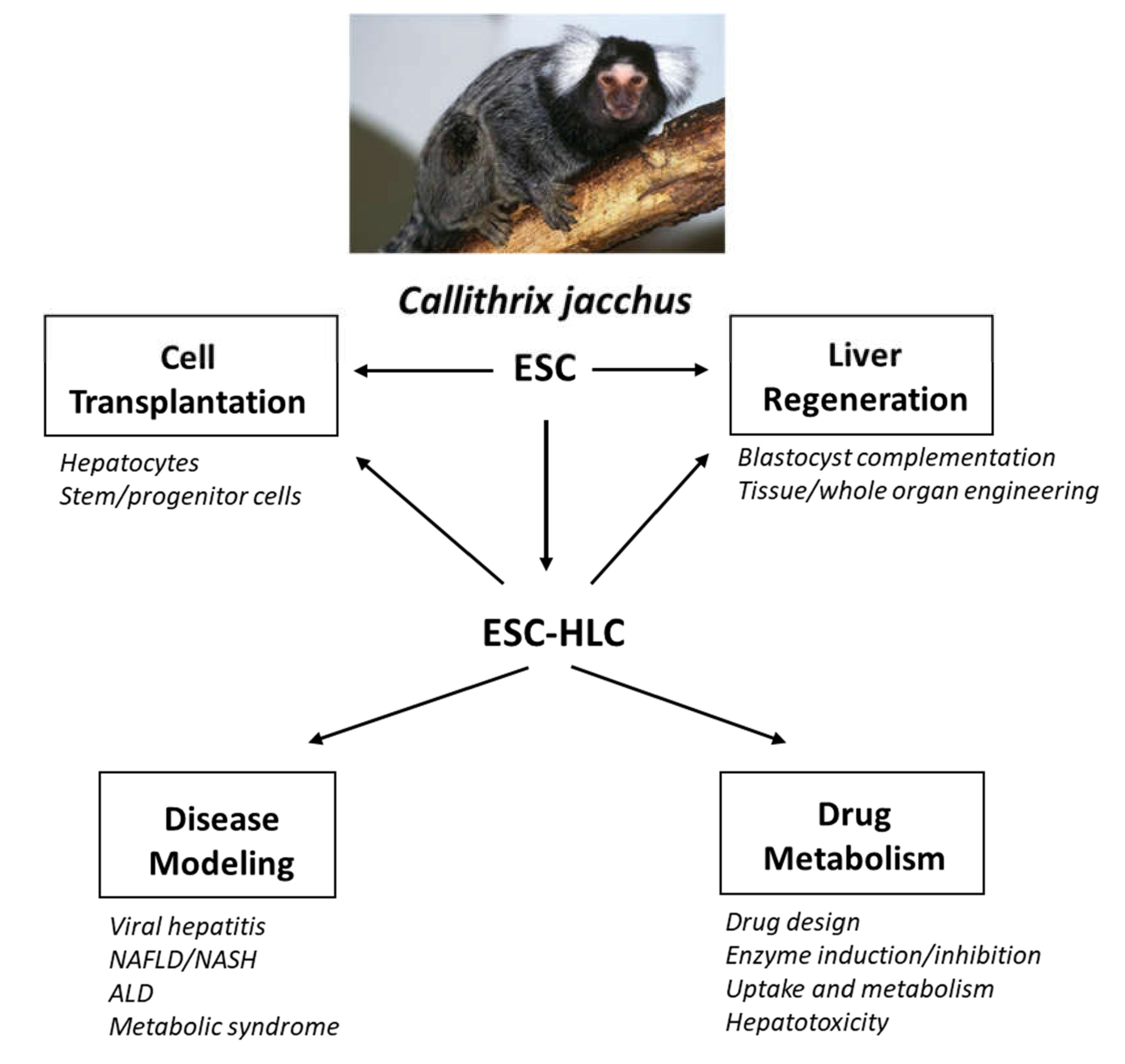
Utility of Common Marmoset (Callithrix jacchus) Embryonic Stem Cells in Liver Disease Modeling, Tissue Engineering and Drug Metabolism



{kind=link}
Abstract
:1. Introduction
2. The Common Marmoset Is an Ideal NHP Model for Research on Liver Diseases
3. Marmoset Embryonic Stem Cells
4. Callithrix jacchus Models of Human Liver Disease
4.1. Viral Hepatitis
4.2. Hepatic Fibrosis
4.3. Non-Alcoholic Fatty Liver Disease
4.4. Metabolic Syndrome
5. Potential Uses for Marmoset ESCs in Liver Regeneration and Tissue Engineering
5.1. Cell Transplantation
5.1.1. Hepatocyte transplantation
5.1.2. Stem Cell Transplantation
5.2. Whole Liver Tissue Engineering
5.3. Blastocyst Complementation
6. Drug Metabolism Studies with Marmoset ESC-Derived HLCs
6.1. Phase I Enzymes
Cytochrome P450 Enzymes
6.2. Phase II Enzymes
6.2.1. Arylamine N-Acetyltransferases
6.2.2. Uridine Diphosphate Glucuronosyltransferases
6.2.3. Other Phase II Enzymes
7. Future Studies and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 11β-HSD1 | 11β-hydroxysteroid dehydrogenase type 1 |
| ALD | Alcoholic liver disease |
| ALT | Alanine aminotransferase |
| AST | Aspartate transaminase |
| cjESC | C. jacchus embryonic stem cell |
| CYP | Cytochrome P450 |
| CN1 | Crigler-Najjar syndrome Type 1 |
| ECM | Extracellular matrix |
| ESC | Embryonic stem cell |
| FFA | Free fatty acid |
| GGT | γ-glutamyl transpeptidase |
| HCC | Hepatocellular carcinoma |
| HAV | Hepatitis A virus |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| HDV | Hepatitis D virus |
| HEV | Hepatitis E virus |
| HF | Hepatic fibrosis |
| HLC | Hepatocyte-like cell |
| IDH | Isocitrate dehydrogenase |
| iPSC | Induced pluripotent stem cell |
| MSC | Mesenchymal stem cell |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NHP | Non-human primate |
| OLT | Orthotopic liver transplantation |
| PH | Partial hepatectomy |
| PHH | Primary human hepatocyte |
| TAA | Thioacetamide |
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Aravalli, R.N.; Belcher, J.D.; Steer, C.J. Liver-targeted gene therapy: Approaches and challenges. Liver Transpl. 2015, 21, 718–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Davis, R.A. Cholesterol and hepatic lipoprotein assembly and secretion. Biochim. Biophys. Acta 2000, 1529, 223–230. [Google Scholar] [CrossRef]
- Brosnan, M.E.; Brosnan, J.T. Hepatic glutamate metabolism: A tale of 2 hepatocytes. Am. J. Clin. Nutr. 2009, 90, 857S–861S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Schloss, R.; Yarmush, M. What came first: Fully functional or metabolically mature liver? Crit. Rev. Biomed. Eng. 2008, 36, 413–439. [Google Scholar]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar]
- Campbell, I. Liver: Metabolic functions. Anaesth. Intens. Care Med. 2006, 7, 51–54. [Google Scholar] [CrossRef]
- Forbes, S.J.; Gupta, S.; Dhawan, A. Cell therapy for liver disease: From liver transplantation to cell factory. J. Hepatol. 2015, 62, S157–S169. [Google Scholar] [CrossRef] [Green Version]
- Li, W.L.; Su, J.; Yao, Y.C.; Tao, X.R.; Yan, Y.B.; Yu, H.Y.; Wang, X.M.; Li, J.X.; Yang, Y.J.; Lau, J.T.; et al. Isolation and characterization of bipotent liver progenitor cells from adult mouse. Stem Cells 2006, 24, 322–332. [Google Scholar] [CrossRef]
- Strick-Marchand, H.; Morosan, S.; Charneau, P.; Kremsdorf, D.; Weiss, M.C. Bipotential mouse embryonic liver stem cell lines contribute to liver regeneration and differentiate as bile ducts and hepatocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 8360–8365. [Google Scholar] [CrossRef] [Green Version]
- Allain, J.E.; Dagher, I.; Mahieu-Caputo, D.; Loux, N.; Andreoletti, M.; Westerman, K.; Briand, P.; Franco, D.; Leboulch, P.; Weber, A. Immortalization of a primate bipotent epithelial liver stem cell. Proc. Natl. Acad. Sci. USA 2002, 99, 3639–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.M.; Vallender, E.J. The resurgence and genetic implications of New World primates in biomedical research. Trends Genet. 2012, 28, 586–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shultz, L.D.; Keck, J.; Burzenski, L.; Jangalwe, S.; Vaidya, S.; Greiner, D.L.; Brehm, M.A. Humanized mouse models of immunological diseases and precision medicine. Mamm. Genome 2019, 30, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Wege, A.K. Humanized mouse models for the preclinical assessment of cancer immunotherapy. BioDrugs 2018, 32, 245–266. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Dawson, P.A. Animal models to study bile acid metabolism. Biochem. Biophys. Acta Mol. Basis Dis. 2019, 1865, 895–911. [Google Scholar] [CrossRef] [PubMed]
- Hanna, C.; Demond, H.; Kelsey, G. Epigenetic regulation in development: Is the mouse a good model for the human? Hum. Reprod. Update 2018, 24, 556–576. [Google Scholar] [CrossRef]
- Cheon, D.J.; Orsulic, S. Mouse models of cancer. Annu. Rev. Pathol 2011, 6, 95–119. [Google Scholar] [CrossRef]
- Burm, R.; Collignon, L.; Mesalam, A.A.; Meuleman, P. Animal models to study hepatitis C virus infection. Front. Immunol. 2018, 9, 1032. [Google Scholar] [CrossRef]
- Abbott, D.H.; Barnett, D.K.; Colman, R.J.; Yamamoto, M.E.; Schultz-Darken, N.J. Aspects of common marmoset basic biology and life history important for biomedical research. Comp. Med. 2003, 53, 339–350. [Google Scholar]
- Carrion, R.J.; Patterson, J.L. An animal model that reflects human disease: The common marmoset (Callithrix jacchus). Curr. Opin. Virol. 2012, 2, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K. Marmoset models commonly used in biomedical research. Comp. Med. 2003, 53, 383–392. [Google Scholar] [PubMed]
- Zühlke, U.; Weinbauer, G. The common marmoset (Callithrix jacchus) as a model in toxicology. Toxicol. Pathol. 2003, 31, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Uno, Y.; Uehara, S.; Yamazaki, H. Utility of non-human primates in drug development: Comparison of non-human primate and human drug-metabolizing cytochrome P450 enzymes. Biochem. Pharmacol. 2016, 121, 1–7. [Google Scholar] [CrossRef]
- Phillips, K.A.; Bales, K.L.; Capitanio, J.P.; Conley, A.; Czoty, P.W.; ’t Hart, B.A.; Hopkins, W.D.; Hu, S.L.; Miller, L.A.; Nader, M.A.; et al. Why primate models matter. Am. J. Primatol. 2014, 76, 801–827. [Google Scholar] [CrossRef] [Green Version]
- Hérodin, F.; Thullier, P.; Garin, D.; Drouet, M. Nonhuman primates are relevant models for research in hematology, immunology and virology. Eur. Cytokine Netw. 2005, 16, 104–116. [Google Scholar]
- Okano, H.; Hikishima, K.; Iriki, A.; Sasaki, E. The common marmoset as a novel animal model system for biomedical and neuroscience research applications. Semin. Fetal Neonatal. Med. 2012, 17, 336–340. [Google Scholar] [CrossRef]
- ’t Hart, B.A.; Abbott, D.H.; Nakamura, K.; Fuchs, E. The marmoset monkey: A multi-purpose preclinical and translational model of human biology and disease. Drug Discov. Today 2012, 17, 1160–1165. [Google Scholar] [CrossRef]
- Kishi, N.; Sato, K.; Sasaki, E.; Okano, H. Common marmoset as a new model animal for neuroscience research and genome editing technology. Dev. Growth Differ. 2014, 56, 53–62. [Google Scholar] [CrossRef]
- Havel, P.J.; Kievit, P.; Comuzzie, A.G.; Bremer, A.A. Use and importance of nonhuman primates in metabolic disease research: Current state of the field. ILAR J. 2017, 58, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Kramer, J.A.; Grindley, J.; Crowell, A.M.; Makaron, L.; Kohli, R.; Kirby, M.; Mansfield, K.G.; Wachtman, L.M. The common marmoset as a model for the study of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Vet. Pathol. 2015, 52, 404–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.; Trennery, P.; Farningham, D. The selection of marmoset monkeys (Callithrix jacchus) in pharmaceutical toxicology. Lab. Animal 2001, 35, 117–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsi, A.; Rees, D.; Andreini, I.; Venturella, S.; Cinelli, S.; Oberto, G. Overview of the marmoset as a model in nonclinical development of pharmaceutical products. Regul. Toxic Pharmacol. 2011, 59, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Akari, H.; Iwasaki, Y.; Yoshida, T.; Iijima, S. Non-human primate model of hepatitis C virus infection. Microbiol. Immunol. 2009, 53, 53–57. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murry, C.E.; Keller, G. Differentiation of embryonic stem cells to clinically relevant populations: Lessons from embryonic development. Cell 2008, 132, 661–680. [Google Scholar] [CrossRef] [Green Version]
- Thomson, J.A.; Kalishman, J.; Golos, T.G.; Durningm, M.; Harris, C.P.; Hearn, J.P. Pluripotent cell lines derived from common marmoset (Callithrix jacchus) blastocysts. Biol. Reprod. 1996, 55, 254–259. [Google Scholar] [CrossRef]
- Zhou, B.; Ho, S.S.; Leung, L.C.; Ward, T.R.; Ho, M.; Plastini, M.J.; Vermilyea, S.C.; Emborg, M.E.; Golos, T.G.; Mourrain, P.; et al. Haplotype-phased common marmoset embryonic stem cells for genome editing using CRISPR/Cas9. bioRxiv 2018. [Google Scholar] [CrossRef]
- Müller, T.; Fleischmann, G.; Eildermann, K.; Mätz-Rensing, K.; Horn, P.A.; Sasaki, E.; Behr, R. A novel embryonic stem cell line derived from the common marmoset monkey (Callithrix jacchus) exhibiting germ cell-like characteristics. Hum. Reprod. 2009, 24, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, E.; Hanazawa, K.; Kurita, R.; Akatsuka, A.; Yoshizaki, T.; Ishii, H.; Tanioka, Y.; Ohnishi, Y.; Suemizu, H.; Sugawara, A.; et al. Establishment of novel embryonic stem cell lines derived from the common marmoset (Callithrix jacchus). Stem Cells 2005, 23, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Debowski, K.; Drummer, C.; Lentes, J.; Cors, M.; Dressel, R.; Lingner, T.; Salinas-Riester, G.; Fuchs, S.; Sasaki, E.; Behr, R. The transcriptomes of novel marmoset monkey embryonic stem cell lines reflect distinct genomic features. Sci. Rep. 2016, 6, 29122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleischmann, G.; Müller, T.; Blasczyk, R.; Sasaki, E.; Horn, P.A. Growth characteristics of the nonhuman primate embryonic stem cell line cjes001 depending on feeder cell treatment. Cloning Stem Cells 2009, 11, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Trettner, S.; Findeisen, A.; Taube, S.; Horn, P.A.; Sasaki, E.; zur Nieden, N.I. Osteogenic induction from marmoset embryonic stem cells cultured in feeder-dependent and feeder-independent conditions. Osteoporos. Int. 2014, 25, 1255–1266. [Google Scholar] [CrossRef]
- Yoshimatsu, S.; Okahara, J.; Sone, T.; Takeda, Y.; Nakamura, M.; Sasaki, E.; Kishi, N.; Shiozawa, S.; Okano, H. Robust and efficient knock-in in embryonic stem cells and early-stage embryos of the common marmoset using the CRISPR-Cas9 system. Sci. Rep. 2019, 9, 1528. [Google Scholar] [CrossRef] [Green Version]
- Yoshimatsu, S.; Sato, T.; Yamamoto, M.; Sasaki, E.; Nakajima, M.; Nakamura, M.; Shiozawa, S.; Noce, T.; Okano, H. Generation of a male common marmoset embryonic stem cell line dsy127-bv8vt1 carrying double reporters specific for the germ cell linage using the CRISPR-Cas9 and PiggyBac transposase systems. Stem Cell Res. 2020, 44, 101740. [Google Scholar] [CrossRef]
- Shiozawa, S.; Nakajima, M.; Okahara, J.; Kuortaki, Y.; Kisa, F.; Yoshimatsu, S.; Nakamura, M.; Koya, I.; Yoshimura, M.; Sasagawa, Y.; et al. Primed to naïve-like conversion of the common marmoset embryonic stem cells. Stem Cells Dev. 2020, 29, 761–773. [Google Scholar] [CrossRef]
- Aravalli, R.N.; Collins, D.P.; Hapke, J.H.; Crane, A.T.; Steer, C.J. Hepatic differentiation of marmoset embryonic stem cells and functional characterization of ESC-derived hepatocyte-like cells. Hepat. Med. 2020, 12, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Morizane, A.; Doi, D.; Kikuchi, T.; Okita, K.; Hotta, A.; Kawasaki, T.; Hayashi, T.; Onoe, H.; Shiina, T.; Yamanaka, S.; et al. Direct comparison of autologous and allogeneic transplantation of iPSC-derived neural cells in the brain of a non-human primate. Stem Cell Rep. 2013, 1, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Iwai, H.; Shimada, H.; Nishimura, S.; Kobayashi, Y.; Itakura, G.; Hori, K.; Hikishima, K.; Ebise, H.; Negishi, N.; Shibata, S.; et al. Allogeneic neural stem/progenitor cells derived from embryonic stem cells promote functional recovery after transplantation into injured spinal cord of nonhuman primates. Stem Cells Transl. Med. 2015, 4, 708–719. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, E. Creating genetically modified marmosets. In The Common Marmoset in Captivity and Biomedical Research, 1st ed.; Marini, R., Wachtman, L., Tardif, S., Mansfield, K., Fox, J., Eds.; Academic Press: San Diego, CA, USA, 2019; pp. 335–353. [Google Scholar]
- Knollmann, B.C. Induced pluripotent stem cell-derived cardiomyocytes: Boutique science or valuable arrhythmia model? Circ. Res. 2013, 112, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Siller, R.; Greenhough, S.; Park, I.H.; Sullivan, G.J. Modelling human disease with pluripotent stem cells. Curr. Gene Ther. 2013, 13, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandy, R.; Tomaz, R.; Vallier, L. Modeling disease with human inducible pluripotent stem cells. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 449–468. [Google Scholar] [CrossRef] [PubMed]
- Rasche, A.; Sander, A.L.; Corman, V.M.; Drexler, J.F. Evolutionary biology of human hepatitis viruses. J. Hepatol. 2019, 70, 501–520. [Google Scholar] [CrossRef] [Green Version]
- Ploss, A.; Kapoor, A. Animal models of hepatitis C virus infection. Cold Spring Harb. Perspect. Med. 2020, 10, a036970. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Strick-Marchand, H. In-vitro and in-vivo models for hepatitis B cure research. Curr. Opin. HIV AIDS 2020, 15, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Dorner, M.; Horwitz, J.A.; Robbins, J.B.; Barry, W.T.; Feng, Q.; Mu, K.; Jones, C.T.; Schoggins, J.W.; Catanese, M.T.; Burton, D.R.; et al. A genetically humanized mouse model for hepatitis C virus infection. Nature 2011, 474, 208–211. [Google Scholar] [CrossRef]
- Tesfaye, A.; Stift, J.; Maric, D.; Cui, Q.; Dienes, H.P.; Feinstone, S.M. Chimeric mouse model for the infection of hepatitis B and C viruses. PLoS ONE 2013, 8, e77298. [Google Scholar] [CrossRef]
- Bility, M.T.; Li, F.; Cheng, L.; Su, L. Liver immune-pathogenesis and therapy of human liver tropic virus infection in humanized mouse models. J. Gastroenterol. Hepatol. 2013, 28, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Wieland, S.F. The chimpanzee model for hepatitis B virus infection. Cold Spring Harb. Perspect. Med. 2015, 5, a021469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanford, R.E.; Walker, C.M.; Lemon, S.M. The chimpanzee model of viral hepatitis: Advances in understanding the immune response and treatment of viral hepatitis. ILAR J. 2017, 58, 172–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, R.H. Hepatitis viruses: Changing patterns of human disease. Proc. Natl. Acad. Sci. USA 1994, 91, 2401–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerson, S.U.; Huang, Y.K.; McRill, C.; Lewis, M.; Shapiro, M.; London, W.T.; Purcell, R.H. Molecular basis of virulence and growth of hepatitis A virus in cell culture. Vaccine 1992, 10, S36–S39. [Google Scholar] [CrossRef]
- Gaspar, A.M.; Vitral, C.L.; Marchevsky, R.S.; Yoshida, C.F.; Schatzmayr, H.G. A Brazilian hepatitis A virus isolated and adapted in primate and primate cell line as a chance for the development of a vaccine. Mem. Inst. Oswaldo Cruz 1992, 87, 449–450. [Google Scholar] [CrossRef] [Green Version]
- Emerson, S.U.; Lewis, M.; Govindarajan, S.; Shapiro, M.; Moskal, T.; Purcell, R.H. cDNA clone of hepatitis A virus encoding a virulent virus: Induction of viral hepatitis by direct nucleic acid transfection of marmosets. J. Virol. 1992, 66, 6649–6654. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.; Marchevsky, R.S.; Pelajo-Machado, M.; Santiago, M.A.; Pissurno, J.W.; França, M.S.; Baptista, M.L.; Gouvea, A.S.; Santana, A.A.; Bertho, A.L.; et al. Inducible nitric oxide synthase (iNOS) expression in liver and splenic T lymphocyte rise are associated with liver histological damage during experimental hepatitis A virus (HAV) infection in Callithrix jacchus. Exp. Toxicol. Pathol. 2000, 52, 3–10. [Google Scholar] [CrossRef]
- Baptista, M.L.; Marchevsky, R.S.; Oliveira, A.V.; Yoshida, C.F.; Schatzmayr, H.G. Histopathological and immunohistochemical studies of hepatitis A virus infection in marmoset Callithrix jacchus. Exp. Toxicol. Pathol. 1993, 45, 7–13. [Google Scholar] [CrossRef]
- Pinto, M.A.; Marchevsky, R.S.; Baptista, M.L.; de Lima, M.A.; Pelajo-Machado, M.; Vitral, C.L.; Kubelka, C.F.; Pissurno, J.W.; Franca, M.S.; Schatzmayr, H.G.; et al. Experimental hepatitis A virus (HAV) infection in Callithrix jacchus: Early detection of HAV antigen and viral fate. Exp. Toxicol. Pathol. 2002, 53, 413–420. [Google Scholar] [CrossRef]
- Vitral, C.L.; Marchevsky, R.S.; Yoshida, C.F.; Coelho, J.M.; Gaspar, A.M.; Schatzmayr, H.G. Intragastric infection induced in marmosets (Callithrix jacchus) by a Brazilian hepatitis A virus (HAF-203). Braz. J. Med. Biol. Res. 1995, 28, 313–321. [Google Scholar]
- Mätz-Rensing, K.; Bleyer, M. Viral diseases of common marmosets. In The Common Marmoset in Captivity and Biomedical Research, 1st ed.; Marini, R., Wachtman, L., Tardif, S., Mansfield, K., Fox, J., Eds.; Academic Press: San Diego, CA, USA, 2019; pp. 251–264. [Google Scholar]
- Cadavid, L.F.; Shufflebotham, C.; Ruiz, F.J.; Yeager, M.; Hughes, A.L.; Watkins, D.I. Evolutionary instability of the major histocompatibility complex class I loci in New World primates. Proc. Natl. Acad. Sci. USA 1997, 94, 14536–14541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antunes, S.G.; de Groot, N.G.; Brok, H.; Doxiadis, G.; Menezes, A.A.; Otting, N.; Bontrop, R.E. The common marmoset: A New World primate species with limited MHC class II variability. Proc. Natl. Acad. Sci. USA 1998, 95, 11745–11750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, J.N.; Pilot-Matias, T.J.; Leary, T.P.; Dawson, G.J.; Desai, S.M.; Schlauder, G.G.; Muerhoff, A.S.; Erker, J.C.; Buijk, S.L.; Chalmers, M.L.; et al. Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc. Natl. Acad. Sci. USA 1995, 92, 3401–3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beames, B.; Chavez, D.; Lanford, R.E. GB virus B as a model for hepatitis C virus. ILAR J. 2001, 42, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bright, H.; Carroll, A.R.; Watts, P.A.; Fenton, R.J. Development of a GB virus B marmoset model and its validation with a novel series of hepatitis C virus NS3 protease inhibitors. J. Virol. 2004, 78, 2062–2071. [Google Scholar] [CrossRef] [Green Version]
- Bukh, J.; Apgar, C.L.; Yanagi, M. Toward a surrogate model for hepatitis C virus: An infectious molecular clone of the GB virus-B hepatitis agent. Virology 1999, 262, 470–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woollard, D.J.; Haqshenas, G.; Dong, X.; Pratt, B.F.; Kent, S.J.; Gowans, E.J. Virus-specific T-cell immunity correlates with control of GB virus B infection in marmosets. J. Virol. 2008, 82, 3054–3060. [Google Scholar] [CrossRef] [Green Version]
- Manickam, C.; Rajakumar, P.; Wachtman, L.; Kramer, J.A.; Martinot, A.J.; Varner, V.; Giavedoni, L.D.; Reeves, R.K. Acute liver damage associated with innate immune activation in a small nonhuman primate model of hepacivirus infection. J. Virol. 2016, 90, 9153–9162. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhu, S.; Shuai, L.; Xu, Y.; Yin, S.; Bian, Y.; Wang, Y.; Zuo, B.; Wang, W.; Zhao, S.; et al. Infection of common marmosets with hepatitis C virus/GB virus-B chimeras. Hepatology 2014, 59, 789–802. [Google Scholar] [CrossRef]
- Zhu, S.; Li, T.; Liu, B.; Xu, Y.; Sun, Y.; Wang, Y.; Wang, Y.; Shuai, L.; Chen, Z.; Allain, J.P.; et al. Infection of common marmosets with GB virus B chimeric virus encoding the major nonstructural proteins NS2 to NS4A of hepatitis C virus. J. Virol. 2016, 90, 8198–8211. [Google Scholar] [CrossRef] [Green Version]
- Catanese, M.T.; Dorner, M. Advances in experimental systems to study hepatitis C virus in vitro and in vivo. Virology 2015, 479–480, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, R.E.; Trehan, K.; Andrus, L.; Sheahan, T.P.; Ploss, A.; Duncan, S.A.; Rice, C.M.; Bhatia, S.N. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2544–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Robotham, J.M.; Lee, E.; Dalton, S.; Kneteman, N.M.; Gilbert, D.M.; Tang, H. Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS Pathog. 2012, 8, e1002617. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Gressner, A.M.; Weiskirchen, R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. J. Cell Mol. Med. 2006, 10, 76–99. [Google Scholar] [CrossRef] [Green Version]
- Delire, B.; Stärkel, P.; Leclercq, I. Animal models for fibrotic liver diseases: What we have, what we need, and what is under development. J. Clin. Transl. Hepatol. 2015, 3, 53–66. [Google Scholar]
- Inoue, T.; Ishizaka, Y.; Sasaki, E.; Lu, J.; Mineshige, T.; Yanase, M.; Sasaki, E.; Shimoda, M. Thioacetamide-induced hepatic fibrosis in the common marmoset. Exp. Anim. 2018, 67, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, K.; Kotaka, M.; Toyohara, T.; Sueta, S.I.; Katakai, Y.; Ageyama, N.; Uemoto, S.; Osafune, K. A nonhuman primate model of liver fibrosis towards cell therapy for liver cirrhosis. Biochem. Biophys. Res. Commun. 2020, 526, 661–669. [Google Scholar] [CrossRef]
- Lau, J.K.; Zhang, X.; Yu, J. Animal models of non-alcoholic fatty liver disease: Current perspectives and recent advances. J. Pathol. 2017, 241, 36–44. [Google Scholar] [CrossRef]
- Nevzorova, Y.A.; Boyer-Diaz, Z.; Cubero, F.J.; Gracia-Sancho, J. Animal models for liver disease—A practical approach for translational research. J. Hepatol. 2020, in press. [Google Scholar] [CrossRef]
- Jahn, D.; Kircher, S.; Hermanns, H.M.; Geier, A. Animal models of NAFLD from a hepatologist’s point of view. Biochem. Biophys. Acta Mol. Basis Dis. 2019, 1865, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Tardif, S.D.; Power, M.L.; Ross, C.N.; Rutherford, J.N. Body mass growth in common marmosets: Toward a model of pediatric obesity. Am. J. Phys. Anthropol. 2013, 150, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.M.; McAloose, D.; Torregrossa, A.M.; Raphael, B.L.; Calle, P.P.; Moore, R.P.; James, S.B. Hematologic iron analyte values as an indicator of hepatic hemosiderosis in callitrichidae. Am. J. Primatol. 2008, 70, 629–633. [Google Scholar] [CrossRef]
- Kostrzewski, T.; Cornforth, T.; Snow, S.A.; Ouro-Gnao, L.; Rowe, C.; Large, E.M.; Hughes, D.J. Three-dimensional perfused human in vitro model of non-alcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Després, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef]
- Esfahani, M.; Baranchi, M.; Goodarzi, M.T. The implication of hepatokines in metabolic syndrome. Diabetes Metab. Syndr. 2019, 13, 2477–2480. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol. 2017, 13, 509–520. [Google Scholar] [CrossRef]
- Dhurandhar, N.V.; Whigham, L.D.; Abbott, D.H.; Schultz-Darken, N.J.; Israel, B.A.; Bradley, S.M.; Kemnitz, J.W.; Allison, D.B.; Atkinson, R.L. Human adenovirus Ad-36 promotes weight gain in male rhesus and marmoset monkeys. J. Nutr. 2002, 132, 3155–3160. [Google Scholar] [CrossRef]
- Manickam, C.; Wachtman, L.; Martinot, A.J.; Giavedoni, L.D.; Reeves, R.K. Metabolic dysregulation in hepacivirus infection of common marmosets (Callithrix jacchus). PLoS ONE 2017, 12, e0170240. [Google Scholar] [CrossRef] [Green Version]
- Wachtman, L.M.; Kramer, J.A.; Miller, A.D.; Hachey, A.M.; Curran, E.H.; Mansfield, K.G. Differential contribution of dietary fat and monosaccharide to metabolic syndrome in the common marmoset (Callithrix jacchus). Obesity 2011, 19, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Nyirenda, M.J.; Carter, R.; Tang, J.I.; de Vries, A.; Schlumbohm, C.; Hillier, S.G.; Streit, F.; Oellerich, M.; Armstrong, V.W.; Fuchs, E.; et al. Prenatal programming of metabolic syndrome in the common marmoset is associated with increased expression of 11β-hydroxysteroid dehydrogenase type 1. Diabetes 2009, 58, 2873–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardif, S.D.; Power, M.L.; Ross, C.N.; Rutherford, J.N.; Layne-Colon, D.G.; Paulik, M.A. Characterization of obese phenotypes in a small nonhuman primate, the common marmoset (Callithrix jacchus). Obesity 2009, 17, 1499–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.R. Cortisol—Cause and cure for metabolic syndrome? Diabet. Med. 2006, 23, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Wallis, M. New insulin-like growth factor (IGF)-precursor sequences from mammalian genomes: The molecular evolution of IGFs and associated peptides in primates. Growth Horm. IGF Res. 2009, 19, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N. Liver regeneration and repair: Hepatocytes, progenitor cells, and stem cells. Hepatology 2004, 39, 1477–1487. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Liver regeneration: Alternative epithelial pathways. Int. J. Biochem. Cell Biol. 2011, 43, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef]
- Luo, J.H.; Ren, B.; Keryanov, S.; Tseng, G.C.; Rao, U.N.; Monga, S.P.; Strom, S.; Demetris, A.J.; Nalesnik, M.; Yu, Y.P.; et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology 2006, 44, 1012–1024. [Google Scholar] [CrossRef]
- Young, H.E.; Black, A.C., Jr. Adult stem cells. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2004, 276, 75–102. [Google Scholar] [CrossRef]
- Sell, S. Liver stem cells. Mod. Pathol. 1994, 7, 105–112. [Google Scholar] [CrossRef]
- Sahin, M.B.; Schwartz, R.E.; Buckley, S.M.; Heremans, Y.; Chase, L.; Hu, W.S.; Verfaillie, C.M. Isolation and characterization of a novel population of progenitor cells from unmanipulated rat liver. Liver Transpl. 2008, 14, 333–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmelzer, E.; Wauthier, E.; Reid, L.M. The phenotypes of pluripotent human hepatic progenitors. Stem Cells 2006, 24, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Segal, J.M.; Kent, D.; Wesche, D.J.; Ng, S.S.; Serra, M.; Oulès, B.; Kar, G.; Emerton, G.; Blackford, S.J.I.; Darmanis, S.; et al. Single cell analysis of human foetal liver captures the transcriptional profile of hepatobiliary hybrid progenitors. Nat. Commun. 2019, 10, 3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskams, T.A.; Libbrecht, L.; Desmet, V.J. Progenitor cells in diseased human liver. Semin. Liver Dis. 2003, 23, 385–396. [Google Scholar]
- Aravalli, R.N. Progress in stem cell-derived technologies for hepatocellular carcinoma. Stem Cells Cloning 2010, 3, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, L.M.D.F.; Moreira, L.F.P.; Pinto, M.A.; Henriques-Pons, A.; Alves, L.A. Domino hepatocyte transplantation: A therapeutic alternative for the treatment of acute liver failure. Can. J. Gastroenterol. Hepatol. 2018, 2018, 2593745. [Google Scholar] [CrossRef] [Green Version]
- Mito, M.; Kusano, M.; Kawaura, Y. Hepatocyte transplantation in man. Transplant. Proc. 1992, 24, 3052–3053. [Google Scholar] [CrossRef]
- Fox, I.J.; Chowdhury, J.R.; Kaufman, S.S.; Goertzen, T.C.; Chowdhury, N.R.; Warkentin, P.I.; Dorko, K.; Sauter, B.V.; Strom, S.C. Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation. N. Engl. J. Med. 1998, 338, 1422–1426. [Google Scholar] [CrossRef]
- Muraca, M.; Gerunda, G.; Neri, D.; Vilei, M.T.; Granato, A.; Feltracco, P.; Meroni, M.; Giron, G.; Burlina, A.B. Hepatocyte transplantation as a treatment for glycogen storage disease type 1a. Lancet 2002, 359, 317–318. [Google Scholar] [CrossRef]
- Vacanti, J.P.; Kulig, K.M. Liver cell therapy and tissue engineering for transplantation. Semin. Pediatr. Surg. 2014, 23, 150–155. [Google Scholar] [CrossRef]
- Gramignoli, R.; Vosough, M.; Kannisto, K.; Srinivasan, R.C.; Strom, S.C. Clinical hepatocyte transplantation: Practical limits and possible solutions. Eur. Surg. Res. 2015, 54, 162–177. [Google Scholar] [CrossRef]
- Ott, M.; Castell, J.V. Hepatocyte transplantation, a step forward? J. Hepatol. 2019, 70, 1049–1050. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.; Groyer-Picard, M.T.; Franco, D.; Dagher, I. Hepatocyte transplantation in animal models. Liver Transpl. 2009, 15, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Torimura, T.; Sakamoto, M.; Hashimoto, O.; Taniguchi, E.; Inoue, K.; Sakata, R.; Kumashiro, R.; Murohara, T.; Ueno, T.; et al. Significance and therapeutic potential of endothelial progenitor cell transplantation in a cirrhotic liver rat model. Gastroenterology 2007, 133, 91–107. [Google Scholar] [CrossRef]
- El-Ansary, M.; Abdel-Aziz, I.; Mogawer, S.; Abdel-Hamid, S.; Hammam, O.; Teaema, S.; Wahdan, M. Phase II trial: Undifferentiated versus differentiated autologous mesenchymal stem cells transplantation in egyptian patients with HCV induced liver cirrhosis. Stem Cell Rev. Rep. 2012, 8, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Mohamadnejad, M.; Alimoghaddam, K.; Bagheri, M.; Ashrafi, M.; Abdollahzadeh, L.; Akhlaghpoor, S.; Bashtar, M.; Ghavamzadeh, A.; Malekzadeh, R. Randomized placebo-controlled trial of mesenchymal stem cell transplantation in decompensated cirrhosis. Liver Int. 2013, 33, 1490–1496. [Google Scholar] [CrossRef]
- Rao, M. Scalable human ES culture for therapeutic use: Propagation, differentiation, genetic modification and regulatory issues. Gene Ther. 2008, 15, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Dashtban, M.; Panchalingam, K.M.; Shafa, M.; Baghbaderani, B.A. Addressing manufacturing challenges for commercialization of iPSC-based therapies. Methods Mol. Biol. 2020, in press. [Google Scholar] [CrossRef]
- Badylak, S.F.; Taylor, D.; Uygun, K. Whole-organ tissue engineering: Decellularization and recellularization of three-dimensional matrix scaffolds. Annu. Rev. Biomed. Eng. 2011, 13, 27–53. [Google Scholar] [CrossRef] [PubMed]
- Uygun, B.E.; Izamis, M.L.; Jaramillo, M.; Chen, Y.; Price, G.; Ozer, S.; Yarmush, M.L. Discarded livers find a new life: Engineered liver grafts using hepatocytes recovered from marginal livers. Artif. Organs 2017, 41, 579–585. [Google Scholar] [CrossRef]
- Uygun, B.E.; Soto-Gutierrez, A.; Yagi, H.; Izamis, M.L.; Guzzardi, M.A.; Shulman, C.; Milwid, J.; Kobayashi, N.; Tilles, A.; Berthiaume, F.; et al. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat. Med. 2010, 16, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.A.; Quintanilha, L.F.; Nonaka, C.K.V.; Souza, B.S.F. Advances in hepatic tissue bioengineering with decellularized liver bioscaffold. Stem Cells Int. 2019, 2019, 2693189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Yan, S.; Gao, J.J.; Wang, Y.Y.; Lu, Z.J.; Cui, C.W.; Zhang, Y.H.; Wang, Y.; Meng, X.Q.; Zhou, L.; et al. In-vivo organ engineering: Perfusion of hepatocytes in a single liver lobe scaffold of living rats. Int. J. Biochem. Cell Biol. 2016, 80, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Fukumitsu, K.; Fukuda, K.; Kitago, M.; Shinoda, M.; Obara, H.; Itano, O.; Kawachi, S.; Tanabe, M.; Coudriet, G.M.; et al. Human-scale whole-organ bioengineering for liver transplantation: A regenerative medicine approach. Cell Transplant. 2013, 22, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Mirmalek-Sani, S.H.; Sullivan, D.C.; Zimmerman, C.; Shupe, T.D.; Petersen, B.E. Immunogenicity of decellularized porcine liver for bioengineered hepatic tissue. Am. J. Pathol. 2013, 183, 558–565. [Google Scholar] [CrossRef] [Green Version]
- Sabetkish, S.; Kajbafzadeh, A.M.; Sabetkish, N.; Khorramirouz, R.; Akbarzadeh, A.; Seyedian, S.L.; Pasalar, P.; Orangian, S.; Beigi, R.S.; Aryan, Z.; et al. Whole-organ tissue engineering: Decellularization and recellularization of three-dimensional matrix liver scaffolds. J. Biomed. Mater. Res. A 2015, 103, 1498–1508. [Google Scholar] [CrossRef]
- Mazza, G.; Al-Akkad, W.; Telese, A.; Longato, L.; Urbani, L.; Robinson, B.; Hall, A.; Kong, K.; Frenguelli, L.; Marrone, G.; et al. Rapid production of human liver scaffolds for functional tissue engineering by high shear stress oscillation-decellularization. Sci. Rep. 2017, 7, 5534. [Google Scholar] [CrossRef]
- Mazza, G.; Rombouts, K.; Hall, A.R.; Urbani, L.; Luong, T.V.; Al-Akkad, W.; Longato, L.; Brown, D.; Maghsoudlou, P.; Dhillon, A.P.; et al. Decellularized human liver as a natural 3D-scaffold for liver bioengineering and transplantation. Sci. Rep. 2015, 5, 13079. [Google Scholar] [CrossRef]
- Jiang, W.C.; Cheng, Y.H.; Yen, M.H.; Chang, Y.; Yang, V.W.; Lee, O.K. Cryo-chemical decellularization of the whole liver for mesenchymal stem cells-based functional hepatic tissue engineering. Biomaterials 2014, 35, 3607–3617. [Google Scholar] [CrossRef]
- Wu, J.; Izpisua Belmonte, J.C. Interspecies chimeric complementation for the generation of functional human tissues and organs in large animal hosts. Transgenic Res. 2016, 25, 375–384. [Google Scholar] [CrossRef]
- Nagashima, H.; Matsunari, H. Growing human organs in pigs—A dream or reality? Theriogenology 2016, 86, 422–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldani, G.; Peloso, A.; Lacotte, S.; Meier, R.; Toso, C. Xenogeneic chimera-generated by blastocyst complementation-as a potential unlimited source of recipient-tailored organs. Xenotransplantation 2017, 24, e12327. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Yamaguchi, T.; Hamanaka, S.; Kato-Itoh, M.; Yamazaki, Y.; Ibata, M.; Sato, H.; Lee, Y.S.; Usui, J.; Knisely, A.S.; et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 2010, 142, 787–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usui, J.; Kobayashi, T.; Yamaguchi, T.; Knisely, A.S.; Nishinakamura, R.; Nakauchi, H. Generation of kidney from pluripotent stem cells via blastocyst complementation. Am. J. Pathol. 2012, 180, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Matsunari, H.; Nagashima, H.; Watanabe, M.; Umeyama, K.; Nakano, K.; Nagaya, M.; Kobayashi, T.; Yamaguchi, T.; Sumazaki, R.; Herzenberg, L.A.; et al. Blastocyst complementation generates exogenic pancreas in vivo in apancreatic cloned pigs. Proc. Natl. Acad. Sci. USA 2013, 110, 4557–4562. [Google Scholar] [CrossRef] [Green Version]
- Matsunari, H.; Watanabe, M.; Hasegawa, K.; Uchikura, A.; Nakano, K.; Umeyama, K.; Masaki, H.; Hamanaka, S.; Yamaguchi, T.; Nagaya, M.; et al. Compensation of disabled organogeneses in genetically modified pig fetuses by blastocyst complementation. Stem Cell Rep. 2020, 14, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Yoshitomi, H.; Rossant, J.; Zaret, K.S. Liver organogenesis promoted by endothelial cells prior to vascular function. Science 2001, 294, 559–563. [Google Scholar] [CrossRef]
- Keng, V.; Yagi, H.; Ikawa, M.; Nagano, T.; Myint, Z.; Yamada, K.; Tanaka, T.; Sato, A.; Muramatsu, I.; Okabe, M.; et al. Homeobox gene hex is essential for onset of mouse embryonic liver development and differentiation of the monocyte lineage. Biochem. Biophys. Res. Commun. 2000, 276, 1155–1161. [Google Scholar] [CrossRef]
- Soufi, A.; Jayaraman, P.S. Prh/hex: An oligomeric transcription factor and multifunctional regulator of cell fate. Biochem. J. 2008, 412, 399–413. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Inazu, T.; Yamada, K.; Myint, Z.; Keng, V.W.; Inoue, Y.; Taniguchi, N.; Noguchi, T. cDNA cloning and expression of rat homeobox gene, hex, and functional characteristics of the protein. Biochem. J. 1999, 339, 111–117. [Google Scholar] [CrossRef]
- Kudo, A.; Kim, Y.H.; Irion, S.; Kasuda, S.; Takeuchi, M.; Ohashi, K.; Iwano, M.; Dohi, Y.; Saito, Y.; Snodgrass, R.; et al. The homeobox gene Hex regulates hepatocyte differentiation from embryonic stem cell-derived endoderm. Hepatology 2010, 51, 633–641. [Google Scholar]
- Bort, R.; Signore, M.; Tremblay, K.; Martinez Barbera, J.P.; Zaret, K.S. Hex homeobox gene controls the transition of the endoderm to a pseudostratified, cell emergent epithelium for liver bud development. Dev. Biol. 2006, 290, 44–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez Barbera, J.P.; Clements, M.; Thomas, P.; Rodriguez, T.; Meloy, D.; Kioussis, D.; Beddington, R.S. The homeobox gene hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development 2000, 127, 2433–2445. [Google Scholar]
- Paz, H.; Lynch, M.R.; Bogue, C.W.; Gasson, J.C. The homeobox gene hhex regulates the earliest stages of definitive hematopoiesis. Blood 2010, 116, 1254–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonnell, A.M.; Dang, C.H. Basic review of the cytochrome P450 system. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar]
- Iyanagi, T. Molecular mechanism of phase I and phase II drug-metabolizing enzymes: Implications for detoxification. Int. Rev. Cytol. 2007, 260, 35–112. [Google Scholar]
- Lakehal, F.; Wendum, D.; Barbu, V.; Becquemont, L.; Poupon, R.; Balladur, P.; Hannoun, L.; Ballet, F.; Beaune, P.H.; Housset, C. Phase I and phase II drug-metabolizing enzymes are expressed and heterogeneously distributed in the biliary epithelium. Hepatology 1999, 30, 1498–1506. [Google Scholar] [CrossRef]
- Prasad, B.; Bhatt, D.K.; Johnson, K.; Chapa, R.; Chu, X.; Salphati, L.; Xiao, G.; Lee, C.; Hop, C.E.C.A.; Mathias, A.; et al. Abundance of phase 1 and 2 drug-metabolizing enzymes in alcoholic and hepatitis C cirrhotic livers: A quantitative targeted proteomics study. Drug Metab. Dispos. 2018, 46, 943–952. [Google Scholar] [CrossRef]
- Swift, B.; Pfeifer, N.D.; Brouwer, K.L. Sandwich-cultured hepatocytes: An in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab. Rev. 2010, 42, 446–471. [Google Scholar] [CrossRef] [Green Version]
- Brandon, E.F.; Raap, C.D.; Meijerman, I.; Beijnen, J.H.; Schellens, J.H. An update on in vitro test methods in human hepatic drug biotransformation research: Pros and cons. Toxicol. Appl. Pharmacol. 2003, 189, 233–246. [Google Scholar] [CrossRef]
- Andersson, T.B.; Kanebratt, K.P.; Kenna, J.G. The HepaRG cell line: A unique in vitro tool for understanding drug metabolism and toxicology in human. Expert Opin. Drug Metab. Toxicol. 2012, 8, 909–920. [Google Scholar] [CrossRef]
- Kvist, A.; Kanebratt, K.P.; Walentinsson, A.; Palmgren, H.; O’Hara, M.; Björkbom, A.; Andersson, L.C.; Ahlqvist, M.; Andersson, T.B. Critical differences in drug metabolic properties of human hepatic cellular models, including primary human hepatocytes, stem cell derived hepatocytes, and hepatoma cell lines. Biochem. Pharmacol. 2018, 155, 124–140. [Google Scholar] [CrossRef]
- Nakamura, N.; Saeki, K.; Mitsumoto, M.; Matsuyama, S.; Nishio, M.; Saeki, K.; Hasegawa, M.; Miyagawa, Y.; Ohkita, H.; Kiyokawa, N.; et al. Feeder-free and serum-free production of hepatocytes, cholangiocytes, and their proliferating progenitors from human pluripotent stem cells: Application to liver-specific functional and cytotoxic assays. Cell Reprogram. 2012, 14, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Zeilinger, K.; Freyer, N.; Damm, G.; Seehofer, D.; Knöspel, F. Cell sources for in vitro human liver cell culture models. Exp. Biol. Med. 2016, 241, 1684–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauschke, V.M.; Hendriks, D.F.G.; Bell, C.C.; Andersson, T.B.; Ingelman-Sundberg, M. Novel 3D culture systems for studies of human liver function and assessments of the hepatotoxicity of drugs and drug candidates. Chem. Res. Toxicol. 2016, 29, 1936–1955. [Google Scholar] [CrossRef] [PubMed]
- Parmentier, Y.; Bossant, M.-J.; Bertrand, M.; Walther, B. In vitro studies of drug metabolism. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 231–257. [Google Scholar]
- Katoh, M.T.C.; Yoshizato, K.; Yokoi, T. Chimeric mice with humanized liver. Toxicolgy 2008, 246, 9–17. [Google Scholar] [CrossRef]
- Shen, H.W.; Jiang, X.L.; Gonzalez, F.J.; Yu, A.M. Humanized transgenic mouse models for drug metabolism and pharmacokinetic research. Curr. Drug Metab. 2011, 12, 997–1006. [Google Scholar] [CrossRef]
- Gotoh, O. Evolution of cytochrome P450 genes from the viewpoint of genome informatics. Biol. Pharm. Bull. 2012, 35, 812–817. [Google Scholar] [CrossRef] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Villeneuve, J.P.; Pichette, V. Cytochrome P450 and liver diseases. Curr. Drug Metab. 2004, 5, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.F.; Zgheib, N.K.; Matzke, G.R.; Chaves-Gnecco, D.; Rabinovitz, M.; Shaikh, O.S.; Branch, R.A. Liver disease selectively modulates cytochrome P450-mediated metabolism. Clin. Pharmacol. Ther. 2006, 80, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Murray, M. P450 enzymes. Inhibition mechanisms, genetic regulation and effects of liver disease. Clin. Pharmacokinet. 1992, 23, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wen, Q.; Li, S.F.; Zhang, Y.F.; Gao, N.; Tian, X.; Fang, Y.; Gao, J.; Cui, M.Z.; He, X.P.; et al. Significant change of cytochrome P450s activities in patients with hepatocellular carcinoma. Oncotarget 2016, 7, 50612–50623. [Google Scholar] [CrossRef]
- Seitz, H.K. The role of cytochrome P4502E1 in the pathogenesis of alcoholic liver disease and carcinogenesis. Chem. Biol. Interact. 2020, 316, 108918. [Google Scholar] [CrossRef]
- Fisher, C.D.; Lickteig, A.J.; Augustine, L.M.; Ranger-Moore, J.; Jackson, J.P.; Ferguson, S.S.; Cherrington, N.J. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab. Dispos. 2009, 37, 2087–2094. [Google Scholar] [CrossRef] [Green Version]
- Uehara, S.; Uno, Y.; Yamazaki, H. The marmoset cytochrome P450 superfamily: Sequence/phylogenetic analyses, genomic structure, and catalytic function. Biochem. Pharmacol. 2020, 171, 113721. [Google Scholar] [CrossRef]
- Sakuma, T.; Igarashi, T.; Hieda, M.; Ohgiya, S.; Isogai, M.; Ninomiya, S.; Nagata, R.; Nemoto, N.; Kamataki, T. Marmoset CYP1A2: Primary structure and constitutive expression in livers. Carcinogenesis 1997, 18, 1985–1991. [Google Scholar] [CrossRef] [Green Version]
- Schulz, T.G.; Thiel, R.; Neubert, D.; Brassil, P.J.; Schulz-Utermoehl, T.; Boobis, A.R.; Edwards, R.J. Assessment of P450 induction in the marmoset monkey using targeted anti-peptide antibodies. Biochim. Biophys. Acta 2001, 1546, 143–155. [Google Scholar] [CrossRef]
- Mayumi, K.; Hanioka, N.; Masuda, K.; Koeda, A.; Naito, S.; Miyata, A.; Narimatsu, S. Characterization of marmoset CYP2B6: cDNA cloning, protein expression and enzymatic functions. Biochem. Pharmacol. 2013, 85, 1182–1194. [Google Scholar] [CrossRef]
- Narimatsu, S.; Nakata, T.; Shimizudani, T.; Nagaoka, K.; Nakura, H.; Masuda, K.; Katsu, T.; Koeda, A.; Naito, S.; Yamano, S.; et al. Regio- and stereoselective oxidation of propranolol enantiomers by human CYP2D6, cynomolgus monkey CYP2D17 and marmoset CYP2D19. Chem. Biol. Interact. 2011, 189, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Uno, Y.; Hagihira, Y.; Murayama, N.; Shimizu, M.; Inoue, T.; Sasaki, E.; Yamazaki, H. Marmoset cytochrome P450 2D8 in livers and small intestines metabolizes typical human P450 2D6 substrates, metoprolol, bufuralol and dextromethorphan. Xenobiotica 2015, 45, 766–772. [Google Scholar] [CrossRef]
- Schulz, T.G.; Thiel, R.; Davies, D.S.; Edwards, R.J. Identification of CYP2E1 in marmoset monkey. Biochim. Biophys. Acta 1998, 1382, 287–294. [Google Scholar] [CrossRef]
- Uehara, S.; Uno, Y.; Inoue, T.; Okamoto, E.; Sasaki, E.; Yamazaki, H. Marmoset cytochrome P450 2J2 mainly expressed in small intestines and livers effectively metabolizes human P450 2J2 probe substrates, astemizole and terfenadine. Xenobiotica 2016, 46, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Uno, Y.; Nakanishi, K.; Ishii, S.; Inoue, T.; Sasaki, E.; Yamazaki, H. Marmoset cytochrome P450 3A4 ortholog expressed in liver and small-intestine tissues efficiently metabolizes midazolam, alprazolam, nifedipine, and testosterone. Drug Metab. Disp. 2017, 45, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Uehara, S.; Uno, Y.; Ishii, S.; Inoue, T.; Sasaki, E.; Yamazaki, H. Marmoset cytochrome P450 4A11, a novel arachidonic acid and lauric acid ω-hydroxylase expressed in liver and kidney tissues. Xenobiotica 2017, 47, 553–561. [Google Scholar] [CrossRef]
- Uehara, S.; Uno, Y.; Yuki, Y.; Inoue, T.; Sasaki, E.; Yamazaki, H. A new marmoset P450 4F12 enzyme expressed in small intestines and livers efficiently metabolizes antihistaminic drug ebastine. Drug Metab. Disp. 2016, 44, 833–841. [Google Scholar] [CrossRef] [Green Version]
- Uehara, S.; Uno, Y.; Inoue, T.; Sasaki, E.; Yamazaki, H. Cloning and tissue expression of cytochrome P450 2S1, 4V2, 7A1, 7B1, 8B1, 24A1, 26A1, 26C1, 27A1, 39A1, and 51A1 in marmosets. Drug Metab. Pharmacokinet. 2020, 35, 244–247. [Google Scholar] [CrossRef]
- Schulz, T.G.; Neubert, D.; Davies, D.S.; Edwards, R.J. Induction of cytochromes P450 by dioxins in liver and lung of marmoset monkeys (Callithrix jacchus). Adv. Exp. Med. Biol. 1996, 387, 443–446. [Google Scholar]
- Uehara, S.; Uno, Y.; Suzuki, T.; Inoue, T.; Utoh, M.; Sasaki, E.; Yamazaki, H. Strong induction of cytochrome P450 1A/3A, but not P450 2B, in cultured hepatocytes from common marmosets and cynomolgus monkeys by typical human P450 inducing agents. Drug Metab. Lett. 2017, 10, 244–253. [Google Scholar] [CrossRef]
- Bhattacharya, C.; Kirby, D.; Van Stipdonk, M.; Stratford, R.E. Comparison of in vitro stereoselective metabolism of bupropion in human, monkey, rat, and mouse liver microsomes. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.F.; Neiner, A.; Kharasch, E.D. Stereoselective bupropion hydroxylation by cytochrome P450 CYP2B6 and cytochrome P450 oxidoreductase genetic variants. Drug Metab. Disp. 2020, 48, 438–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uehara, S.; Murayama, N.; Yamazaki, H.; Suemizu, H. Regioselective hydroxylation of an antiarrhythmic drug, propafenone, mediated by rat liver cytochrome P450 2D2 differs from that catalyzed by human P450 2D6. Xenobiotica 2019, 49, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Bammler, T.K.; Slone, D.H.; Eaton, D.L. Effects of dietary oltipraz and ethoxyquin on aflatoxin B1 biotransformation in non-human primates. Toxicol. Sci. 2000, 54, 30–41. [Google Scholar] [CrossRef]
- Sridhar, J.; Goyal, N.; Liu, J.; Foroozesh, M. Review of ligand specificity factors for CYP1A subfamily enzymes from molecular modeling studies reported to-date. Molecules 2017, 22, 1143. [Google Scholar] [CrossRef] [Green Version]
- Sevrioukova, I.F.; Poulos, T.L. Current approaches for investigating and predicting cytochrome P450 3A4-ligand interactions. Adv. Exp. Med. Biol. 2015, 851, 83–105. [Google Scholar]
- Novak, R.F.; Woodcroft, K.J. The alcohol-inducible form of cytochrome P450 (CYP2E1): Role in toxicology and regulation of expression. Arch. Pharm. Res. 2000, 23, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Ha, S.K.; Choi, Y.; Akbar, M.; Song, B.J. Role of cyp2e1 in mitochondrial dysfunction and hepatic injury by alcohol and non-alcoholic substances. Curr. Mol. Pharmacol. 2017, 10, 207–225. [Google Scholar] [CrossRef] [Green Version]
- Sim, E.; Lack, N.; Wang, C.J.; Long, H.; Westwood, I.; Fullam, E.; Kawamura, A. Arylamine N-acetyltransferases: Structural and functional implications of polymorphisms. Toxicology 2008, 254, 170–183. [Google Scholar] [CrossRef]
- Martell, K.J.; Levy, G.N.; Weber, W.W. Cloned mouse N-acetyltransferases: Enzymatic properties of expressed NAT-1 and NAT-2 gene products. Mol. Pharmacol. 1992, 42, 265–272. [Google Scholar]
- Jones, R.F.; Land, S.J.; King, C.M. Recombinant rat and hamster N-acetyltransferases-1 and -2: Relative rates of N-acetylation of arylamines and N,O-acyltransfer with arylhydroxamic acids. Carcinogenesis 1996, 17, 1729–1733. [Google Scholar] [CrossRef] [PubMed]
- Uno, Y.; Murayama, N.; Yamazaki, H. Molecular and functional characterization of N-acetyltransferases NAT1 and NAT2 in cynomolgus macaque. Chem. Res. Toxicol. 2018, 31, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Uno, Y.; Murayama, N.; Yamazaki, H. Genetic variants of N-acetyltransferases 1 and 2 (NAT1 and NAT2) in cynomolgus and rhesus macaques. Biochem. Pharmacol. 2020, 177, 113996. [Google Scholar] [CrossRef]
- The Marmoset Genome Sequencing and Analysis Consortium. The common marmoset genome provides insight into primate biology and evolution. Nat. Genet. 2014, 46, 850–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meech, R.; Mackenzie, P.I. Structure and function of uridine diphosphate glucuronosyltransferases. Clin. Exp. Pharmacol. Physiol. 1997, 24, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, P.I.; Bock, K.W.; Burchell, B.; Guillemette, C.; Ikushiro, S.; Iyanagi, T.; Miners, J.O.; Owens, I.S.; Nebert, D.W. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet. Genom. 2005, 15, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Strassburg, C.P.; Kalthoff, S.; Ehmer, U. Variability and function of family 1 uridine-5’-diphosphate glucuronosyltransferases (UGT1A). Crit Rev. Clin. Lab. Sci. 2008, 45, 485–530. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Rahim, F. Crigler-Najjar syndrome: Current perspectives and the application of clinical genetics. Endocr. Metab. Immune. Disord. Drug Targets 2018, 18, 201–211. [Google Scholar] [CrossRef]
- Soars, M.G.; Riley, R.J.; Burchell, B. Evaluation of the marmoset as a model species for drug glucuronidation. Xenobiotica 2001, 31, 849–860. [Google Scholar] [CrossRef]
- Uno, Y.; Uehara, S.; Inoue, T.; Kawamura, S.; Murayama, N.; Nishikawa, M.; Ikushiro, S.; Sasaki, E.; Yamazaki, H. Molecular characterization of functional UDP-glucuronosyltransferases 1A and 2B in common marmosets. Biochem. Pharmacol. 2020, 172, 113748. [Google Scholar] [CrossRef]
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II drug metabolizing enzymes. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 2010, 154, 103–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uno, Y.; Uehara, S.; Tanaka, S.; Murayama, N.; Yamazaki, H. Systematic characterization of glutathione S-transferases in common marmosets. Biochem. Pharmacol. 2020, 174, 113835. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.G.; Wiebel, F.A.; Thier, R.; Neubert, D.; Davies, D.S.; Edwards, R.J. Identification of theta-class glutathione S-transferase in liver cytosol of the marmoset monkey. Arch. Toxicol. 2000, 74, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Lautala, P.; Ulmanen, I.; Taskinen, J. Molecular mechanisms controlling the rate and specificity of catechol O-methylation by human soluble catechol O-methyltransferase. Mol. Pharmacol. 2001, 59, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Uehara, S.; Uno, Y.; Inoue, T.; Sasaki, E.; Yamazaki, H. Cloning and expression of a novel catechol-O-methyltransferase in common marmosets. J. Vet. Med. Sci. 2017, 79, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, T.E.; Colman, R.J.; Tardif, S.D.; Sosa, M.E.; Wegner, F.H.; Wittwer, D.J.; Shrestha, H. Development of metabolic function biomarkers in the common marmoset, Callithrix jacchus. Am. J. Primatol. 2013, 75, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Nii, T.; Marumoto, T.; Kohara, H.; Yamaguchi, S.; Kawano, H.; Sasaki, E.; Kametani, Y.; Tani, K. Improved hematopoietic differentiation of primate embryonic stem cells by inhibition of the PI3K-AKT pathway under defined conditions. Exp. Hematol. 2015, 43, 901–911. [Google Scholar] [CrossRef]
- Sato, K.; Oiwa, R.; Kumita, W.; Henry, R.; Sakuma, T.; Ito, R.; Nozu, R.; Inoue, T.; Katano, I.; Sato, K.; et al. Generation of a nonhuman primate model of severe combined immunodeficiency using highly efficient genome editing. Cell Stem Cell 2016, 19, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhang, Y.; Mishra, A.; Tardif, S.D.; Hornsby, P.J. Generation of induced pluripotent stem cells from newborn marmoset skin fibroblasts. Stem Cell Res. 2010, 4, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Tomioka, I.; Maeda, T.; Shimada, H.; Kawai, K.; Okada, Y.; Igarashi, H.; Oiwa, R.; Iwasaki, T.; Aoki, M.; Kimura, T.; et al. Generating induced pluripotent stem cells from common marmoset (Callithrix jacchus) fetal liver cells using defined factors, including lin28. Genes Cells 2010, 15, 959–969. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, A.; Hemmer, K.; Bernemann, I.; Göhring, G.; Pogozhykh, O.; Figueiredo, C.; Glage, S.; Schambach, A.; Schwamborn, J.C.; Blasczyk, R.; et al. Induced pluripotent stem cells generated from adult bone marrow-derived cells of the nonhuman primate (Callithrix jacchus) using a novel quad-cistronic and excisable lentiviral vector. Cell Reprogram. 2012, 14, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debowski, K.; Warthemann, R.; Lentes, J.; Salinas-Riester, G.; Dressel, R.; Langenstroth, D.; Gromoll, J.; Sasaki, E.; Behr, R. Non-viral generation of marmoset monkey iPS cells by a six-factor-in-one-vector approach. PLoS ONE 2015, 10, e0118424. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Jing, R.; Rao, Q.; Zhang, L.; Gao, Y.; Liu, F.; Wang, X.; Hui, L.; Yin, H. Immortalized common marmoset (Callithrix jacchus) hepatic progenitor cells possess bipotentiality in vitro and in vivo. Cell Discov. 2018, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Erker, L.; Grompe, M. Signaling networks in hepatic oval cell activation. Stem Cell Res. 2007, 1, 90–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivertsson, L.; Synnergren, J.; Jensen, J.; Björquist, P.; Ingelman-Sundberg, M. Hepatic differentiation and maturation of human embryonic stem cells cultured in a perfused three-dimensional bioreactor. Stem Cells Dev. 2013, 22, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Miki, T.; Ring, A.; Gerlach, J. Hepatic differentiation of human embryonic stem cells is promoted by three-dimensional dynamic perfusion culture conditions. Tissue Eng. Part. C Methods 2011, 17, 557–568. [Google Scholar] [CrossRef]
- Goulart, E.; de Caires-Junior, L.C.; Telles-Silva, K.A.; Araujo, B.H.S.; Rocco, S.A.; Sforca, M.; de Sousa, I.L.; Kobayashi, G.S.; Musso, C.M.; Assoni, A.F.; et al. 3D bioprinting of liver spheroids derived from human induced pluripotent stem cells sustain liver function and viability in vitro. Biofabrication 2019, 12, 015010. [Google Scholar] [CrossRef]
- Ma, X.; Qu, X.; Zhu, W.; Li, Y.S.; Yuan, S.; Zhang, H.; Liu, J.; Wang, P.; Lai, C.S.; Zanella, F.; et al. Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting. Proc. Natl. Acad. Sci. USA 2016, 113, 2206–2211. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aravalli, R.N.; Steer, C.J. Utility of Common Marmoset (Callithrix jacchus) Embryonic Stem Cells in Liver Disease Modeling, Tissue Engineering and Drug Metabolism. Genes 2020, 11, 729. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11070729
Aravalli RN, Steer CJ. Utility of Common Marmoset (Callithrix jacchus) Embryonic Stem Cells in Liver Disease Modeling, Tissue Engineering and Drug Metabolism. Genes. 2020; 11(7):729. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11070729
Chicago/Turabian StyleAravalli, Rajagopal N., and Clifford J. Steer. 2020. "Utility of Common Marmoset (Callithrix jacchus) Embryonic Stem Cells in Liver Disease Modeling, Tissue Engineering and Drug Metabolism" Genes 11, no. 7: 729. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11070729