Transcriptome Analysis Reveals Important Candidate Genes Related to Nutrient Reservoir, Carbohydrate Metabolism, and Defence Proteins during Grain Development of Hexaploid Bread Wheat and Its Diploid Progenitors

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Growing Condition:
2.3. RNA Extraction and Illumina Sequencing
2.4. RNA-Seq Data Processing and Differential Gene Expression Analysis
2.5. Transcript Annotation, Gene Ontology and KEGG Pathway Analysis
2.6. Quantitative PCR Analysis
2.7. Statistical Analysis
3. Results
3.1. Comparative Analyses of Transcriptome Profiles during Grain Development
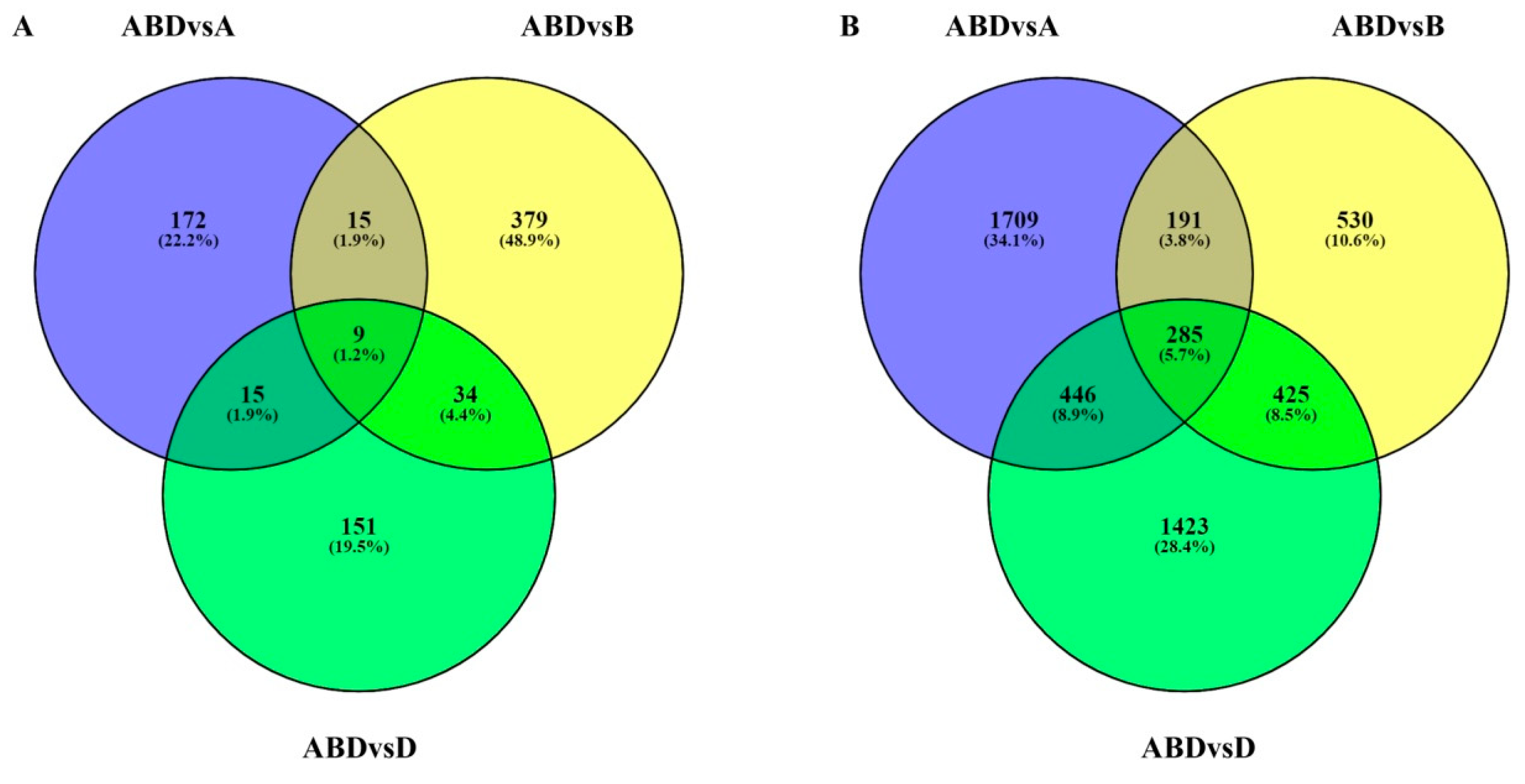
3.2. Identification of Differentially Expressed Transcripts (DETs) during Grain Development
3.3. Validation of Transcriptome Data
3.4. Functional Analysis of DEGs
3.5. Highly Up- and Down-Regulated Transcripts
3.6. Stage-Specific Expression Analysis of Highly Up- and Down-Regulated Genes
4. Discussion
4.1. RNA-Seq Analysis of Hexaploid Wheat and Its Diploid Progenitors
4.1.1. Transcriptome Data: Global View
4.1.2. Functional Analysis of DEGs
4.1.3. Highly Up- and Down-Regulated Transcripts
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kucek, L.K.; Veenstra, L.D.; Amnuaycheewa, P.; Sorrells, M.E. A grounded guide to gluten: How modern genotypes and processing impact wheat sensitivity. Compr. Rev. Food Sci. Food Saf. 2015, 1414, 285–302. [Google Scholar] [CrossRef]
- Wan, Y.; Poole, R.L.; Huttly, A.K.; Toscano-Underwood, C.; Feeney, K.; Welham, S.; Mitchell, R.A. Transcriptome analysis of grain development in hexaploid wheat. BMC Genom. 2008, 99, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Li, G.; Chu, Z.; Ru, Z.; Jiang, X.; Wen, Z.; Wei, W. Transcriptome analysis reveals important candidate genes involved in grain-size formation at the stage of grain enlargement in common wheat cultivar “Bainong 4199”. PLoS ONE 2019, 1414, e0214149. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.E.I.; Zhihui, W.U.; Yufeng ZHANG, D.G.; Yuzhou, X.U.; Weixia, C.H.E.N.; Haiying, Z.H.O.U.; Baoyun, L.I. Transcriptome analysis of wheat grain using RNA-Seq. Front. Agric. Sci. Eng. 2015, 11, 214–222. [Google Scholar]
- Shewry, P.R. Wheat. J. Exp. Bot. 2009, 6060, 1537–1553. [Google Scholar] [CrossRef]
- Yu, Y.; Zhu, D.; Ma, C.; Cao, H.; Wang, Y.; Xu, Y.; Yan, Y. Transcriptome analysis reveals key differentially expressed genes involved in wheat grain development. Crop J. 2016, 44, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Chen, F.; Cui, D. Proteomic analysis of middle and late stages of bread wheat (Triticum aestivum L.) grain development. Front. Plant Sci. 2015, 6, 735. [Google Scholar] [CrossRef] [Green Version]
- Kranz, B.; Koch, M.; Schapfl, M.; Fischer, L. Investigation of the Germination of Barley and Wheat Grains with a Design of Experiments for the Production of Hydrolases. Food Technol. Biotechnol. 2015, 5353, 127. [Google Scholar]
- Li, Z.; Mouille, G.; Kosar-Hashemi, B.; Rahman, S.; Clarke, B.; Gale, K.R.; Morell, M.K. The structure and expression of the wheat starch synthase III gene. Motifs in the expressed gene define the lineage of the starch synthase III gene family. Plant Physiol. 2000, 123123, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Bowles, D.J. Defense-related proteins in higher plants. Annu. Rev. Biochem. 1990, 5959, 873–907. [Google Scholar] [CrossRef]
- Ziegler, K.; Neumann, J.; Liu, F.; Fröhlich-Nowoisky, J.; Cremer, C.; Saloga, J.; Lucas, K. Nitration of Wheat Amylase Trypsin Inhibitors Increases their Innate and Adaptive Immunostimulatory Potential in vitro. Front. Immunol. 2018, 9, 3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junker, Y.; Zeissig, S.; Kim, S.J.; Barisani, D.; Wieser, H.; Leffler, D.A.; Kelly, C.P. Wheat amylase trypsin inhibitors drive intestinal inflammation via activation of toll-like receptor 4. J. Exp. Med. 2012, 209209, 2395–2408. [Google Scholar] [CrossRef] [PubMed]
- Wielkopolan, B.; Krawczyk, K.; Obrępalska-Stęplowska, A. Gene expression of serine and cysteine proteinase inhibitors during cereal leaf beetle larvae feeding on wheat: The role of insect-associated microorganisms. Arthropod-Plant Interact. 2018, 1212, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Rangan, P.; Furtado, A.; Henry, R.J. The transcriptome of the developing grain: A resource for understanding seed development and the molecular control of the functional and nutritional properties of wheat. BMC Genom. 2017, 1818, 766. [Google Scholar] [CrossRef] [Green Version]
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.; D’Amore, R.; Allen, A.M.; Kay, S. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 2012, 491491, 705. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. Babraham Bioinformatics-FastQC a Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 December 2018).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 1717, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 1212, 357. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Chter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 2828, 511. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Shankar, R.; Bhattacharjee, A.; Jain, M. Transcriptome analysis in different rice cultivars provides novel insights into desiccation and salinity stress responses. Sci. Rep. 2016, 6, 23719. [Google Scholar] [CrossRef] [Green Version]
- Seifert, O.; Bayat, A.; Geffers, R.; Dienus, K.; Buer, J.; Löfgren, S.; Matussek, A. Identification of unique gene expression patterns within different lesional sites of keloids. Wound Repair Regen. 2008, 1616, 254–265. [Google Scholar] [CrossRef]
- Curran, K.A.; Crook, N.C.; Karim, A.S.; Gupta, A.; Wagman, A.M.; Alper, H.S. Design of synthetic yeast promoters via tuning of nucleosome architecture. Nat. Commun. 2014, 5, 4002. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, M.; Li, T.; Liu, W.; Liu, Y.; Li, Y.; Chang, J. Overexpression of avenin-like b proteins in bread wheat (Triticum aestivum L.) improves dough mixing properties by their incorporation into glutenin polymers. PLoS ONE 2013, 88, e66758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrill, P.; Adamski, N.; Uauy, C. Genomics as the key to unlocking the polyploid potential of wheat. New Phytol. 2015, 208208, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Scossa, F.; Laudencia-Chingcuanco, D.; Anderson, O.D.; Vensel, W.H.; Lafiandra, D.; D’Ovidio, R.; Masci, S. Comparative proteomic and transcriptional profiling of a bread wheat cultivar and its derived transgenic line overexpressing a low molecular weight glutenin subunit gene in the endosperm. Proteomics 2008, 88, 2948–2966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, G.; Liu, D.; Wang, D.; Yang, W.; Sun, J.; Zhan, K. Genome-, transcriptome-and proteome-wide analyses of the gliadin gene families in Triticum urartu. PLoS ONE 2015, 1010, e0131559. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.F.; Geniza, M.; Hanumappa, M.; Naithani, S.; Sullivan, C.; Preece, J.; Tiwari, V.K.; Elser, J.; Leonard, J.M.; Sage, A. De Novo Transcriptome Assembly and Analyses of Gene Expression during Photomorphogenesis in Diploid Wheat Triticum Monococcum. PLoS ONE 2014, 9, e95855. [Google Scholar] [CrossRef]
- Luo, M.C.; Gu, Y.Q.; Puiu, D.; Wang, H.; Twardziok, S.O.; Deal, K.R.; McGuire, P.E. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017, 551551, 498. [Google Scholar] [CrossRef]
- Bread Wheat’s Large and Complex Genome is Revealed.”. ScienceDaily. 2012. Available online: www.sciencedaily.com/releases/2012/11/121128132357.htm (accessed on 15 April 2020).
- El Baidouri, M.; Murat, F.; Veyssiere, M.; Molinier, M.; Flores, R.; Burlot, L.; Salse, J. Reconciling the evolutionary origin of bread wheat (Triticum aestivum). New Phytol. 2017, 213213, 1477–1486. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Zhen, S.; Wang, S.; Wang, Y.; Cao, H.; Zhang, Y.; Yan, Y. Comparative transcriptome analysis of wheat embryo and endosperm responses to ABA and H2O2 stresses during seed germination. BMC Genom. 2016, 1717, 97. [Google Scholar] [CrossRef] [Green Version]
- Appels, R.; Eversole, K.; Feuillet, C.; Keller, B.; Rogers, J.; Stein, N.; Ronen, G. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361361, eaar7191. [Google Scholar]
- Miki, Y.; Yoshida, K.; Mizuno, N.; Nasuda, S.; Sato, K.; Takumi, S. Origin of wheat B-genome chromosomes inferred from RNA sequencing analysis of leaf transcripts from section Sitopsis species of Aegilops. DNA Res. 2019, 2626, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paux, E.; Roger, D.; Badaeva, E.; Gay, G.; Bernard, M.; Sourdille, P.; Feuillet, C. Characterizing the composition and evolution of homoeologous genomes in hexaploid wheat through BAC-end sequencing on chromosome 3B. Plant J. 2006, 4848, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Payne, P.I. Genetics of wheat storage proteins and the effect of allelic variation on bread-making quality. Annu. Rev. Plant Physiol. 1987, 3838, 141–153. [Google Scholar] [CrossRef]
- Shewry, P.R.; Napier, J.A.; Tatham, A.S. Seed storage proteins: Structures and biosynthesis. Plant Cell 1995, 77, 945. [Google Scholar]
- Shewry, P.R.; Halford, N.G. Cereal seed storage proteins: Structures, properties and role in grain utilization. J. Exp. Bot. 2002, 5353, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salentijn, E.M.; Esselink, D.G.; Goryunova, S.V.; van der Meer, I.M.; Gilissen, L.J.; Smulders, M.J. Quantitative and qualitative differences in celiac disease epitopes among durum wheat varieties identified through deep RNA-amplicon sequencing. BMC Genom. 2013, 1414, 905. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, M.; Masuda, T.; Tani, F.; Kitabatake, N. Expression and development of wheat proteins during maturation of wheat kernel and the rheological properties of dough prepared from the flour of mature and immature wheat. Food Sci. Technol. Res. 2011, 1717, 111–120. [Google Scholar] [CrossRef] [Green Version]
- McMaugh, S.J.; Thistleton, J.L.; Anschaw, E.; Luo, J.; Konik-Rose, C.; Wang, H.; Morell, M.K. Suppression of starch synthase I expression affects the granule morphology and granule size and fine structure of starch in wheat endosperm. J. Exp. Bot. 2014, 6565, 2189–2201. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Cabrera, A.; Hoffstetter, A.; Griffey, C.; Van Sanford, D.; Costa, J.; Sneller, C. Genomic selection for wheat traits and trait stability. Theor. Appl. Genet. 2016, 129, 1697–1710. [Google Scholar] [CrossRef]
- Details of New Wheat Variety Released. Available online: http://dwd.dacnet.nic.in/Variety/variety.pdf (accessed on 5 January 2020).
- Tetlow, I.; Emes, M. Starch biosynthesis in the developing endosperms of grasses and cereals. Agronomy 2017, 7, 81. [Google Scholar] [CrossRef] [Green Version]
- Stanley, D.; Rejzek, M.; Naested, H.; Smedley, M.; Otero, S.; Fahy, B.; Denyer, K. The role of α-glucosidase in germinating barley grains. Plant Physiol. 2011, 155, 932–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whan, A.; Dielen, A.S.; Mieog, J.; Bowerman, A.F.; Robinson, H.M.; Byrne, K.; Ral, J.P. Engineering α-amylase levels in wheat grain suggests a highly sophisticated level of carbohydrate regulation during development. J. Exp. Bot. 2014, 65, 5443–5457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackig, M.; Corbineau, F.; Grzesikit, M.; Guyi, P.; Come, D. Carbohydrate metabolism in the developing and maturing wheat embryo in relation to its desiccation tolerance. J. Exp. Bot. 1996, 47, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.H. Nutrient Uptake, Transport and Translocation in Cereals: Influences of Environmental and Farming Conditions; No. 2009: 1; Faculty of Landscape Planning, Horticulture and Agricultural Science, Swedish University of Agricultural Sciences: Alnarp, Sweden, 2009. [Google Scholar]
- Altpeter, F.; Diaz, I.; McAuslane, H.; Gaddour, K.; Carbonero, P.; Vasil, I.K. Increased insect resistance in transgenic wheat stably expressing trypsin inhibitor CMe. Mol. Breed. 1999, 5, 53–63. [Google Scholar] [CrossRef]
- Florack, D.E.A.; Stiekema, W.J. Thionins: Properties, possible biological roles and mechanisms of action. Plant Mol. Biol. 1994, 26, 25–37. [Google Scholar] [CrossRef]
- Plattner, S.; Gruber, C.; Stadlmann, J.; Widmann, S.; Gruber, C.W.; Altmann, F.; Bohlmann, H. Isolation and characterization of a thionin proprotein-processing enzyme from barley. J. Biol. Chem. 2015, 290, 18056–18067. [Google Scholar] [CrossRef] [Green Version]
- Qu, L.J.; Chen, J.; Liu, M.; Pan, N.; Okamoto, H.; Lin, Z.; Zhao, X. Molecular cloning and functional analysis of a novel type of Bowman-Birk inhibitor gene family in rice. Plant Physiol. 2003, 133, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Chilosi, G.; Caruso, C.; Caporale, C.; Leonardi, L.; Bertini, L.; Buzi, A.; Buonocore, V. Antifungal Activity of a Bowman–Birk-type Trypsin Inhibitor from Wheat Kernel. J. Phytopathol. 2000, 148, 477–481. [Google Scholar] [CrossRef]
- Tedeschi, F.; Di Maro, A.; Facchiano, A.; Costantini, S.; Chambery, A.; Bruni, N.; Poerio, E. Wheat Subtilisin/Chymotrypsin Inhibitor (WSCI) as a scaffold for novel serine protease inhibitors with a given specificity. Mol. Biosyst. 2012, 8, 3335–3343. [Google Scholar] [CrossRef]
- Tundo, S.; Lupi, R.; Lafond, M.; Giardina, T.; Larré, C.; Denery-Papini, S.; Masci, S. Wheat ATI CM3, CM16 and 0.28 Allergens Produced in Pichia Pastoris Display a Different Eliciting Potential in Food Allergy to Wheat. Plants 2018, 7, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, N.M.; Redestig, H.; Dessimoz, C. Homoeologs: What are they and how do we infer them? Trends Plant Sci. 2016, 21, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Lu, S. Biosynthesis and regulation of phenylpropanoids in plants. Crit. Rev. Plant Sci. 2017, 36, 257–290. [Google Scholar] [CrossRef]
- Fraser, C.M.; Chapple, C. The phenylpropanoid pathway in Arabidopsis. Arab. Book/Am. Soc. Plant Biol. 2011, 9, e0152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Wang, A.; Li, X.; Dong, K.; Wang, K.; Appels, R.; Yan, Y. Wheat quality related differential expressions of albumins and globulins revealed by two-dimensional difference gel electrophoresis (2-D DIGE). J. Proteom. 2009, 73, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zheng, Q. Dynamic rheological properties of wheat flour dough and proteins. Trends Food Sci. Technol. 2007, 18, 132–138. [Google Scholar] [CrossRef]
- Hill, K.; Horváth-Szanics, E.; Hajós, G.; Kiss, É. Surface and interfacial properties of water-soluble wheat proteins. Colloids Surf. A Physicochem. Eng. Asp. 2008, 319, 180–187. [Google Scholar] [CrossRef]
- Li, Q.; Wang, W.; Wang, W.; Zhang, G.; Liu, Y.; Wang, Y.; Wang, W. Wheat F-box protein gene TaFBA1 is involved in plant tolerance to heat stress. Front. Plant Sci. 2018, 9, 521. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.M.; Kong, X.Z.; Kang, H.H.; Sun, X.D.; Wang, W. The involvement of wheat F-box protein gene TaFBA1 in the oxidative stress tolerance of plants. PLoS ONE 2015, 10, e0122117. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Kuroda, M.; Yamakawa, H.; Ashizawa, T.; Hirayae, K.; Kurimoto, L.; Shibuya, N. Suppression of a phospholipase D gene, OsPLDβ1, activates defense responses and increases disease resistance in rice. Plant Physiol. 2009, 150, 308–319. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Xu, W.; Xiang, Y.; Jia, H.; Zhang, L.; Ma, Z. Association of jacalin-related lectins with wheat responses to stresses revealed by transcriptional profiling. Plant Mol. Biol. 2014, 84, 95–110. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Li, L.; Xu, H.; Xi, J.; Cao, X.; Xu, H.; Xu, J. A rice jacalin-related mannose-binding lectin gene, OsJRL, enhances Escherichia coli viability under high salinity stress and improves salinity tolerance of rice. Plant Biol. 2017, 19, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Esch, L.; Schaffrath, U. An update on jacalin-like lectins and their role in plant defense. Int. J. Mol. Sci. 2017, 18, 1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacerda, A.; Vasconcelos, É.A.R.; Pelegrini, P.B.; Grossi-de-Sa, M.F. Antifungal defensins and their role in plant defense. Front. Microbiol. 2014, 5, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotypes | Genome | Variety Name/Accession No. |
|---|---|---|
| Triticum monococcum | AA | AN104 |
| Aegilops speltoides | BB | PN84 |
| Aegilops tauschii | DD | PN95 |
| Triticum aestivum | AABBDD | PBW343 |
| Wheat Genotype | Total Raw Reads (in millions) | Processed Reads (in millions) | Alignment (%) | Expressed Transcripts |
|---|---|---|---|---|
| AA: T.monococcum (AN104&PKM1) | 55.63 | 50.81 | 45.73 | 79328 |
| BB: Ae. speltoides (PN84&PKM2) | 55.23 | 49.55 | 54.48 | 113733 |
| DD: Ae. tauschii (PN95&PKM3) | 68.13 | 61.98 | 64.64 | 90640 |
| AABBDD: T. aestivum (PBW343&PKM5) | 103.31 | 93.19 | 71.44 | 121295 |
| Comparison | ABD vs. A Up Down | ABD vs. B Up Down | ABD vs. D Up Down | |||
|---|---|---|---|---|---|---|
| Number of DETs | 7993 | 22052 | 8405 | 16354 | 14657 | 24616 |
| Comparison | Molecular Function DEGs Up Down | Enriched GO Terms (Up-Regulated) at FDR<0.05 | Enriched GO (Gene Ontology) Terms (Down-Regulated) at FDR<0.05 | |
|---|---|---|---|---|
| ABD vs. A | 4549 | 2526 | 127 | 76 |
| ABD vs. B | 4099 | 2507 | 95 | 52 |
| ABD vs. D | 8296 | 9577 | 132 | 131 |
| Enriched GO Terms Activity | ABD vs. A Up Down | ABD vs. B Up Down | ABD vs. D Up Down | |||
|---|---|---|---|---|---|---|
| Nutrient reservoir (GO:0045735) | 36 | 30 | 35 | 0 | 60 | 0 |
| Transcription factor protein binding(GO:0000968) | 50 | 0 | 27 | 0 | 63 | 61 |
| Catalytic activity (GO:0003824) | 2121 | 1228 | 1969 | 1186 | 3613 | 4563 |
| Signal transducer (GO:0004871) | 42 | 42 | 55 | 26 | 122 | 116 |
| Molecular function regulator (GO:0098772) | 91 | 56 | 142 | 56 | 187 | 230 |
| Molecular transducer (GO:0060089) | 43 | 41 | 56 | 27 | 119 | 118 |
| Carbohydrate transporter (GO:1901476) | 14 | 12 | 16 | 9 | 35 | 0 |
| ABD vs. A Up Down | ABD vs. B Up Down | ABD vs. D Up Down | ||||
|---|---|---|---|---|---|---|
| Total number of DEGs | 1628 | 2625 | 1441 | 1617 | 2810 | 3014 |
| Total Pathways involved | 209 | 303 | 167 | 258 | 398 | 317 |
| Pathways | ABD vs. A | ABD vs. B | ABD vs. D |
|---|---|---|---|
| Pehnylpropanoid biosynthesis | 247 | 208 | 304 |
| Starch and sucrose metabolism | 202 | 160 | 248 |
| Oxidative Phosphorylation | 139 | 97 | 166 |
| Galactose metabolism | 120 | 99 | 202 |
| Ubiquinone and other terpenoid-quinone biosynthesis | 36 | 34 | 63 |
| Cutin, suberine, and wax biosynthesis, glycerolipid metabolism | 47 | 31 | 42 |
| Purine metabolism | 28 | 308 | 677 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaushik, M.; Rai, S.; Venkadesan, S.; Sinha, S.K.; Mohan, S.; Mandal, P.K. Transcriptome Analysis Reveals Important Candidate Genes Related to Nutrient Reservoir, Carbohydrate Metabolism, and Defence Proteins during Grain Development of Hexaploid Bread Wheat and Its Diploid Progenitors. Genes 2020, 11, 509. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050509
Kaushik M, Rai S, Venkadesan S, Sinha SK, Mohan S, Mandal PK. Transcriptome Analysis Reveals Important Candidate Genes Related to Nutrient Reservoir, Carbohydrate Metabolism, and Defence Proteins during Grain Development of Hexaploid Bread Wheat and Its Diploid Progenitors. Genes. 2020; 11(5):509. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050509
Chicago/Turabian StyleKaushik, Megha, Shubham Rai, Sureshkumar Venkadesan, Subodh Kumar Sinha, Sumedha Mohan, and Pranab Kumar Mandal. 2020. "Transcriptome Analysis Reveals Important Candidate Genes Related to Nutrient Reservoir, Carbohydrate Metabolism, and Defence Proteins during Grain Development of Hexaploid Bread Wheat and Its Diploid Progenitors" Genes 11, no. 5: 509. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11050509