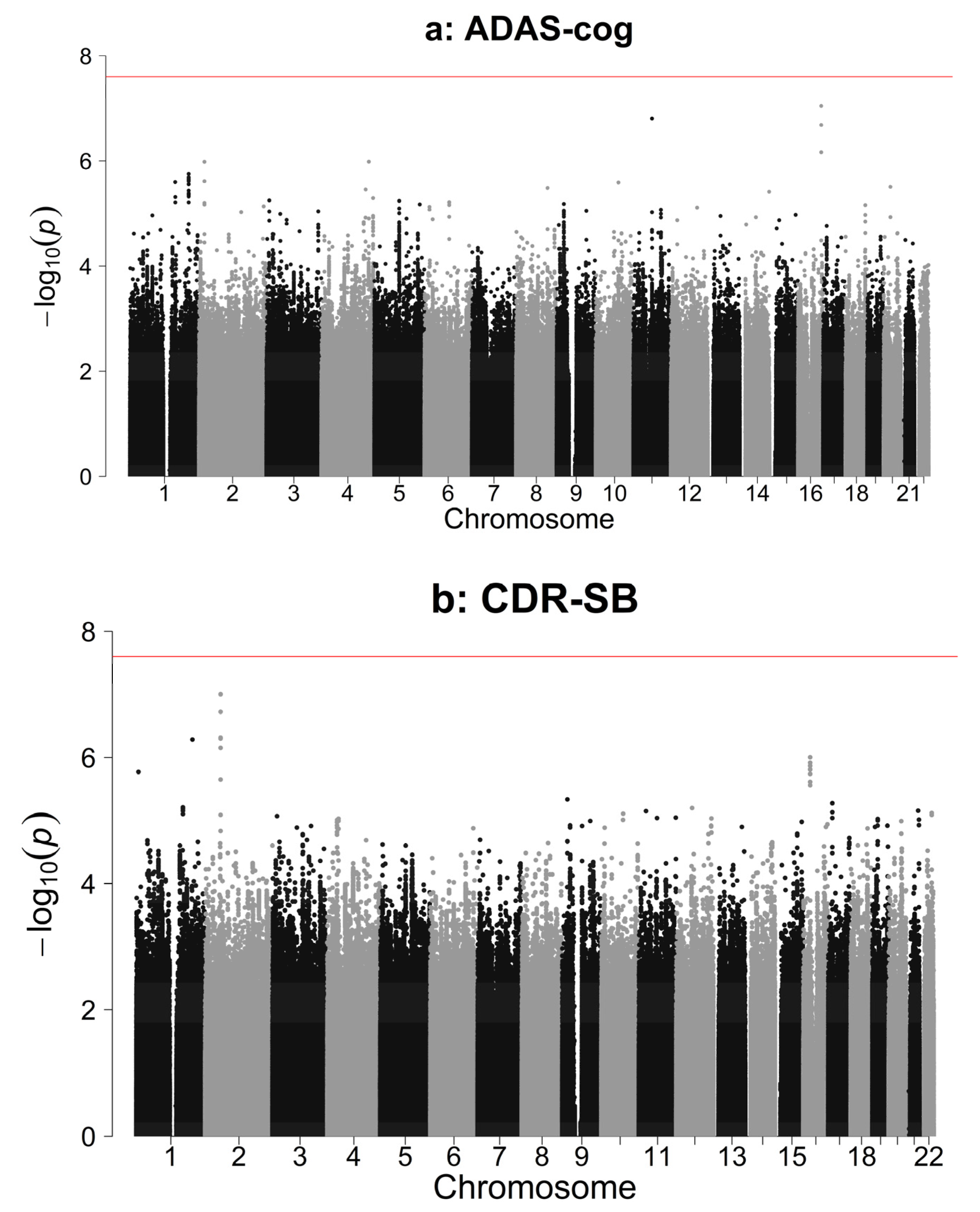{kind=link}
| Measure | ITT | Genotyped |
|---|---|---|
| Randomised treatment, Pooled Group, n (%) | 2532 (100%) | 2164 (100%) |
| Sex, F:M (%F) | 1447:1085 (57%) | 1234:930 (57%) |
| Age, Years, Mean (SD) | 73.71 (8.14) | 73.66 (8.09) |
| Ethnicity n (%) | ||
| Hispanic or Latino | 284 (11%) | 249 (12%) |
| Not Hispanic or Latino | 2238 (88%) | 1910 (88%) |
| Missing, n (%) | 10 (<1%) | 5 (<1%) |
| ADAS-cog change from baseline W48 LOCF, Mean (SD) | 2.98 (6.71) | 2.94 (6.78) |
| Missing, n (%) | 99 (3.9%) | 71 (3.3%) |
| CDR-SB change from baseline W48 LOCF, Mean (SD) | 1.50 (2.46) | 1.47 (2.45) |
| Missing, n (%) | 196 (7.7%) | 149 (6.9%) |
| Baseline ADAS-cog, Mean (SD) | 24.65 (9.86) | 24.49 (9.86) |
| Missing, n (%) | 5 (<1%) | 4 (<1%) |
| Baseline CDR-SB, Mean (SD) | 6.79 (3.52) | 6.73 (3.52) |
| Missing, n (%) | 41 (1.6%) | 32 (1.5%) |
| BMI, Mean (SD) | 25.65 (4.03) | 25.68 (4.01) |
| Missing, n (%) | 6 (<1%) | 4 (<1%) |
| Number of copies of ApoE4, Mean (SD) | 0.71 (0.69) | 0.72 (0.69) |
| Screening Mini Mental State Examination (MMSE), Mean (SD) | 19.78 (4.11) | 19.85 (4.08) |
| Missing, n (%) | 2 (<1%) | 2 (<1%) |
| Years of Education, Mean (SD) | 10.76 (4.02) | 10.76 (4.03) |
| Missing, n (%) | 7 (<1%) | 4 (<1%) |
| Time in treatment (days), Mean (SD) | 296.86 (88.23) | 299.93 (84.73) |
| Missing, n (%) | 26 (1%) | 21 (<1%) |
| Phenotype | ADAS-cog | CDR-SB |
|---|---|---|
| Threshold PT | 0.0707 | 0.00005 |
| Variance explained by PRS (R2) | 0.0054 | 0.0014 |
| Variance explained by all covariates (R2) | 0.1172 | 0.1034 |
| Variance explained by all covariates other than PRS—“Null Model” (R2) | 0.112 | 0.1021 |
| Effect of 1 SD increase in PRS on cognitive decline | 0.529 | 0.092 |
| Coefficient for PRS | 3646.94 | 11.99 |
| Standard Error for PRS | 1026.53 | 11.99 |
| p-value for |Coefficient| = 0 | 0.0004 | 0.0817 |
| Number of SNPs in PRS (<PT) | 15960 | 93 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Euesden, J.; Gowrisankar, S.; Qu, A.X.; St. Jean, P.; Hughes, A.R.; Pulford, D.J. Cognitive Decline in Alzheimer’s Disease: Limited Clinical Utility for GWAS or Polygenic Risk Scores in a Clinical Trial Setting. Genes 2020, 11, 501. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050501
Euesden J, Gowrisankar S, Qu AX, St. Jean P, Hughes AR, Pulford DJ. Cognitive Decline in Alzheimer’s Disease: Limited Clinical Utility for GWAS or Polygenic Risk Scores in a Clinical Trial Setting. Genes. 2020; 11(5):501. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050501
Chicago/Turabian StyleEuesden, Jack, Sivakumar Gowrisankar, Angela Xiaoyan Qu, Pamela St. Jean, Arlene R. Hughes, and David J. Pulford. 2020. "Cognitive Decline in Alzheimer’s Disease: Limited Clinical Utility for GWAS or Polygenic Risk Scores in a Clinical Trial Setting" Genes 11, no. 5: 501. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050501