MYC DNA Methylation in Prostate Tumor Tissue is Associated with Gleason Score

Abstract
:1. Introduction
2. Materials and Methods
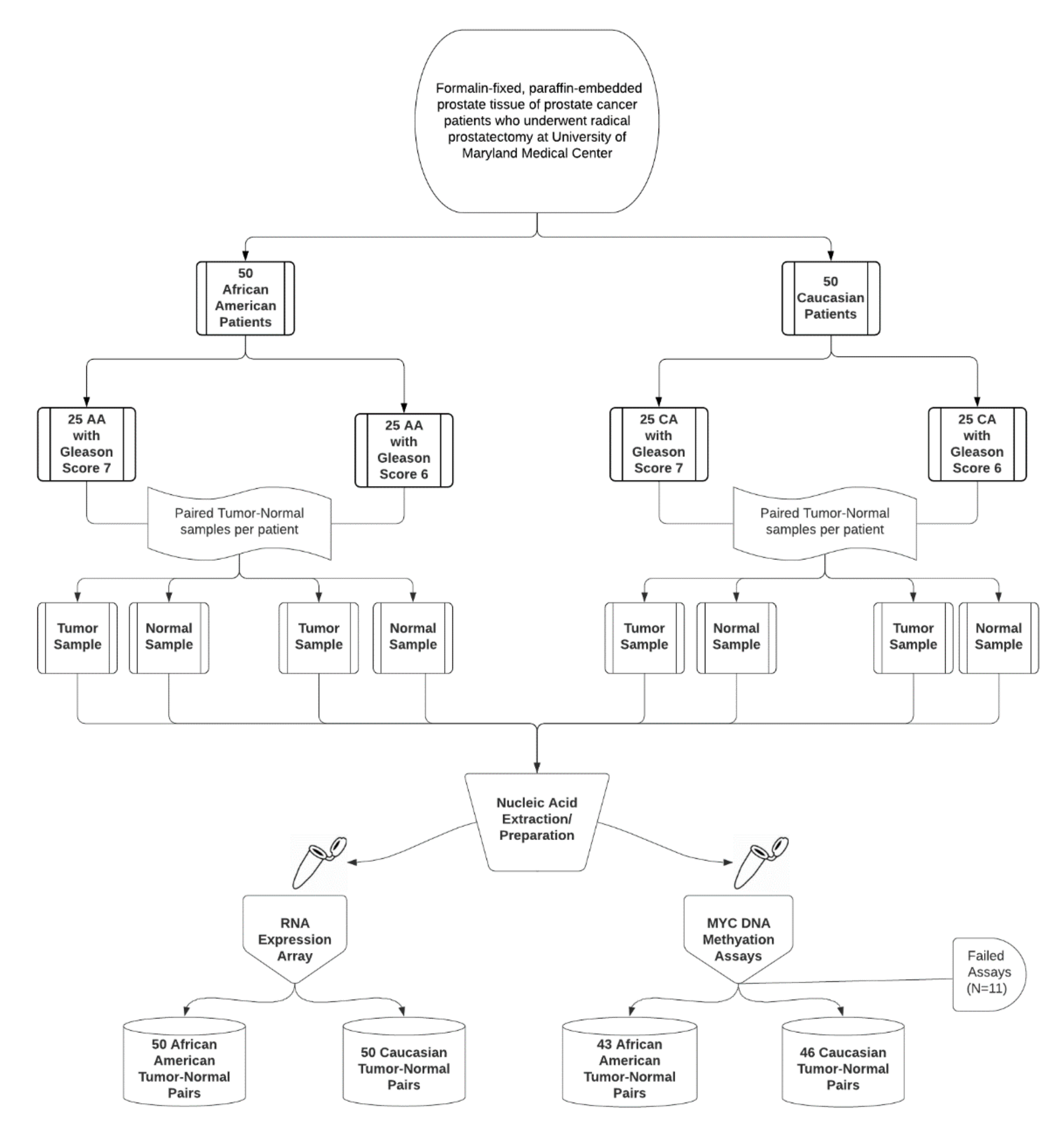
2.1. Study Samples
2.2. Nucleic Acid Extraction/Preparation and RNA Quality Assessment
2.3. MYC DNA Methylation Assays
2.4. RNA Expression Data
2.5. The Cancer Genome Atlas (TCGA) DNA Methylation Data
2.6. Statistical Analysis
3. Results
3.1. Study Population Characteristics
3.2. MYC DNA Methylation CpG-CpG Correlations and Prostate Tumor–Normal Differences
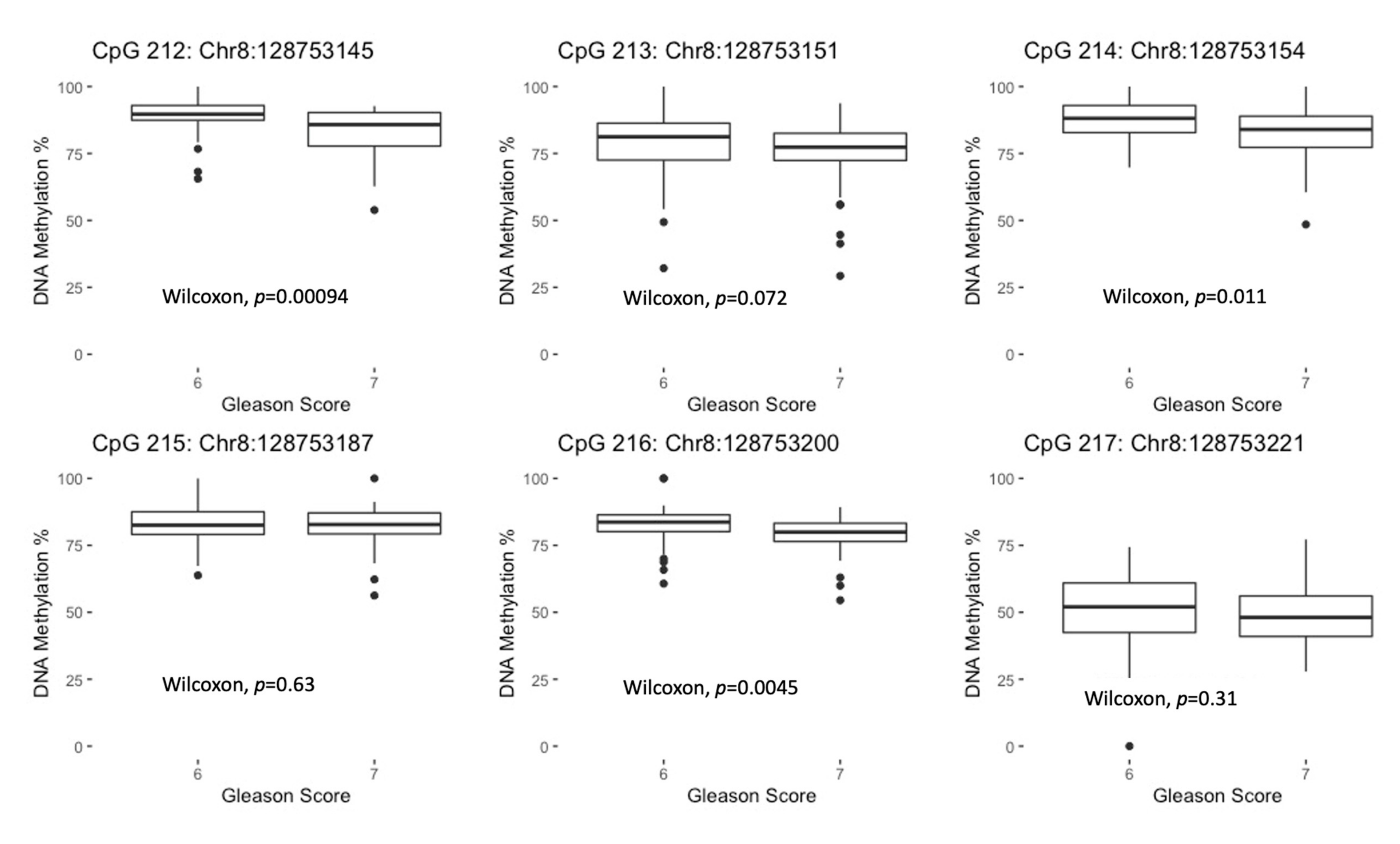
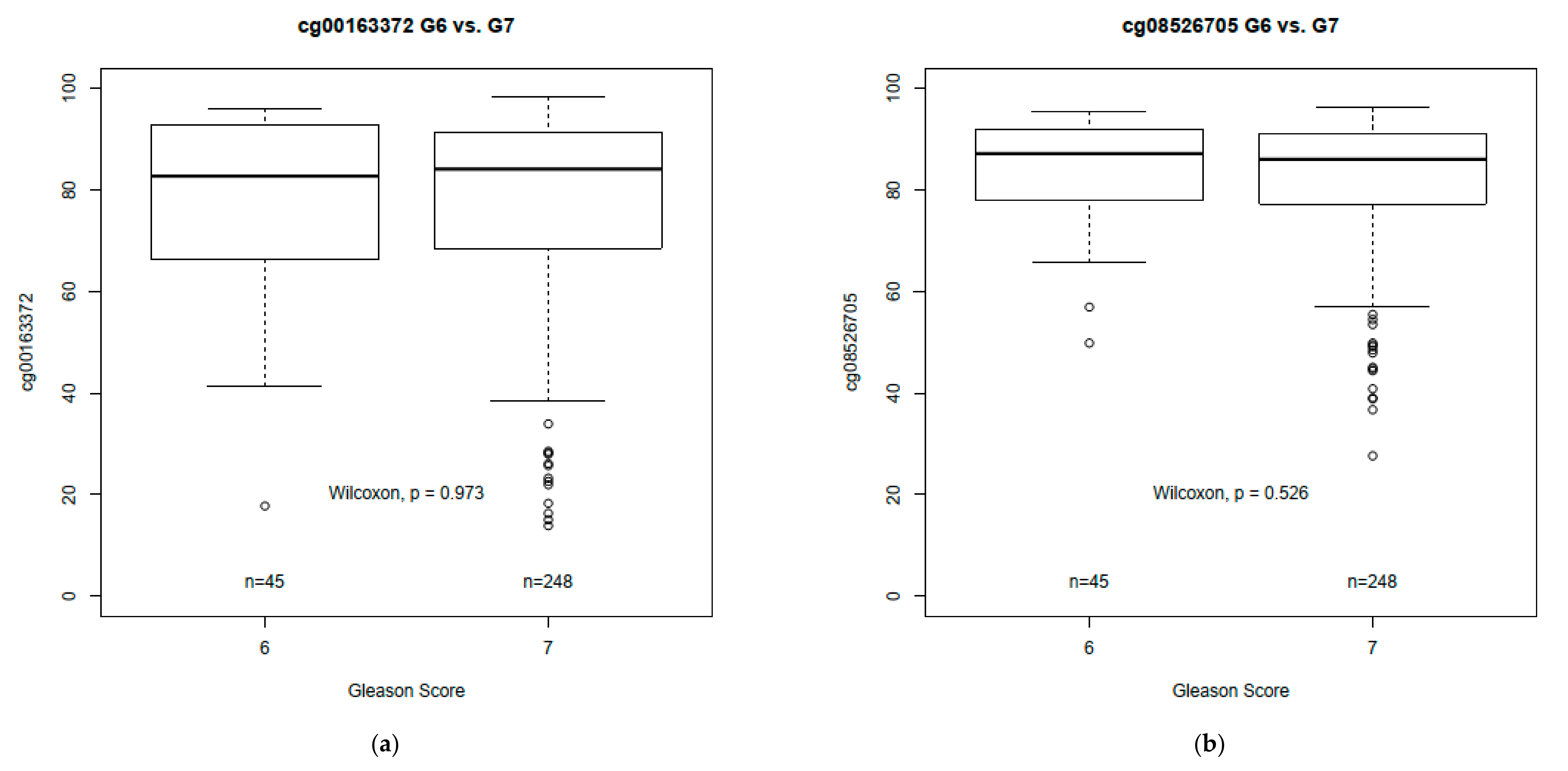
3.3. MYC DNA Methylation Differences by GS in Prostate Tumor Tissue
3.4. RNA Expression Tumor-Normal Differences and Differences by GS in Prostate Tumor Tissue
3.5. Prostate tumor MYC DNA methylation and RNA expression in prostate tumor tissue
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Assay ID | Genomic Target Sequence | Bisulfite Converted Target Sequence | Pyrosequencing Dispensation Order |
|---|---|---|---|
| ADS3573-FS | ACTTGTTGCGGAAACGACGAGAACAG TTGAAACACAAACTTGAACAGCTACGG AACTCTTGTGCGTAAGGAAAAGTAAGG AAAACGATTCCTTCTAACAGAAATG | ATTTGTTGYGGAAAYGAYGAGAATA GTTGAAATATAAATTTGAATAGTTA YGGAATTTTTGTGYGTAAGGAAAAG TAAGGAAAAYGATTTTTTTTAATAG AAATG | ACTGTAGTCGATCGATCGAGATCAGTGA TCATCACTGATCAGCTGATCGACTCTGT GTCGTCAGAAGTCAGATCGA |
| ADS3573-FS2 | AACTTGAACAGCTACGGAACTCTTGTG CGTAAGGAAAAGTAAGGAAAACGATT CCTTCTAACAGAAATGTCCTGA | AATTTGAATAGTTAYGGAATTTTTGT GYGTAAGGAAAAGTAAGGAAAAYG ATTTTTTTTAATAGAAATGTTTTGA | ACTGACTAGCTGATCGATCTGTAGTCGT AGAAGTAGAATCGA |

References
- Amundadottir, L.T.; Sulem, P.; Gudmundsson, J.; Helgason, A.; Baker, A.; Agnarsson, B.; Sigurdsson, A.; Benediktsdottir, K.R.; Cazier, J.-B.; Sainz, J.; et al. A common variant associated with prostate cancer in European and African populations. Nat. Genet. 2006, 38, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, J.; Sulem, P.; Manolescu, A.; Amundadottir, L.T.; Gudbjartsson, D.; Helgason, A.; Rafnar, T.; Bergthorsson, J.T.; Agnarsson, B.; Baker, A.; et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat. Genet. 2007, 39, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Haiman, C.; Patterson, N.; Freedman, M.L.; Myers, S.R.; Pike, M.C.; Waliszewska, A.; Neubauer, J.; Tandon, A.; Schirmer, C.; McDonald, G.J.; et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat. Genet. 2007, 39, 638–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeager, M.; Orr, N.; Hayes, R.B.; Jacobs, K.B.; Kraft, P.; Wacholder, S.; Minichiello, M.J.; Fearnhead, P.; Yu, K.; Chatterjee, N.; et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat. Genet. 2007, 39, 645–649. [Google Scholar] [CrossRef]
- Eeles, R.A.; Kote-Jarai, Z.; Giles, G.G.; Al Olama, A.A.; Guy, M.; Jugurnauth, S.K.; Mulholland, S.; Leongamornlert, D.A.; Edwards, S.M.; Field, H.I.; et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat. Genet. 2008, 40, 316–321. [Google Scholar] [CrossRef]
- Thomas, G.; Jacobs, K.B.; Yeager, M.; Kraft, P.; Wacholder, S.; Orr, N.; Yu, K.; Chatterjee, N.; Welch, R.W.; Hutchinson, A.; et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet. 2008, 40, 310–315. [Google Scholar] [CrossRef]
- Al Olama, A.A.; Kote-Jarai, Z.; Giles, G.G.; Guy, M.; Morrison, J.; Severi, G.; Leongamornlert, D.; Tymrakiewicz, M.; Jhavar, S.; Hopper, J.L.; et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat. Genet. 2009, 41, 1058–1060. [Google Scholar] [CrossRef]
- Schumacher, F.R.; Berndt, S.I.; Siddiq, A.; Jacobs, K.B.; Wang, Z.; Lindstrom, S.; Stevens, V.L.; Chen, C.; Mondul, A.M.; Travis, R.C.; et al. Genome-wide association study identifies new prostate cancer susceptibility loci. Hum. Mol. Genet. 2011, 20, 3867–3875. [Google Scholar] [CrossRef] [Green Version]
- Gudmundsson, J.; Sulem, P.; Gudbjartsson, D.F.; Masson, G.; Agnarsson, B.A.; Benediktsdottir, K.R.; Sigurdsson, A.; Magnusson, O.T.; Gudjonsson, S.A.; Magnusdottir, D.N.; et al. A study based on whole-genome sequencing yields a rare variant at 8q24 associated with prostate cancer. Nat. Genet. 2012, 44, 1326–1329. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Rand, K.A.; Hazelett, D.J.; Ingles, S.A.; Kittles, R.A.; Strom, S.S.; Rybicki, B.A.; Nemesure, B.; Isaacs, W.B.; Stanford, J.L.; et al. Prostate Cancer Susceptibility in Men of African Ancestry at 8q24. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Matejcic, M.; Saunders, E.J.; Dadaev, T.; Brook, M.N.; Wang, K.; Sheng, X.; Al Olama, A.A.; Schumacher, F.R.; Ingles, S.A.; Benlloch, S.; et al. Germline variation at 8q24 and prostate cancer risk in men of European ancestry. Nat. Commun. 2018, 9, 4616. [Google Scholar] [CrossRef] [PubMed]
- Darst, B.F.; Wan, P.; Sheng, X.; Bensen, J.T.; Ingles, S.A.; Rybicki, B.A.; Nemesure, B.; John, E.M.; Fowke, J.H.; Stevens, V.L.; et al. A Germline Variant at 8q24 Contributes to Familial Clustering of Prostate Cancer in Men of African Ancestry. Eur. Urol. 2020, 78, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Bieberich, C.J.; Van Dang, C.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastler, S.; Honold, L.; Luedeke, M.; Kuefer, R.; Möller, P.; Hoegel, J.; Vogel, W.; Maier, C.; Assum, G. POU5F1P1, a putative cancer susceptibility gene, is overexpressed in prostatic carcinoma. Prostate 2009, 70. [Google Scholar] [CrossRef]
- Chung, S.; Nakagawa, H.; Uemura, M.; Piao, L.; Ashikawa, K.; Hosono, N.; Takata, R.; Akamatsu, S.; Kawaguchi, T.; Morizono, T.; et al. Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 2010, 102, 245–252. [Google Scholar] [CrossRef]
- Bawa, P.; Zackaria, S.; Verma, M.; Gupta, S.; Srivatsan, R.; Chaudhary, B.; Srinivasan, S. Integrative Analysis of Normal Long Intergenic Non-Coding RNAs in Prostate Cancer. PLoS ONE 2015, 10, e0122143. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Zhao, S.; He, X.; Zheng, Z.; Bai, W.; Duan, Y.; Cheng, S.; Wang, J.; Liu, X.; Zhang, G. The up-regulation of long non-coding RNA CCAT2 indicates a poor prognosis for prostate cancer and promotes metastasis by affecting epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 2016, 480, 508–514. [Google Scholar] [CrossRef]
- Jia, L.; Landan, G.; Pomerantz, M.; Jaschek, R.; Herman, P.; Reich, D.; Yan, C.; Khalid, O.; Kantoff, P.W.; Oh, W.K.; et al. Functional Enhancers at the Gene-Poor 8q24 Cancer-Linked Locus. PLoS Genet. 2009, 5, e1000597. [Google Scholar] [CrossRef]
- Ahmadiyeh, N.; Pomerantz, M.M.; Grisanzio, C.; Herman, P.; Jia, L.; Almendro, V.; He, H.H.; Brown, M.; Liu, X.S.; Davis, M.; et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar] [CrossRef] [Green Version]
- Sotelo, J.; Esposito, D.; Duhagon, M.A.; Banfield, K.; Mehalko, J.; Liao, H.; Stephens, R.M.; Harris, T.J.R.; Munroe, D.J.; Wu, X. Long-range enhancers on 8q24 regulate c-Myc. Proc. Natl. Acad. Sci. USA 2010, 107, 3001–3005. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Yuan, T.; Schilter, K.F.; Dittmar, R.; MacKinnon, A.; Huang, X.; Tschannen, M.; Worthey, E.; Jacob, H.; Xia, S.; et al. Prostate cancer risk locus at 8q24 as a regulatory hub by physical interactions with multiple genomic loci across the genome. Hum. Mol. Genet. 2014, 24, 154–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Kim, S.; Wang, K.; Farnham, P.J.; Coetzee, G.A.; Lu, W. 4C-seq revealed long-range interactions of a functional enhancer at the 8q24 prostate cancer risk locus. Sci. Rep. 2016, 6, 22462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M.; Herman, J.G. Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J. Pathol. 2002, 196, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Mills, I.; Lynch, A.G. The importance of DNA methylation in prostate cancer development. J. Steroid Biochem. Mol. Biol. 2017, 166, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, G.H.; Lee, S.; Lee, H.J.; Hwang, K.S. Aberrant CpG island hypermethylation of multiple genes in prostate cancer and prostatic intraepithelial neoplasia. J. Pathol. 2004, 202, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Yegnasubramanian, S.; Kowalski, J.; Gonzalgo, M.L.; Zahurak, M.; Piantadosi, S.; Walsh, P.C.; Bova, G.S.; De Marzo, A.M.; Isaacs, W.; Nelson, W.G. Hypermethylation of CpG Islands in Primary and Metastatic Human Prostate Cancer. Cancer Res. 2004, 64, 1975–1986. [Google Scholar] [CrossRef] [Green Version]
- Yegnasubramanian, S.; Haffner, M.C.; Zhang, Y.; Gurel, B.; Cornish, T.C.; Wu, Z.; Irizarry, R.A.; Morgan, J.; Hicks, J.; Deweese, T.L.; et al. DNA Hypomethylation Arises Later in Prostate Cancer Progression than CpG Island Hypermethylation and Contributes to Metastatic Tumor Heterogeneity. Cancer Res. 2008, 68, 8954–8967. [Google Scholar] [CrossRef] [Green Version]
- Barry, K.H.; Moore, L.; Sampson, J.N.; Koutros, S.; Yan, L.; Meyer, A.; Reddy, M.; Oler, A.J.; Cook, M.B.; Fraumeni, J.F., Jr.; et al. Prospective study of DNA methylation at chromosome 8q24 in peripheral blood and prostate cancer risk. Br. J. Cancer 2017, 116, 1470–1479. [Google Scholar] [CrossRef] [Green Version]
- Catalona, W.J.; Smith, D.S. 5-Year Tumor Recurrence Rates after Anatomical Radical Retropubic Prostatectomy for Prostate Cancer. J. Urol. 1994, 152, 1837–1842. [Google Scholar] [CrossRef]
- Ohtsuki, T.; Nishitani, K.; Hatamochi, A.; Yavvata, Y.; Namba, M. Analysis of methylation in the c-MYC gene in five human myeloma cell lines. Br. J. Haematol. 1991, 77, 172–179. [Google Scholar] [CrossRef]
- Sharrard, R.M.; Royds, J.; Rogers, S.N.; Shorthouse, A.J. Patterns of methylation of the c-myc gene in human colorectal cancer progression. Br. J. Cancer 1992, 65, 667–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A. Racial disparities in prostate cancer a molecular perspective. Front. Biosci. 2017, 22, 772–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khani, F.; Mosquera, J.M.; Park, K.; Blattner, M.; O’Reilly, C.; Macdonald, T.Y.; Chen, Z.; Srivastava, A.; Tewari, A.K.; Barbieri, C.E.; et al. Evidence for Molecular Differences in Prostate Cancer between African American and Caucasian Men. Clin. Cancer Res. 2014, 20, 4925–4934. [Google Scholar] [CrossRef] [Green Version]
- Singh, R. Molecular basis for prostate cancer racial disparities. Front. Biosci. 2017, 22, 428–450. [Google Scholar] [CrossRef] [Green Version]
- Barry, K.H.; Moore, L.E.; Sampson, J.; Yan, L.; Meyer, A.; Oler, A.J.; Chung, C.C.; Wang, Z.; Yeager, M.; Amundadottir, L.T.; et al. DNA methylation levels at chromosome 8q24 in peripheral blood are associated with 8q24 cancer susceptibility loci. Cancer Prev. Res. 2014, 7, 1282–1292. [Google Scholar] [CrossRef] [Green Version]
- NetAffx. Available online: https://www.affymetrix.com/analysis/index.affx (accessed on 21 June 2020).
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Ribeiro, F.R.; Jerónimo, C.; Henrique, R.; Fonseca, D.; Oliveira, J.; Lothe, R.; Teixeira, M.R. 8q Gain Is an Independent Predictor of Poor Survival in Diagnostic Needle Biopsies from Prostate Cancer Suspects. Clin. Cancer Res. 2006, 12, 3961–3970. [Google Scholar] [CrossRef] [Green Version]
- Devaney, J.M.; Wang, S.; Funda, S.; Long, J.; Taghipour, D.J.; Tbaishat, R.; Furbert-Harris, P.; Ittmann, M.; Kwabi-Addo, B. Identification of novel DNA-methylated genes that correlate with human prostate cancer and high-grade prostatic intraepithelial neoplasia. Prostate Cancer Prostatic Dis. 2013, 16, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Dhanasekaran, S.M.; Prensner, J.R.; Cao, X.; Robinson, D.; Kalyana-Sundaram, S.; Huang, C.; Shankar, S.; Jing, X.; Iyer, M.; et al. Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome Res. 2011, 21, 1028–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Kim, S.-T.; Turner, A.R.; Young, T.; Smith, S.; Liu, W.; Lindberg, J.; Egevad, L.; Gronberg, H.; Isaacs, W.B.; et al. Identification of New Differentially Methylated Genes That Have Potential Functional Consequences in Prostate Cancer. PLoS ONE 2012, 7, e48455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Kelly, W.K.; Fu, A.; Haines, K.; Hoffman, A.; Zheng, T.; Zhu, Y. Genome-wide methylation analysis identifies involvement of TNF-α mediated cancer pathways in prostate cancer. Cancer Lett. 2011, 302, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Absher, D.M.; Gulzar, Z.G.; Young, S.R.; McKenney, J.K.; Peehl, D.M.; Brooks, J.D.; Myers, R.M.; Sherlock, G. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res. 2011, 21, 1017–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kron, K.; Pethe, V.; Briollais, L.; Sadikovic, B.; Ozcelik, H.; Sunderji, A.; Venkateswaran, V.; Pinthus, J.; Fleshner, N.; Van Der Kwast, T.; et al. Discovery of Novel Hypermethylated Genes in Prostate Cancer Using Genomic CpG Island Microarrays. PLoS ONE 2009, 4, e4830. [Google Scholar] [CrossRef]
- Luo, J.-H.; Ding, Y.; Chen, R.; Michalopoulos, G.; Nelson, J.; Tseng, G.; Yu, Y.P. Genome-Wide Methylation Analysis of Prostate Tissues Reveals Global Methylation Patterns of Prostate Cancer. Am. J. Pathol. 2013, 182, 2028–2036. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, S.; Klee, E.W.; Young, C.Y.F.; Sun, Z.; Jimenez, R.E.; Klee, G.G.; Tindall, D.J.; Donkena, K.V. Global Methylation Profiling for Risk Prediction of Prostate Cancer. Clin. Cancer Res. 2012, 18, 2882–2895. [Google Scholar] [CrossRef] [Green Version]
- Kirby, M.K.; Ramaker, R.C.; Roberts, B.S.; Lasseigne, B.N.; Gunther, D.S.; Burwell, T.C.; Davis, N.S.; Gulzar, Z.G.; Absher, D.; Cooper, S.J.; et al. Genome-wide DNA methylation measurements in prostate tissues uncovers novel prostate cancer diagnostic biomarkers and transcription factor binding patterns. BMC Cancer 2017, 17, 273. [Google Scholar] [CrossRef] [Green Version]
- Paziewska, A.; Dabrowska, M.; Goryca, K.; Antoniewicz, A.; Dobruch, J.; Mikula, M.; Jarosz, D.F.; Zapala, L.; Borowka, A.; Ostrowski, J. DNA methylation status is more reliable than gene expression at detecting cancer in prostate biopsy. Br. J. Cancer 2014, 111, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Aref-Eshghi, E.; Schenkel, L.C.; Ainsworth, P.; Lin, H.; Rodenhiser, D.I.; Cutz, J.-C.; Sadikovic, B. Genomic DNA Methylation-Derived Algorithm Enables Accurate Detection of Malignant Prostate Tissues. Front. Oncol. 2018, 8, 100. [Google Scholar] [CrossRef] [Green Version]
- Geybels, M.S.; Wright, J.L.; Bibikova, M.; Klotzle, B.; Fan, J.-B.; Zhao, S.; Feng, Z.; Ostrander, E.A.; Lin, D.W.; Nelson, P.S.; et al. Epigenetic signature of Gleason score and prostate cancer recurrence after radical prostatectomy. Clin. Epigenet. 2016, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Bhasin, J.M.; Lee, B.H.; Matkin, L.; Taylor, M.G.; Hu, B.; Xu, Y.; Magi-Galluzzi, C.; Klein, E.A.; Ting, A.H. Methylome-wide Sequencing Detects DNA Hypermethylation Distinguishing Indolent from Aggressive Prostate Cancer. Cell Rep. 2015, 13, 2135–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldrup, C.; Mundbjerg, K.; Vestergaard, E.M.; Lamy, P.; Wild, P.; Schulz, W.A.; Arsov, C.; Visakorpi, T.; Borre, M.; Høyer, S.; et al. DNA Methylation Signatures for Prediction of Biochemical Recurrence After Radical Prostatectomy of Clinically Localized Prostate Cancer. J. Clin. Oncol. 2013, 31, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Schuijers, J.; Manteiga, J.C.; Weintraub, A.S.; Day, D.S.; Zamudio, A.V.; Hnisz, D.; Lee, T.I.; Young, R.A. Transcriptional Dysregulation of MYC Reveals Common Enhancer-Docking Mechanism. Cell Rep. 2018, 23, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, K.H.; Moore, L.E.; Liao, L.M.; Huang, W.-Y.; Andreotti, G.; Poulin, M.; Berndt, S.I. Prospective study of DNA methylation at LINE-1 and Alu in peripheral blood and the risk of prostate cancer. Prostate 2015, 75, 1718–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros-Silva, D.; Costa-Pinheiro, P.; Duarte, H.; Sousa, E.J.; Evangelista, A.F.; Graça, I.; Carneiro, I.; Martins, A.T.; Oliveira, J.; Carvalho, A.L.; et al. MicroRNA-27a-5p regulation by promoter methylation and MYC signaling in prostate carcinogenesis. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Zhang, A.; Zhang, J.; Kaipainen, A.; Lucas, J.M.; Yang, H. Long non-coding RNA: A newly deciphered "code" in prostate cancer. Cancer Lett. 2016, 375, 323–330. [Google Scholar] [CrossRef]
- El Bezawy, R.; Cominetti, D.; Fenderico, N.; Zuco, V.; Beretta, G.L.; Dugo, M.; Arrighetti, N.; Stucchi, C.; Rancati, T.; Valdagni, R.; et al. miR-875-5p counteracts epithelial-to-mesenchymal transition and enhances radiation response in prostate cancer through repression of the EGFR-ZEB1 axis. Cancer Lett. 2017, 395, 53–62. [Google Scholar] [CrossRef]
- Meyer, K.B.; Maia, A.-T.; O’Reilly, M.; Ghoussaini, M.; Prathalingam, R.; Porter-Gill, P.; Ambs, S.; Prokunina-Olsson, L.; Carroll, J.; Ponder, B.A.J. A Functional Variant at a Prostate Cancer Predisposition Locus at 8q24 Is Associated with PVT1 Expression. PLoS Genet. 2011, 7, e1002165. [Google Scholar] [CrossRef] [Green Version]


| Characteristic | Overall (n = 89) | GS 6 (n = 40) | GS 7 (n = 49) |
|---|---|---|---|
| Age at surgery, median (range) | 58 (42, 75) | 56.5 (42, 72) | 58 (47, 75) |
| Age at diagnosis, median (range) | 57 (42, 75) | 56 (42, 72) | 57.5 (47, 75) |
| Surgery year, n (col %) | |||
| 2001–2009 | 18 (20.2) | 18 (45.0) | 0 (0.0) |
| 2010–2017 | 71 (79.8) | 22 (55.0) | 49 (100.0) |
| Race, n (col %) | |||
| Caucasian | 46 (51.7) | 21 (52.5) | 25 (51.0) |
| African American | 43 (48.3) | 19 (47.5) | 24 (49.0) |
| Gleason pattern, n (col %) | |||
| 3 + 3 | 40 (44.9) | 40 (100.0) | 0 (0.0) |
| 3 + 4 | 35 (39.3) | 0 (0) | 35 (71.4) |
| 4 + 3 | 14 (15.7) | 0 (0) | 14 (28.6) |
| pT stage, n (col %) | |||
| 2 | 60 (67.4) | 29 (72.5) | 31 (63.3) |
| 3a | 11 (12.4) | 2 (5.0) | 9 (18.4) |
| 3b | 7 (7.9) | 1 (2.5) | 6 (12.2) |
| 3—not specified | 4 (4.5) | 1 (2.5) | 3 (6.1) |
| Unknown | 7 (7.9) | 7 (17.5) | 0 (0.0) |
| pN status, n (col %) | |||
| N0 | 55 (61.8) | 15 (37.5) | 40 (81.6) |
| N1 | 3 (3.4) | 0 (0.0) | 3 (6.1) |
| Unknown | 31 (34.8) | 25 (62.5) | 6 (12.2) |
| PSA (ng/mL), median (IQR) | 6.2 (4.8, 8.0) | 6.2 (4.7, 7.2) | 6.6 (5.0, 9.0) |
| CpG Site (GRCH37/hg19 Coordinate) | Median % DNA methylation (IQR) for Normal Prostate Tissue Samples | Median % DNA methylation (IQR) for Prostate Tumor Tissue Samples | Median Tumor-Normal Difference in % DNA Methylation (IQR) | Wilcoxon Signed-Rank p-Value a |
|---|---|---|---|---|
| University of Maryland samples (n = 89 pairs) | ||||
| CpG 212 (Chr8:128753145) | 88.27 (85.32, 91.48) | 88.29 (82.35, 91.07) | −0.20 (−7.58, 2.91) | 0.06 |
| CpG 213 (Chr8:128753151) | 85.65 (82.00, 88.48) | 78.72 (72.39, 84.20) | −7.36 (−18.40, 0.43) | 1.8 × 10−8 |
| CpG 214 (Chr8:128753154) | 89.72 (87.03, 93.23) | 86.09 (78.82, 90.38) | −2.98 (−11.38, 2.04) | 4.9 × 10−5 |
| CpG 215 (Chr8:128753187) | 86.65 (83.90, 89.65) | 82.68 (79.23, 87.12) | −2.84 (−9.79, 0.67) | 1.7 × 10−6 |
| CpG 216 (Chr8:128753200) | 83.39 (80.67, 86.29) | 81.41 (77.56, 85.22) | −1.88 (−5.70, 2.00) | 3.9 × 10−3 |
| CpG 217 (Chr8:128753221) | 66.66 (60.81, 71.15) | 49.09 (41.30, 58.63) | −14.74 (−25.53, −6.86) | 9.1 × 10−19 |
| Across all 6 CpG sites | 83.23 (81.77, 84.82) | 77.40 (73.43, 81.46) | −4.01 (−12.12, 1.32) | 5.9 × 10−14 |
| TCGA samples (n = 50 pairs) | ||||
| cg00163372 (Chr8:128752987) | 94.52 (91.57, 95.50) | 84.62 (78.10, 92.01) | −6.89 (−16.92, −2.47) | 9.4 × 10−7 |
| cg08526705 (Chr8:128753028) | 93.01 (91.03, 94.42) | 87.30 (80.87, 91.82) | −5.46 (−13.41, −0.32) | 1.3 × 10−5 |
| Decreasing DNA Methylation (GRCH37/hg19 Coordinate) | Overall, n = 89 OR (95% CI) | African American, n = 43 OR (95% CI) | Caucasian, n = 46 OR (95% CI) | p-int b |
|---|---|---|---|---|
| CpG 212 (Chr8:128753145) | 1.10 (1.03–1.18) | 1.23 (1.04–1.45) | 1.07 (1.00–1.15) | 0.15 |
| CpG 213 (Chr8:128753151) | 1.02 (0.99–1.06) | 1.06 (0.99–1.14) | 1.01 (0.97–1.05) | 0.20 |
| CpG 214 (Chr8:128753154) | 1.07 (1.02–1.13) | 1.08 (0.99–1.17) | 1.07 (1.00–1.14) | 0.87 |
| CpG 215 (Chr8:128753187) | 1.03 (0.97–1.09) | 1.09 (0.98–1.20) | 1.00 (0.94–1.07) | 0.19 |
| CpG 216 (Chr8:128753200) | 1.08 (1.01–1.16) | 1.15 (0.99–1.32) | 1.06 (0.98–1.15) | 0.36 |
| CpG 217 (Chr8:128753221) | 1.01 (0.98–1.04) | 1.01 (0.96–1.06) | 1.01 (0.97–1.06) | 0.99 |
| Across all 6 sites | 1.01 (1.00–1.02) | 1.01 (1.00–1.02) | 1.01 (0.99–1.03) | 0.89 |
| Tumor-Normal Difference (n = 100 Pairs a) | Differences by GS (n = 100 Tumor Tissue Samples a) | ||||||
|---|---|---|---|---|---|---|---|
| Gene/ncRNA | GRCH37/hg19 Coordinate from UCSC Genome Browser | Human Clariom D TC | Median Difference in log2 Expression (IQR) | Wilcoxon Signed-Rank p | GS 6, Median log2 Expression (IQR) | GS 7, Median log2 Expression (IQR) | Wilcoxon Rank Sum p |
| MYC | chr8:128748315–128753680 | TC0800008845.hg.1 | 0.2 (−0.2, 0.5) | 0.01 | 5.9 (5.6, 6.2) | 5.9 (5.7, 6.4) | 0.21 |
| UPSTREAM of MYC | |||||||
| PCAT1 | chr8:128025399–128033259 | TC0800008832.hg.1 | 0.1 (−0.2, 0.4) | 0.06 | 4.2 (4.0, 4.4) | 4.1 (3.9, 4.3) | 0.18 |
| PCAT2 | chr8:128084939–128094466 | TC0800012460.hg.1 | 0.1 (−0.5, 0.7) | 0.39 | 4.0 (3.6, 4.3) | 3.9 (3.5, 4.3) | 0.69 |
| PRNCR1 | chr8:128092119–128104840 | TC0800008833.hg.1 | 0.2 (−0.2, 0.7) | 1.6 × 10−4 | 4.3 (4.1, 4.6) | 4.4 (4.1, 4.7) | 0.64 |
| RP11-255B23.1 | chr8:128098508–128099755 | TC0800012461.hg.1 | −0.7 (−0.3, 0.2) | 0.04 | 4.3 (4.1, 4.6) | 4.4 (4.2, 4.8) | 0.08 |
| CASC19 | chr8:128200030–128209872 | TC0800012462.hg.1 | 0.0 (−0.2, 0.3) | 0.52 | 3.4 (3.3, 3.7) | 3.4 (3.2, 3.7) | 0.22 |
| CCAT1 | chr8:128219627–128231513 | TC0800012463.hg.1 | 0.1 (−0.3, 0.5) | 0.10 | 3.5 (3.3, 3.9) | 3.2 (3.0, 3.6) | 1.5 × 10−3 |
| CASC21 | chr8:128256882–128404876 | TC0800008836.hg.1 | 0.0 (−0.2, 0.4) | 0.17 | 3.7 (3.5, 4.1) | 3.7 (3.3, 3.9) | 0.35 |
| RP11-382A18.3 | chr8:128304478–128304685 | TC0800008837.hg.1 | 0.2 (−0.2, 0.8) | 6.8 × 10−4 | 5.5 (5.3, 5.8) | 5.6 (5.1, 6.1) | 0.81 |
| CCAT2 | chr8:128412644–128414395 | TC0800008838.hg.1 | 0.1 (−0.3, 0.3) | 0.53 | 3.4 (3.2, 3.5) | 3.3 (3.2, 3.6) | 0.83 |
| POU5F1B | chr8:128428070–128429455 | TC0800008839.hg.1 | 0.1 (−0.2, 0.5) | 0.02 | 5.1 (4.8, 5.4) | 5.0 (4.8, 5.2) | 0.13 |
| CASC8 | chr8:128455595–128494384 | TC0800012464.hg.1 | 0.0 (−0.3, 0.5) | 0.34 | 3.2 (3.0, 3.5) | 3.3 (3.1, 3.5) | 0.70 |
| LOC727677 | chr8:128455595–128494384 | (same as above) | |||||
| CASC11 | chr8:128712853–128746213 | TC0800011782.hg.1 | 0.0 (−0.3, 0.4) | 0.40 | 3.8 (3.4, 4.1) | 3.7 (3.4, 3.9) | 0.56 |
| DOWNSTREAM OF MYC | |||||||
| PVT1 | chr8:128806779–129113499 | TC0800008848.hg.1 | 0.0 (−0.5, 0.5) | 0.75 | 4.6 (4.2, 4.9) | 4.4 (4.0, 5.0) | 0.72 |
| miR-1204 | chr8:128808208–128808274 | (same as above) | |||||
| TMEM75 | chr8:128958805–128960969 | TC0800011789.hg.1 | 0.0 (−0.3, 0.4) | 0.49 | 3.9 (3.7, 4.1) | 3.8 (3.6, 4.1) | 0.33 |
| miR-1205 | chr8:128972879–128972941 | TC0800008855.hg.1 | 0.0 (−0.7, 0.8) | 0.92 | 4.8 (4.2, 5.4) | 4.8 (4.5, 5.6) | 0.34 |
| RNU1-106P | chr8:129011377–129011540 | TC0800008856.hg.1 | 0.0 (−0.4, 0.5) | 0.96 | 13.9 (13.3, 14.5) | 14.2 (13.6, 14.9) | 0.09 |
| RNU4-25P | chr8:129022628–129022857 | TC0800011790.hg.1 | 0.2 (−0.2, 0.5) | 0.02 | 5.8 (5.5, 6.2) | 5.3 (4.9, 5.6) | 3.6 × 10−5 |
| RN7SKP226 | chr8:129232750–129232998 | TC0800008864.hg.1 | −0.1 (−0.7, 0.3) | 0.12 | 5.2 (4.7, 5.7) | 5.4 (4.9, 5.9) | 0.31 |
| miR-1206 | chr8:129021144–129021202 | TC0800008857.hg.1 | 0.4 (−0.9, 1.6) | 0.07 | 5.9 (4.1, 7.8) | 6.5 (4.9, 8.0) | 0.38 |
| miR-1207 | chr8:129061398–129061484 | TC0800008858.hg.1 | 0.1 (−0.1, 0.3) | 0.05 | 4.2 (4.1, 4.4) | 4.2 (4.1, 4.5) | 0.86 |
| miR-1208 | chr8:129162362–129162434 | TC0800008862.hg.1 | 0.0 (−0.2, 0.3) | 0.44 | 4.6 (4.5, 4.9) | 4.6 (4.4, 4.8) | 0.06 |
| Non-coding RNA | Description | Coordinates (GRCh37/hg19) | MYC CpG Site and Position (hg19) | Linear regression β (s.e.); p-Value (q-Value) a |
|---|---|---|---|---|
| PRNCR1 | prostate cancer associated non-coding RNA 1 | chr8:128092119–128104840 | CpG 216 (chr8:128753200) CpG 217 (chr8:128753221) | β = −0.022 (0.007); p = 2.0 × 10−3 (q = 0.04) β = −0.0094 (0.0039); p = 0.02 (q = 0.20) |
| CASC11 | cancer susceptibility candidate 11 (non-protein coding), transcript variant 2 | chr8:128712853–128746213 | CpG 212 (chr8:128753145) CpG 217 (chr8:128753221) | β = −0.011 (0.005); p = 0.05 (q = 0.59) β = −0.010 (0.003); p = 4.1 × 10−3 (q = 0.09) |
| miR-1206 | microRNA 1206 | chr8:129021144–129021202 | CpG 212 (chr8:128753145) | β = 0.053 (0.025); p = 0.03 (q = 0.59) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barry, K.H.; Mohanty, K.; Erickson, P.A.; Wang, D.; Shi, J.; Rose, G.; Cellini, A.; Clark, K.; Ambulos, N., Jr.; Yin, J.; et al. MYC DNA Methylation in Prostate Tumor Tissue is Associated with Gleason Score. Genes 2021, 12, 12. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010012
Barry KH, Mohanty K, Erickson PA, Wang D, Shi J, Rose G, Cellini A, Clark K, Ambulos N Jr., Yin J, et al. MYC DNA Methylation in Prostate Tumor Tissue is Associated with Gleason Score. Genes. 2021; 12(1):12. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010012
Chicago/Turabian StyleBarry, Kathryn Hughes, Kareshma Mohanty, Patricia A. Erickson, Difei Wang, Jianxin Shi, Gary Rose, Ashley Cellini, Kimberly Clark, Nicholas Ambulos, Jr., Jing Yin, and et al. 2021. "MYC DNA Methylation in Prostate Tumor Tissue is Associated with Gleason Score" Genes 12, no. 1: 12. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010012