Whole Mitochondrial Genome Analysis in Serbian Cases of Leber’s Hereditary Optic Neuropathy

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. DNA Extraction
2.3. DNA Amplification by Polymerase Chain Reaction
2.4. Capillary Electrophoresis
2.5. Bioinformatics Analysis
2.6. Haplogroup Analysis
3. Results
3.1. Determination of Primary and Secondary LHON Pathogenic Mutations
3.2. Bioinformatics Analysis for LHON Associated Mutations
3.3. MITOMASTER Analysis for Associated Mitochondrial Haplotypes
3.4. Phenotypic Characterization in Symptomatic and Asymptomatic Subjects
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Borrelli, E.; Triolo, G.; Cascavilla, M.L.; La Morgia, C.; Rizzo, G.; Savini, G.; Balducci, N.; Nucci, P.; Giglio, R.; Darvizeh, F.; et al. Changes in Choroidal Thickness follow the RNFL Changes in Leber’s Hereditary Optic Neuropathy. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, W. The foundation of experimental ophthalmology by Theodor Leber. Doc. Ophthalmol. 1988, 68, 71–77. [Google Scholar] [CrossRef]
- Khan, N.A.; Govindaraj, P.; Soumittra, N.; Sharma, S.; Srilekha, S.; Ambika, S.; Vanniarajan, A.; Meena, A.K.; Uppin, M.S.; Sundaram, C.; et al. Author Response: Penetrance of the LHON Mutation m.11778G>A May Depend on Factors Other Than Haplotype or Heteroplasmy Rate. Investig. Ophthalmol. Vis. Sci. 2018, 59, 382. [Google Scholar] [CrossRef]
- Van der Walt, E.M.; Smuts, I.; Taylor, R.W.; Elson, J.L.; Turnbull, D.M.; Louw, R.; Van der Westhuizen, F.H. Characterization of mtDNA variation in a cohort of South African paediatric patients with mitochondrial disease. Eur. J. Hum. Genet. 2012, 20, 650–656. [Google Scholar] [CrossRef] [Green Version]
- Erickson, R.P. Leber’s optic atrophy, a possible example of maternal inheritance. Am. J. Hum. Genet. 1972, 24, 348–349. [Google Scholar]
- Formosa, L.E.; Dibley, M.G.; Stroud, D.A.; Ryan, M.T. Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin. Cell Dev. Biol. 2018, 76, 154–162. [Google Scholar] [CrossRef]
- Nass, M.M.K.; Nass, S. Intramitochondrial fibers with DNA characteristics. J. Cell Biol. 1963, 19, 593–611. [Google Scholar] [CrossRef] [Green Version]
- Leone, G.; Abla, H.; Gasparre, G.; Porcelli, A.; Iommarini, L. The Oncojanus Paradigm of Respiratory Complex I. Genes 2018, 9, 243. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Kwok, S.S.; Chan, Y.K.; Ming Lai, J.S.; Pan, W.; Nie, L.; Shih, K.C. Therapeutic Strategies for Attenuation of Retinal Ganglion Cell Injury in Optic Neuropathies: Concepts in Translational Research and Therapeutic Implications. BioMed Res. Int. 2019, 2019, 8397521. [Google Scholar] [CrossRef] [Green Version]
- Mansergh, F.C.; Chadderton, N.; Kenna, P.F.; Gobbo, O.L.; Farrar, G.J. Cell therapy using retinal progenitor cells shows therapeutic effect in a chemically-induced rotenone mouse model of Leber hereditary optic neuropathy. Eur. J. Hum. Genet. 2014, 22, 1314–1320. [Google Scholar] [CrossRef]
- Falabella, M.; Forte, E.; Magnifico, M.C.; Santini, P.; Arese, M.; Giuffrè, A.; Radić, K.; Chessa, L.; Coarelli, G.; Buscarinu, M.C.; et al. Evidence for Detrimental Cross Interactions between Reactive Oxygen and Nitrogen Species in Leber’s Hereditary Optic Neuropathy Cells. Available online: https://new.hindawi.com/journals/omcl/2016/3187560/ (accessed on 19 January 2020).
- Carelli, V. Optic nerve degeneration and mitochondrial dysfunction: Genetic and acquired optic neuropathies. Neurochem. Int. 2002, 40, 573–584. [Google Scholar] [CrossRef]
- Catarino, C.B.; Ahting, U.; Gusic, M.; Iuso, A.; Repp, B.; Peters, K.; Biskup, S.; Von Livonius, B.; Prokisch, H.; Klopstock, T. Characterization of a Leber’s hereditary optic neuropathy (LHON) family harboring two primary LHON mutations m.11778G>A and m.14484T>C of the mitochondrial DNA. Mitochondrion 2017, 36, 15–20. [Google Scholar] [CrossRef]
- Emperador, S.; Vidal, M.; Hernández-Ainsa, C.; Ruiz-Ruiz, C.; Woods, D.; Morales-Becerra, A.; Arruga, J.; Artuch, R.; López-Gallardo, E.; Bayona-Bafaluy, M.P.; et al. The Decrease in Mitochondrial DNA Mutation Load Parallels Visual Recovery in a Leber Hereditary Optic Neuropathy Patient. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Gan, D.; Li, M.; Wu, J.; Sun, X.; Tian, G. Analysis of Genetic Mutations in a Cohort of Hereditary Optic Neuropathy in Shanghai, China. Available online: https://new.hindawi.com/journals/joph/2017/6186052/ (accessed on 19 January 2020).
- Shemesh, A.; Margolin, E. Leber Optic Atrophy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Maass, J.; Matthé, E. Bilateral vision loss due to Leber’s hereditary optic neuropathy after long-term alcohol, nicotine and drug abuse. Doc. Ophthalmol. 2018, 136, 145–153. [Google Scholar] [CrossRef]
- Kodroń, A.; Krawczyński, M.R.; Tońska, K.; Bartnik, E. m.3635G>;A mutation as a cause of Leber hereditary optic neuropathy. J. Clin. Pathol. 2014, 67, 639–641. [Google Scholar] [CrossRef]
- Kim, J.Y.; Hwang, J.-M.; Park, S.S. Mitochondrial DNA C4171A/ND1 is a novel primary causative mutation of Leber’s hereditary optic neuropathy with a good prognosis. Ann. Neurol. 2002, 51, 630–634. [Google Scholar] [CrossRef]
- Tavares, W.C.; Seuánez, H.N. Disease-associated mitochondrial mutations and the evolution of primate mitogenomes. PLoS ONE 2017, 12, e0177403. [Google Scholar] [CrossRef]
- Brown, M.D.; Starikovskaya, E.; Derbeneva, O.; Hosseini, S.; Allen, J.C.; Mikhailovskaya, I.E.; Sukernik, R.I.; Wallace, D.C. The role of mtDNA background in disease expression: A new primary LHON mutation associated with Western Eurasian haplogroup. J. Hum. Genet. 2002, 110, 130–138. [Google Scholar] [CrossRef]
- Zhao, F.; Guan, M.; Zhou, X.; Yuan, M.; Liang, M.; Liu, Q.; Liu, Y.; Zhang, Y.; Yang, L.; Tong, Y.; et al. Leber’s hereditary optic neuropathy is associated with mitochondrial ND6 T14502C mutation. Biochem. Biophys. Res. Commun. 2009, 389, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Krylova, T.D.; Sheremet, N.L.; Tabakov, V.Y.; Lyamzaev, K.G.; Itkis, Y.S.; Tsygankova, P.G.; Andreeva, N.A.; Shmelkova, M.S.; Nevinitsyna, T.A.; Kadyshev, V.V.; et al. Three rare pathogenic mtDNA substitutions in LHON patients with low heteroplasmy. Mitochondrion 2020, 50, 139–144. [Google Scholar] [CrossRef]
- López-Gallardo, E.; Emperador, S.; Hernández-Ainsa, C.; Montoya, J.; Bayona-Bafaluy, M.P.; Ruiz-Pesini, E. Food derived respiratory complex I inhibitors modify the effect of Leber hereditary optic neuropathy mutations. Food Chem. Toxicol. 2018, 120, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rong, E.; Wang, H.; Hao, S.; Fu, Y.; Ma, Y.; Wang, T. Heteroplasmy Detection of Mitochondrial DNA A3243G Mutation Using Quantitative Real-Time PCR Assay Based on TaqMan-MGB Probes. BioMed Res. Int. 2018, 2018, 1286480. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Valletti, A.; Longo, G.; Bisceglia, L.; Montoya, J.; Emperador, S.; Guerriero, S.; Petruzzella, V. Mitochondrial DNA copy number in affected and unaffected LHON mutation carriers. BMC Res. Notes 2018, 11, 911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirches, E. LHON: Mitochondrial Mutations and More. Curr Genom. 2011, 12, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Duan, S.; Qin, Y.; Lin, S.; Zheng, K.; Li, X.; Zhang, L.; Gu, X.; Yao, K.; Wang, B. Leber’s Hereditary Optic Neuropathy–Specific Heteroplasmic Mutation m.14495A>G Found in a Chinese Family. Trans. Vis. Sci. Technol. 2019, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ji, Y.; Lu, Y.; Fu, R.; Xu, M.; Liu, X.; Guan, M.-X. Leber’s hereditary optic neuropathy (LHON)-associated ND5 12338T > C mutation altered the assembly and function of complex I, apoptosis and mitophagy. Hum. Mol. Genet. 2018, 27, 1999–2011. [Google Scholar] [CrossRef]
- Fauser, S.; Luberichs, J.; Besch, D.; Leo-Kottler, B. Sequence analysis of the complete mitochondrial genome in patients with Leber’s hereditary optic neuropathy lacking the three most common pathogenic DNA mutations. Biochem. Biophys. Res. Commun. 2002, 295, 342–347. [Google Scholar] [CrossRef]
- Ding, Y.; Ye, Y.-F.; Li, M.-Y.; Xia, B.-H.; Leng, J.-H. Mitochondrial tRNAAla 5601C>T variant may affect the clinical expression of the LHON-related ND4 11778G>A mutation in a family. Mol. Med. Rep. 2020, 21, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Liang, M.; Zhang, J.; Zhu, L.; Zhang, Z.; Fu, R.; Liu, X.; Zhang, M.; Fu, Q.; Zhao, F.; et al. Mitochondrial ND1 Variants in 1281 Chinese Subjects With Leber’s Hereditary Optic Neuropathy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2377–2389. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Qu, J.; Zhou, X.; Tong, Y.; Hu, Y.; Qian, Y.; Lu, F.; Mo, J.Q.; West, C.E.; Guan, M.-X. The mitochondrial tRNAThr A15951G mutation may influence the phenotypic expression of the LHON-associated ND4 G11778A mutation in a Chinese family. Gene 2006, 376, 79–86. [Google Scholar] [CrossRef]
- Qu, J.; Li, R.; Zhou, X.; Tong, Y.; Lu, F.; Qian, Y.; Hu, Y.; Mo, J.Q.; West, C.E.; Guan, M.-X. The Novel A4435G Mutation in the Mitochondrial tRNAMet May Modulate the Phenotypic Expression of the LHON-Associated ND4 G11778A Mutation. Investig. Ophthalmol. Vis. Sci. 2006, 47, 475–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporali, L.; Iommarini, L.; Morgia, C.L.; Olivieri, A.; Achilli, A.; Maresca, A.; Valentino, M.L.; Capristo, M.; Tagliavini, F.; Dotto, V.D.; et al. Peculiar combinations of individually non-pathogenic missense mitochondrial DNA variants cause low penetrance Leber’s hereditary optic neuropathy. PLoS Genet. 2018, 14, e1007210. [Google Scholar] [CrossRef]
- Jancic, J.; Rovcanin, B.; Djuric, V.; Pepic, A.; Samardzic, J.; Nikolic, B.; Novakovic, I.; Kostic, V.S. Analysis of secondary mtDNA mutations in families with Leber’s hereditary optic neuropathy: Four novel variants and their association with clinical presentation. Mitochondrion 2020, 50, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Jančić, J.; Dejanović, I.; Samardžić, J.; Radovanović, S.; Pepić, A.; Kosanović-Jaković, N.; Ćetković, M.; Kostić, V. Leber hereditary optic neuropathy in the population of Serbia. Eur. J. Paediatr. Neurol. 2014, 18, 354–359. [Google Scholar] [CrossRef]
- Taylor, R.W.; Taylor, G.A.; Durham, S.E.; Turnbull, D.M. The determination of complete human mitochondrial DNA sequences in single cells: Implications for the study of somatic mitochondrial DNA point mutations. Nucleic Acids Res. 2001, 29, e74. [Google Scholar] [CrossRef]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Ruiz-Pesini, E.; Lott, M.T.; Procaccio, V.; Poole, J.C.; Brandon, M.C.; Mishmar, D.; Yi, C.; Kreuziger, J.; Baldi, P.; Wallace, D.C. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007, 35, D823–D828. [Google Scholar] [CrossRef] [Green Version]
- Ingman, M. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006, 34, D749–D751. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [Green Version]
- UniProt Consortium. The Universal Protein Resource (UniProt). Nucleic Acids Res. 2007, 35, D193–D197. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandon, M.C.; Ruiz-Pesini, E.; Mishmar, D.; Procaccio, V.; Lott, M.T.; Nguyen, K.C.; Spolim, S.; Patil, U.; Baldi, P.; Wallace, D.C. MITOMASTER—A Bioinformatics Tool For the Analysis of Mitochondrial DNA Sequences. Hum. Mutat. 2009, 30, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Oven, M. Van PhyloTree Build 17: Growing the human mitochondrial DNA tree. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e392–e394. [Google Scholar] [CrossRef] [Green Version]
- Ji, F.; Sharpley, M.S.; Derbeneva, O.; Alves, L.S.; Qian, P.; Wang, Y.; Chalkia, D.; Lvova, M.; Xu, J.; Yao, W.; et al. Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans. Proc. Natl. Acad. Sci. USA 2012, 109, 7391–7396. [Google Scholar] [CrossRef] [Green Version]
- Fiedorczuk, K.; Sazanov, L.A. Mammalian Mitochondrial Complex I Structure and Disease-Causing Mutations. Trends Cell Biol. 2018, 28, 835–867. [Google Scholar] [CrossRef]
- Shinde, S.; Bhadra, U. A complex genome-microRNA interplay in human mitochondria. BioMed Res. Int. 2015, 2015, 206382. [Google Scholar] [CrossRef] [Green Version]
- Mascialino, B.; Leinonen, M.; Meier, T. Meta-analysis of the prevalence of Leber hereditary optic neuropathy mtDNA mutations in Europe. Eur. J. Ophthalmol. 2012, 22, 461–465. [Google Scholar] [CrossRef]
- Ghelli, A.; Porcelli, A.M.; Zanna, C.; Vidoni, S.; Mattioli, S.; Barbieri, A.; Iommarini, L.; Pala, M.; Achilli, A.; Torroni, A.; et al. The Background of Mitochondrial DNA Haplogroup J Increases the Sensitivity of Leber’s Hereditary Optic Neuropathy Cells to 2,5-Hexanedione Toxicity. PLoS ONE 2009, 4, e07922. [Google Scholar] [CrossRef]
- Blanc, C.; Heran, F.; Habas, C.; Bejot, Y.; Sahel, J.; Vignal-Clermont, C. MRI of the Optic Nerves and Chiasm in Patients with Leber Hereditary Optic Neuropathy. J. Neuro-Ophthalmol. 2018, 38, 434–437. [Google Scholar] [CrossRef]
- Brown, M.D.; Torroni, A.; Reckord, C.L.; Wallace, D.C. Phylogenetic analysis of Leber’s hereditary optic neuropathy mitochondrial DNA’s indicates multiple independent occurrences of the common mutations. Hum. Mutat. 2018, 6, 311–325. [Google Scholar] [CrossRef]
- Zhang, M.; Zhou, X.; Li, C.; Zhao, F.; Zhang, J.; Yuan, M.; Sun, Y.-H.; Wang, J.; Tong, Y.; Liang, M.; et al. Mitochondrial haplogroup M9a specific variant ND1 T3394C may have a modifying role in the phenotypic expression of the LHON-associated ND4 G11778A mutation. Mol. Genet. Metab. 2010, 101, 192–199. [Google Scholar] [CrossRef]
- Du, W.-D.; Chen, G.; Cao, H.-M.; Jin, Q.-H.; Liao, R.-F.; He, X.-C.; Chen, D.-B.; Huang, S.-R.; Zhao, H.; Lv, Y.-M. A simple oligonucleotide biochip capable of rapidly detecting known mitochondrial DNA mutations in Chinese patients with Leber’s hereditary optic neuropathy (LHON). Dis. Markers 2011, 30, 181–190. [Google Scholar] [CrossRef]
- Zgonjanin, D.; Veselinović, I.; Kubat, M.; Furač, I.; Antov, M.; Lončar, E.; Tasić, M.; Vuković, R.; Omorjan, R. Sequence polymorphism of the mitochondrial DNA control region in the population of Vojvodina Province, Serbia. Leg. Med. 2010, 12, 104–107. [Google Scholar] [CrossRef]
- Davidovic, S.; Malyarchuk, B.; Aleksic, J.M.; Derenko, M.; Topalovic, V.; Litvinov, A.; Stevanovic, M.; Kovacevic-Grujicic, N. Mitochondrial DNA perspective of Serbian genetic diversity: Serbian mitochondrial DNA diversity. Am. J. Phys. Anthropol. 2015, 156, 449–465. [Google Scholar] [CrossRef]
- Davidovic, S.; Malyarchuk, B.; Grzybowski, T.; Aleksic, J.M.; Derenko, M.; Litvinov, A.; Rogalla-Ładniak, U.; Stevanovic, M.; Kovacevic-Grujicic, N. Complete mitogenome data for the Serbian population: The contribution to high-quality forensic databases. Int. J. Leg. Med. 2020. [Google Scholar] [CrossRef]
- Šarac, J.; Havaš Auguštin, D.; Metspalu, E.; Novokmet, N.; Missoni, S.; Rudan, P. Maternal Genetic Profile of Serbian and Montenegrin Populations from Southeastern Europe. GenApp 2018, 1, 14. [Google Scholar] [CrossRef]
- Herrnstadt, C.; Howell, N. An evolutionary perspective on pathogenic mtDNA mutations: Haplogroup associations of clinical disorders. Mitochondrion 2004, 4, 791–798. [Google Scholar] [CrossRef]
- Man, P.Y.W.; Howell, N.; Mackey, D.A.; Nørby, S.; Rosenberg, T.; Turnbull, D.M.; Chinnery, P.F. Mitochondrial DNA haplogroup distribution within Leber hereditary optic neuropathy pedigrees. J. Med. Genet. 2004, 41, e41. [Google Scholar] [CrossRef] [Green Version]
- Hudson, G.; Carelli, V.; Spruijt, L.; Gerards, M.; Mowbray, C.; Achilli, A.; Pyle, A.; Elson, J.; Howell, N.; La Morgia, C. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA–haplogroup background. Am. J. Hum. Genet. 2007, 81, 228–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Durán, A.; Pacheu-Grau, D.; Martínez-Romero, Í.; López-Gallardo, E.; López-Pérez, M.J.; Montoya, J.; Ruiz-Pesini, E. Oxidative phosphorylation differences between mitochondrial DNA haplogroups modify the risk of Leber’s hereditary optic neuropathy. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1216–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starikovskaya, E.B.; Shalaurova, S.A.; Dryomov, S.V.; Nazhmidenova, A.M.; Volodko, N.V.; Bychkov, I.Y.; Mazunin, I.O.; Sukernik, R.I. Mitochondrial DNA variation of Leber’s Hereditary Optic Neuropathy (LHON) in Western Siberia. bioRxiv 2019, 744219. [Google Scholar] [CrossRef]
- Torroni, A.; Petrozzi, M.; D’Urbano, L.; Sellitto, D.; Zeviani, M.; Carrara, F.; Carducci, C.; Leuzzi, V.; Carelli, V.; Barboni, P.; et al. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am. J. Hum. Genet. 1997, 60, 1107–1121. [Google Scholar]
- Scorrano, G.; Finocchio, A.; Angelis, F.D.; Martínez-Labarga, C.; Šarac, J.; Contini, I.; Scano, G.; Frezza, D.; Novokmet, N.; Rickards, O. The Genetic Landscape of Serbian Populations through Mitochondrial DNA Sequencing and Non-Recombining Region of the Y Chromosome Microsatellites. Coll. Antropol. 2017, 41, 275–296. [Google Scholar]
- Abu-Amero, K.K.; Bosley, T.M. Mitochondrial Abnormalities in Patients with LHON-like Optic Neuropathies. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4211. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Gomez-Duran, A.; Hudson, G.; Chinnery, P.F. Background sequence characteristics influence the occurrence and severity of disease-causing mtDNA mutations. PLoS Genet. 2017, 13, e1007126. [Google Scholar] [CrossRef]
- Dautant, A.; Meier, T.; Hahn, A.; Tribouillard-Tanvier, D.; di Rago, J.-P.; Kucharczyk, R. ATP Synthase Diseases of Mitochondrial Genetic Origin. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef]
- Kahloun, R.; Abroug, N.; Ksiaa, I.; Mahmoud, A.; Zeghidi, H.; Zaouali, S.; Khairallah, M. Infectious optic neuropathies: A clinical update. Eye Brain 2015, 7, 59–81. [Google Scholar] [CrossRef] [Green Version]
- Hoorbakht, H.; Bagherkashi, F. Optic Neuritis, its Differential Diagnosis and Management. Open Ophthalmol. J. 2012, 6, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Korkiamäki, P.; Kervinen, M.; Karjalainen, K.; Majamaa, K.; Uusimaa, J.; Remes, A.M. Prevalence of the primary LHON mutations in Northern Finland associated with bilateral optic atrophy and tobacco-alcohol amblyopia. Acta Ophthalmol. 2013, 91, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Sajjadi, H.; Poorsalman, H. Previously Diagnosed Leber’s Hereditary Optic Neuropathy with Clinical Signs of Idiopathic Intracranial Hypertension Responsive to Acetazolamide Therapy. J. Ophthalmic Vis. Res. 2019, 14, 109–113. [Google Scholar] [CrossRef]
- Kwittken, J.; Barest, H.D. The Neuropathology of Hereditary Optic Atrophy (Leber’s Disease): The First Complete Anatomic Study. Am. J. Pathol. 1958, 34, 185–207. [Google Scholar] [PubMed]
- Leber, T.H. Ueber hereditäre und congenital-angelegte Sehnervenleiden. Graefe’s Arhiv für Ophthalmologie 1871, 17, 249–291. [Google Scholar] [CrossRef]
- Rozen, T.D. Can the effects of the mitochondrial DNA mutations found in Leber’s hereditary optic neuropathy be protective against the development of cluster headache in smokers? Cephalalgia Rep. 2020, 3. [Google Scholar] [CrossRef]



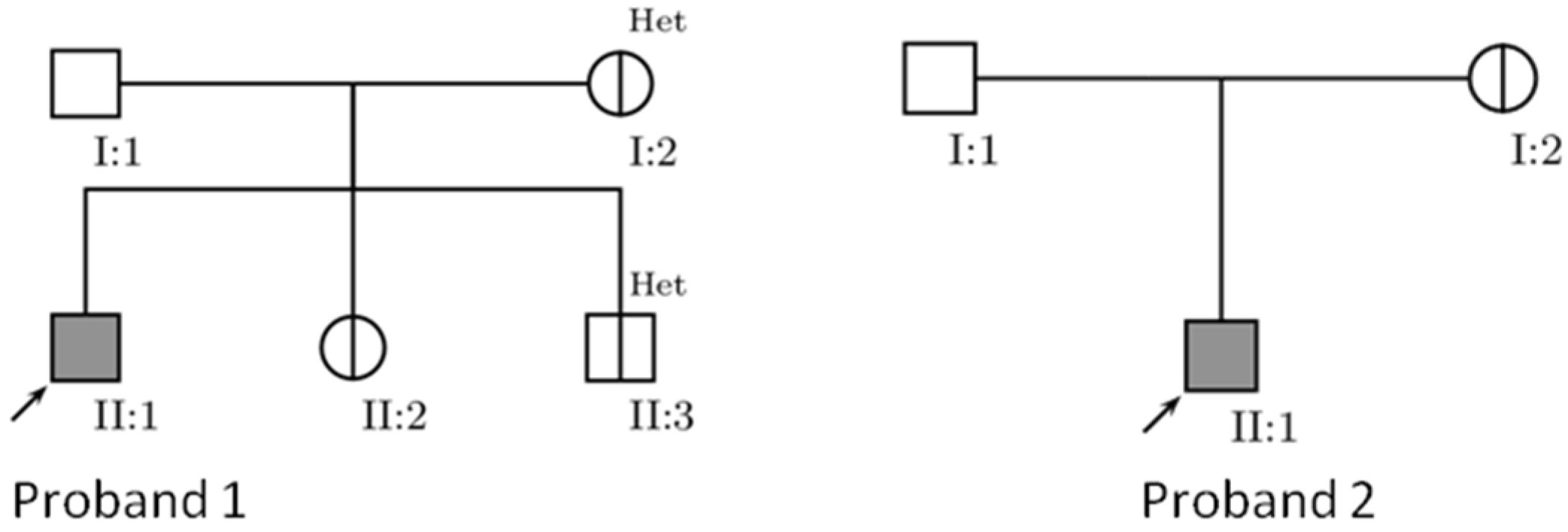{kind=link}
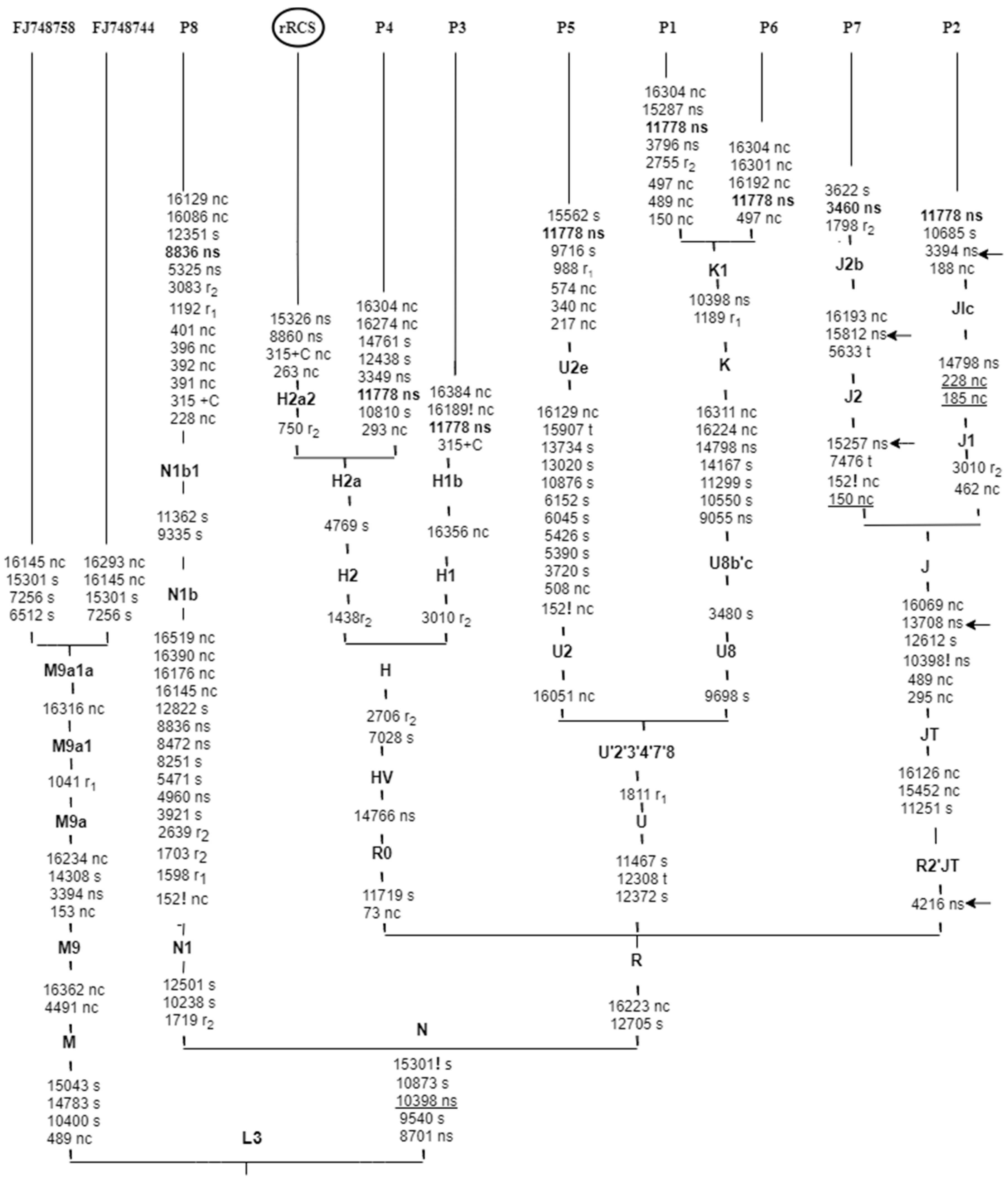{kind=link}
| Subjects | Primary Mutations | Secondary Mutations | Associated Mutations | Haplogroup |
|---|---|---|---|---|
| P1, mother, sibs | 11778G>A | − | − | K1 |
| P2, mother | 11778G>A | 3394T>C, 4216T>C, 13708G>A | 15287T>C, 2755A>G, 3796A>G | J1c |
| P3 | 11778G>A | − | − | H1b |
| P4 | 11778G>A | − | − | H2a |
| P5 | 11778G>A | − | 988G>A | U2e |
| P6 | 11778G>A | − | − | K1 |
| P7 | 3460G>A | 4216T>C, 13708G>A, 15257G>A,15812G>A | − | J2b |
| P8 | 8836A>G | − | − | N1b |
| Variant | Gene | Codon | A.A Change | Status | UniProt ID | Polyphen Prediction | PANTHER | PROVEAN | Previous Reported |
|---|---|---|---|---|---|---|---|---|---|
| m.3394T>C | MT-ND1 | 30 | Y-H | Secondary | P03886 | Benign | Probably damaging | Deleterious (−4.40) | Yes |
| m.3460G>A | MT-ND1 | 52 | A-T | Primary | P03886 | Probably damaging | Probably damaging | Neutral (−2.36) | Yes |
| m.4216T>C | MT-ND1 | 304 | Y-H | Secondary | P03886 | Benign | Probably damaging | Neutral (3.51) | Yes |
| m.8836A>G | MT-ATP6 | 104 | M-V | Rare LHON mutation | P00846 | Possibly damaging | Possibly damaging | Neutral (−2.46) | Yes |
| m.11778G>A | MT-ND4 | 340 | R-H | Primary | P03905 | Probably damaging | Probably damaging | Deleterious (−4.74) | Yes |
| m.13708G>A | MT-ND5 | 458 | A-T | Secondary | P03915 | Benign | Probably benign | Neutral (−1.50) | Yes |
| m.15257G>A | MT-CYB | 171 | D-N | Secondary | P00156 | Benign | Probably damaging | Deleterious (−3.56) | Yes |
| m.15812G>A | MT-CYB | 356 | V-M | Secondary | P00156 | Benign | Probably benign | Neutral (−0.73) | Yes |
| Variant | Haplogroup (HG) | Frequency in HG Branch | Conservation |
|---|---|---|---|
| m.3394T>C | J1c | 12.60 | 93.33% |
| m.3460G>A | J2b | 0 | 91.11% |
| m.4216T>C | J1c | 99.07 | 24.44% |
| J2b | 99.14 | 24.44% | |
| m.8836A>G | N1b | 97.97 | 88.89% |
| m.11778G>A | J1c | 0.77 | 100.00% |
| K1 | 0 | 100.00% | |
| H2a | 0 | 100.00% | |
| H1b | 0.42 | 100.00% | |
| U2e | 0.60 | 100.00% | |
| m.13708G>A | J1c | 98.76 | 33.33% |
| J2b | 98.85 | 33.33% | |
| m.15257G>A | J2b | 99.14 | 95.56% |
| m.15812G>A | J2b | 98.85 | 24.44% |
| Subject | Gender | Age at Evaluation | Clinical Picture | Ocular Evaluation | Fundoscopy | Environmental Factors |
|---|---|---|---|---|---|---|
| Mother of P1 | Female | 45 | - | VOS: 1.0-VOD: 1.0. | Yellow optic nerve papilla, tortuous vessels. | - |
| PR-VEP: no abnormalities. | ||||||
| P1 | Male | 15 | Simultaneous binocularly vision loss. | VOS: 2–3/60-VOD: 0.2–0.3/60. Absolute central scotoma. Color vision defects. PR-VEP: bilateral lesion of optic pathways, more right. | Optic disc: clearly demarcated, pale, dilated capillaries peripapillary, blood vessels slightly tortuous flow. Five months later: Optic nerve atrophy. | - |
| Sister of P1 | Female | 12 | - | VOS: 1.0-VOD: 1.0. | Normal disc appearance. | - |
| PR-VEP: high amplitude. | ||||||
| Bother of P1 | Male | 4 | - | VOS: 1.0-VOD: 1.0. | Normal disc appearance. | - |
| PR-VEP: high amplitude. | ||||||
| Mother of P2 | Female | 39 | - | VOS: 1.0-VOD: 1.0. | Several tortuous capillaries along the optic nerve disc. | - |
| P2 | Male | 13 | Poor vision of both eyes, right eye then left eye five months later. | VOS: 1.0/60-VOD: 0.1/60. Centrocecal scotoma. Color vision defects. | Optic disc: blurred edges, yellow, peripapillary tortuous dilated capillaries, circumpapillary telangiectatic microangiopathy, swelling of the nerve fiber layer around the disc. | - |
| PR-VEP: Bilateral extension of P100 latencies. | ||||||
| P3 | Male | 30 | Painless gradually low vision in both eyes, first on right eye. | VOS: 3/60-VOD: 4/60. | Pallor of optic nerve papilla. | - |
| Central scotoma. | ||||||
| PR-VEP: Bilateral extension of P100 latencies on both sides. | ||||||
| P4 | Male | 55 | Simultaneous vision loss in eyes. | VOS: 2–3/60-VOD: 0.05–0.1/60. Central scotoma. | Left optic disk slightly paler, circumpapillary telangiectasia, and vessel tortuosity. | Alcohol: occasionally consumed; |
| PR-VEP: Bilateral extension of P100 latencies on both sides. | Post-infection. | |||||
| V | Male | 20 | Sudden loss of vision on left eye then on the right one. | VOS: 4/60-VOD: 0.10/60. | Normal disc appearance. | - |
| P6 | Male | 17 | Blurred vision, right eye then left eye month later. | VOS: 1.50–2/60-VOD: 0.5–0.75/60. Absolute central scotoma. PR-VEP: bilateral lesion of optic pathways, more right. | Optic nerve atrophy. | - |
| P7 | Female | 17 | Impaired vision in eyes, left eye then right eye three weeks later. | VOS: 0.5/60-VOD: 0.8–1/60. Bilateral amblyopia, the left non-reactive to light. PR-VEP: decreased amplitude of cortical, prolonged P100 latency only on left eye. | Pale, clear borders, blood vessel tortuosity, numerous striated reflexes in the macula, macular relief disturbed, and no pain when moving the bulbus on left eye. | Smoking: up to ten cigarettes a day. Alcohol: occasionally consumed |
| P8 | Male | 33 | Sequentially poor vision of eyes within weeks, more pronounced on the left. Bad vision in the darkness. Headache. | VOS 0.5/60-VOD 3/60. Both eyes are non-reactive to light. IOP: 14 mmHg. | Bilateral papilledema. Retinal detachment. Later on optic nerve atrophy. | - |
| Subject | Neurological Evaluation | Non-Neurological Evaluation |
|---|---|---|
| Mother of P1 | AEP: extension of conduction along intra-axial acoustic pathways | Trivial mitral and aortic regurgitation. |
| P1 | MRI: no abnormalities. | Mild mitral valve prolapse with trivial regurgitation. |
| SSEP: giant SEPs above the primary SS cortex of the right hemisphere. | ||
| AEP: no abnormalities. | ||
| Sister of P1 | AEP: extension of conduction along intra-axial acoustic pathways. | - |
| Brother of P1 | AEP: no abnormalities. | - |
| Mother of P2 | MRI: no abnormalities. | QT interval slightly prolonged. |
| P2 | MRI: hyperintense lesion of the right optic nerve | Normal ECG. |
| P3 | MRI: initial cortical reductive changes of the brain supratentorially. | - |
| TCD: the optic nerves are thinner in diameter on both sides. | ||
| P4 | MRI: chronic microangiopathic changes and periventricular ischemic. | Hypertension. |
| P5 | MRI: increased diameter of the retrobulbar segment of the right optic nerve. | ECG: sinus tachycardia. |
| P6 | MRI: supra-and infratentorial hyper intensive changes and demyelination. | - |
| SSEP: asymmetry of latencies of cortical responses to the damage of the left hemisphere. | ||
| AEP: lower amplitude V wave left. | ||
| P7 | MRI: no abnormalities. | Enlarged spleen, aortic effusion. |
| Bicuspid aortic valve with mild aortic insufficiency. | ||
| ECG: short PR interval with delta wave. | ||
| P8 | MRI: no abnormalities. | Gastric ulcer. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawod, P.G.A.; Jancic, J.; Marjanovic, A.; Brankovic, M.; Jankovic, M.; Samardzic, J.; Potkonjak, D.; Djuric, V.; Mesaros, S.; Novakovic, I.; et al. Whole Mitochondrial Genome Analysis in Serbian Cases of Leber’s Hereditary Optic Neuropathy. Genes 2020, 11, 1037. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11091037
Dawod PGA, Jancic J, Marjanovic A, Brankovic M, Jankovic M, Samardzic J, Potkonjak D, Djuric V, Mesaros S, Novakovic I, et al. Whole Mitochondrial Genome Analysis in Serbian Cases of Leber’s Hereditary Optic Neuropathy. Genes. 2020; 11(9):1037. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11091037
Chicago/Turabian StyleDawod, Phepy G. A., Jasna Jancic, Ana Marjanovic, Marija Brankovic, Milena Jankovic, Janko Samardzic, Dario Potkonjak, Vesna Djuric, Sarlota Mesaros, Ivana Novakovic, and et al. 2020. "Whole Mitochondrial Genome Analysis in Serbian Cases of Leber’s Hereditary Optic Neuropathy" Genes 11, no. 9: 1037. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11091037