Immune-Based Therapies and the Role of Microsatellite Instability in Pancreatic Cancer

 ,
,
Abstract
:1. Introduction
2. Lynch Syndrome and Microsatellite Instability (MSI)
3. MSI and Pancreatic Cancer
4. Chemotherapeutic Options in MSI Pancreatic Cancer Patients
5. Ongoing Studies
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, S.; Maisonneuve, P.; Lowenfels, A.B. Epidemiology of pancreatic cancer: An overview. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Kastrinos, F.; Mukherjee, B.; Tayob, N.; Wang, F.; Sparr, J.; Raymond, V.M.; Bandipalliam, P.; Stoffel, E.M.; Gruber, S.B.; Syngal, S. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009, 302, 1790–1795. [Google Scholar] [CrossRef]
- Amundadottir, L.; Kraft, P.; Stolzenberg-Solomon, R.Z.; Fuchs, C.S.; Petersen, G.M.; Arslan, A.A.; Bueno-de-Mesquita, H.B.; Gross, M.; Helzlsouer, K.; Jacobs, E.J.; et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat. Genet. 2009, 41, 986–990. [Google Scholar] [CrossRef] [Green Version]
- Wolpin, B.M.; Chan, A.T.; Hartge, P.; Chanock, S.J.; Kraft, P.; Hunter, D.J.; Giovannucci, E.L.; Fuchs, C.S. ABO blood group and the risk of pancreatic cancer. J. Natl. Cancer Inst. 2009, 101, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Wolpin, B.M.; Kraft, P.; Gross, M.; Helzlsouer, K.; Bueno-de-Mesquita, H.B.; Steplowski, E.; Stolzenberg-Solomon, R.Z.; Arslan, A.A.; Jacobs, E.J.; Lacroix, A.; et al. Pancreatic cancer risk and ABO blood group alleles: Results from the pancreatic cancer cohort consortium. Cancer Res. 2010, 70, 1015–1023. [Google Scholar] [CrossRef] [Green Version]
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694–9705. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [Green Version]
- Greer, J.B.; Whitcomb, D.C.; Brand, R.E. Genetic predisposition to pancreatic cancer: A brief review. Am. J. Gastroenterol. 2007, 102, 2564–2569. [Google Scholar] [CrossRef]
- Slebos, R.J.; Hoppin, J.A.; Tolbert, P.E.; Holly, E.A.; Brock, J.W.; Zhang, R.H.; Bracci, P.M.; Foley, J.; Stockton, P.; McGregor, L.M.; et al. K-ras and p53 in pancreatic cancer: Association with medical history, histopathology, and environmental exposures in a population-based study. Cancer Epidemiol. Biomark. Prev. 2000, 9, 1223–1232. [Google Scholar]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet. 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, T.P. Demographics, epidemiology, and inheritance of pancreatic ductal adenocarcinoma. Semin Oncol. 2015, 42, 8–18. [Google Scholar] [CrossRef]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Lohr, J.M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Teague, A.; Lim, K.H.; Wang-Gillam, A. Advanced pancreatic adenocarcinoma: A review of current treatment strategies and developing therapies. Ther. Adv. Med. Oncol. 2015, 7, 68–84. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ma, Q.; Xu, Q.; Duan, W.; Lei, J.; Wu, E. Targeting the cancer-stroma interaction: A potential approach for pancreatic cancer treatment. Curr. Pharm Des. 2012, 18, 2404–2415. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Laguna, I.; Hidalgo, M. Pancreatic cancer: From state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 2015, 12, 319–334. [Google Scholar] [CrossRef]
- Spanknebel, K.; Conlon, K.C. Advances in the surgical management of pancreatic cancer. Cancer J. 2001, 7, 312–323. [Google Scholar]
- Malvezzi, M.; Bertuccio, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2014. Ann. Oncol. 2014, 25, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.S.; Kennedy, E.B.; Khorana, A.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Krishnamurthi, S.; Moravek, C.; O’Reilly, E.M.; Philip, P.A.; et al. Metastatic Pancreatic Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 2545–2556. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Isawa, T.; Koike, M.; Tsuruta, K.; Okamoto, A. Hematogenous metastases of pancreatic ductal carcinoma. Pancreas 1995, 11, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. Canadian Cancer Trials G the Unicancer GIPG FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zulke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA 2013, 310, 1473–1481. [Google Scholar] [CrossRef] [Green Version]
- Suker, M.; Beumer, B.R.; Sadot, E.; Marthey, L.; Faris, J.E.; Mellon, E.A.; El-Rayes, B.F.; Wang-Gillam, A.; Lacy, J.; Hosein, P.J.; et al. FOLFIRINOX for locally advanced pancreatic cancer: A systematic review and patient-level meta-analysis. Lancet Oncol. 2016, 17, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Taieb, J.; Abdallah, R. How I treat pancreatic cancer. ESMO Open 2020, 4 (Suppl. 2). [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Lupinacci, R.M.; Goloudina, A.; Buhard, O.; Bachet, J.B.; Marechal, R.; Demetter, P.; Cros, J.; Bardier-Dupas, A.; Collura, A.; Cervera, P.; et al. Prevalence of Microsatellite Instability in Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gastroenterology 2018, 154, 1061–1065. [Google Scholar] [CrossRef]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A., Jr.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakata, B.; Yashiro, M.; Nishioka, N.; Aya, M.; Yamada, S.; Takenaka, C.; Ohira, M.; Ishikawa, T.; Nishino, H.; Wakasa, K.; et al. Very low incidence of microsatellite instability in intraductal papillary-mucinous neoplasm of the pancreas. Int. J. Cancer 2002, 102, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Sparr, J.A.; Bandipalliam, P.; Redston, M.S.; Syngal, S. Intraductal papillary mucinous neoplasm of the pancreas with loss of mismatch repair in a patient with Lynch syndrome. Am. J. Surg. Pathol. 2009, 33, 309–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, M.R.; Jayaraj, A.; Xiong, W.; Yeh, M.M.; Raskind, W.H.; Pillarisetty, V.G. Pancreatic intraductal papillary mucinous neoplasm in a patient with Lynch syndrome. World J. Gastroenterol. 2015, 21, 2820–2825. [Google Scholar] [CrossRef]
- Warthin, A.S. Heredity with reference to carcinoma. Arch. Intern. Med. 1913, XII, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Lynch, H.T.; Lynch, P.M.; Lanspa, S.J.; Snyder, C.L.; Lynch, J.F.; Boland, C.R. Review of the Lynch syndrome: History, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin. Genet. 2009, 76, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W.; American College of Gastroenterology. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratti, M.; Lampis, A.; Hahne, J.C.; Passalacqua, R.; Valeri, N. Microsatellite instability in gastric cancer: Molecular bases, clinical perspectives, and new treatment approaches. Cell Mol. Life Sci. 2018, 75, 4151–4162. [Google Scholar] [CrossRef]
- Valeri, N.; Gasparini, P.; Braconi, C.; Paone, A.; Lovat, F.; Fabbri, M.; Sumani, K.M.; Alder, H.; Amadori, D.; Patel, T.; et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc. Natl. Acad. Sci. USA 2010, 107, 21098–21103. [Google Scholar] [CrossRef] [Green Version]
- Valeri, N.; Gasparini, P.; Fabbri, M.; Braconi, C.; Veronese, A.; Lovat, F.; Adair, B.; Vannini, I.; Fanini, F.; Bottoni, A.; et al. Modulation of mismatch repair and genomic stability by miR-155. Proc. Natl. Acad. Sci. USA 2010, 107, 6982–6987. [Google Scholar] [CrossRef] [Green Version]
- Humphris, J.L.; Patch, A.M.; Nones, K.; Bailey, P.J.; Johns, A.L.; McKay, S.; Chang, D.K.; Miller, D.K.; Pajic, M.; Kassahn, K.S.; et al. Hypermutation In Pancreatic Cancer. Gastroenterology 2017, 152, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FCDAS; Wernhoff, P.; Dominguez-Barrera, C.; Dominguez-Valentin, M. Update on Hereditary Colorectal Cancer. Anticancer Res. 2016, 36, 4399–4405. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujanda, L.; Herreros-Villanueva, M. Pancreatic Cancer in Lynch Syndrome Patients. J. Cancer 2017, 8, 3667–3674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banville, N.; Geraghty, R.; Fox, E.; Leahy, D.T.; Green, A.; Keegan, D.; Geoghegan, J.; O’Donoghue, D.; Hyland, J.; Sheahan, K. Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum. Pathol. 2006, 37, 1498–1502. [Google Scholar] [CrossRef]
- Grover, S.; Syngal, S. Hereditary pancreatic cancer. Gastroenterology 2010, 139, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Geary, J.; Sasieni, P.; Houlston, R.; Izatt, L.; Eeles, R.; Payne, S.J.; Fisher, S.; Hodgson, S.V. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC). Fam. Cancer 2008, 7, 163–172. [Google Scholar] [CrossRef]
- Liu, W.; Shia, J.; Gonen, M.; Lowery, M.A.; O’Reilly, E.M.; Klimstra, D.S. DNA mismatch repair abnormalities in acinar cell carcinoma of the pancreas: Frequency and clinical significance. Pancreas 2014, 43, 1264–1270. [Google Scholar] [CrossRef]
- Karamurzin, Y.; Zeng, Z.; Stadler, Z.K.; Zhang, L.; Ouansafi, I.; Al-Ahmadie, H.A.; Sempoux, C.; Saltz, L.B.; Soslow, R.A.; O’Reilly, E.M.; et al. Unusual DNA mismatch repair-deficient tumors in Lynch syndrome: A report of new cases and review of the literature. Hum. Pathol. 2012, 43, 1677–1687. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, W.Y.; Hwang, D.Y.; Han, H.S. Intraductal papillary mucinous neoplasm of the ileal heterotopic pancreas in a patient with hereditary non-polyposis colorectal cancer: A case report. World J. Gastroenterol. 2015, 21, 7916–7920. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Ouyang, H.; Migita, T.; Kato, Y.; Kimura, M.; Shiiba, K.; Sunamura, M.; Matsuno, S.; Horii, A. The somatic mutation frequency of the transforming growth factor β receptor type II gene varies widely among different cancers with microsatellite instability. Eur. J. Surg. Oncol. 1996, 22, 474–477. [Google Scholar] [CrossRef]
- Yamamoto, H.; Itoh, F.; Nakamura, H.; Fukushima, H.; Sasaki, S.; Perucho, M.; Imai, K. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001, 61, 3139–3144. [Google Scholar] [PubMed]
- Abraham, S.C.; Wu, T.T.; Hruban, R.H.; Lee, J.H.; Yeo, C.J.; Conlon, K.; Brennan, M.; Cameron, J.L.; Klimstra, D.S. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: Frequent allelic loss on chromosome 11p and alterations in the APC/β-catenin pathway. Am. J. Pathol. 2002, 160, 953–962. [Google Scholar] [CrossRef]
- Tomaszewska, R.; Okon, K.; Stachura, J. Expression of the DNA mismatch repair proteins (hMLH1 and hMSH2) in infiltrating pancreatic cancer and its relation to some phenotypic features. Pol. J. Pathol. 2003, 54, 31–37. [Google Scholar] [PubMed]
- Luttges, J.; Beyser, K.; Pust, S.; Paulus, A.; Ruschoff, J.; Kloppel, G. Pancreatic mucinous noncystic (colloid) carcinomas and intraductal papillary mucinous carcinomas are usually microsatellite stable. Mod. Pathol. 2003, 16, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Maple, J.T.; Smyrk, T.C.; Boardman, L.A.; Johnson, R.A.; Thibodeau, S.N.; Chari, S.T. Defective DNA mismatch repair in long-term (> or =3 years) survivors with pancreatic cancer. Pancreatology 2005, 5, 220–227. [Google Scholar] [CrossRef]
- Nakata, B.; Wang, Y.Q.; Yashiro, M.; Nishioka, N.; Tanaka, H.; Ohira, M.; Ishikawa, T.; Nishino, H.; Hirakawa, K. Prognostic value of microsatellite instability in resectable pancreatic cancer. Clin. Cancer Res. 2002, 8, 2536–2540. [Google Scholar]
- Fujii, K.; Miyashita, K.; Yamada, Y.; Eguchi, T.; Taguchi, K.; Oda, Y.; Oda, S.; Yoshida, M.A.; Tanaka, M.; Tsuneyoshi, M. Simulation-based analyses reveal stable microsatellite sequences in human pancreatic cancer. Cancer Genet. Cytogenet. 2009, 189, 5–14. [Google Scholar] [CrossRef]
- Laghi, L.; Beghelli, S.; Spinelli, A.; Bianchi, P.; Basso, G.; Di Caro, G.; Brecht, A.; Celesti, G.; Turri, G.; Bersani, S.; et al. Irrelevance of microsatellite instability in the epidemiology of sporadic pancreatic ductal adenocarcinoma. PLoS ONE 2012, 7, e46002. [Google Scholar] [CrossRef]
- Han, H.J.; Yanagisawa, A.; Kato, Y.; Park, J.G.; Nakamura, Y. Genetic instability in pancreatic cancer and poorly differentiated type of gastric cancer. Cancer Res. 1993, 53, 5087–5089. [Google Scholar] [PubMed]
- Seymour, A.B.; Hruban, R.H.; Redston, M.; Caldas, C.; Powell, S.M.; Kinzler, K.W.; Yeo, C.J.; Kern, S.E. Allelotype of pancreatic adenocarcinoma. Cancer Res. 1994, 54, 2761–2764. [Google Scholar] [PubMed]
- Brentnall, T.A.; Chen, R.; Lee, J.G.; Kimmey, M.B.; Bronner, M.P.; Haggitt, R.C.; Kowdley, K.V.; Hecker, L.M.; Byrd, D.R. Microsatellite instability and K-ras mutations associated with pancreatic adenocarcinoma and pancreatitis. Cancer Res. 1995, 55, 4264–4267. [Google Scholar] [PubMed]
- Venkatasubbarao, K.; Ahmed, M.M.; Swiderski, C.; Harp, C.; Lee, E.Y.; McGrath, P.; Mohiuddin, M.; Strodel, W.; Freeman, J.W. Novel mutations in the polyadenine tract of the transforming growth factor β type II receptor gene are found in a subpopulation of human pancreatic adenocarcinomas. Genes Chromosomes Cancer. 1998, 22, 138–144. [Google Scholar] [CrossRef]
- Ouyang, H.; Shiwaku, H.O.; Hagiwara, H.; Miura, K.; Abe, T.; Kato, Y.; Ohtani, H.; Shiiba, K.; Souza, R.F.; Meltzer, S.J.; et al. The insulin-like growth factor II receptor gene is mutated in genetically unstable cancers of the endometrium, stomach, and colorectum. Cancer Res. 1997, 57, 1851–1854. [Google Scholar]
- Ouyang, H.; Furukawa, T.; Abe, T.; Kato, Y.; Horii, A. The BAX gene, the promoter of apoptosis, is mutated in genetically unstable cancers of the colorectum, stomach, and endometrium. Clin. Cancer Res. 1998, 4, 1071–1074. [Google Scholar]
- Goggins, M.; Offerhaus, G.J.; Hilgers, W.; Griffin, C.A.; Shekher, M.; Tang, D.; Sohn, T.A.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am. J. Pathol. 1998, 152, 1501–1507. [Google Scholar]
- Ghimenti, C.; Tannergard, P.; Wahlberg, S.; Liu, T.; Giulianotti, P.G.; Mosca, F.; Fornaciari, G.; Bevilacqua, G.; Lindblom, A.; Caligo, M.A. Microsatellite instability and mismatch repair gene inactivation in sporadic pancreatic and colon tumours. Br. J. Cancer 1999, 80, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Wilentz, R.E.; Goggins, M.; Redston, M.; Marcus, V.A.; Adsay, N.V.; Sohn, T.A.; Kadkol, S.S.; Yeo, C.J.; Choti, M.; Zahurak, M.; et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: A newly described and characterized entity. Am. J. Pathol. 2000, 156, 1641–1651. [Google Scholar] [CrossRef]
- Ueki, T.; Toyota, M.; Sohn, T.; Yeo, C.J.; Issa, J.P.; Hruban, R.H.; Goggins, M. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000, 60, 1835–1839. [Google Scholar]
- Nakata, B.; Wang, Y.Q.; Yashiro, M.; Ohira, M.; Ishikawa, T.; Nishino, H.; Seki, S.; Hirakawa, K. Negative hMSH2 protein expression in pancreatic carcinoma may predict a better prognosis of patients. Oncol. Rep. 2003, 10, 997–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottenhof, N.A.; Morsink, F.H.; Ten Kate, F.; van Noorden, C.J.; Offerhaus, G.J. Multivariate analysis of immunohistochemical evaluation of protein expression in pancreatic ductal adenocarcinoma reveals prognostic significance for persistent Smad4 expression only. Cell Oncol. 2012, 35, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuhashi, K.; Nosho, K.; Sukawa, Y.; Matsunaga, Y.; Ito, M.; Kurihara, H.; Kanno, S.; Igarashi, H.; Naito, T.; Adachi, Y.; et al. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget 2015, 6, 7209–7220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riazy, M.; Kalloger, S.E.; Sheffield, B.S.; Peixoto, R.D.; Li-Chang, H.H.; Scudamore, C.H.; Renouf, D.J.; Schaeffer, D.F. Mismatch repair status may predict response to adjuvant chemotherapy in resectable pancreatic ductal adenocarcinoma. Mod. Pathol. 2015, 28, 1383–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, R.C.; Selander, I.; Connor, A.A.; Selvarajah, S.; Borgida, A.; Briollais, L.; Petersen, G.M.; Lerner-Ellis, J.; Holter, S.; Gallinger, S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015, 148, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Electronic address aadhe; Cancer Genome Atlas Research, N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: Histology, molecular pathology and clinical implications. Gut 2020. [Google Scholar] [CrossRef]
- Cortes-Ciriano, I.; Lee, S.; Park, W.Y.; Kim, T.M.; Park, P.J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef] [Green Version]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis Oncol. 2017, 2017. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.W.; Yang, S.Z.; Gao, J.; Pan, K.; Chen, J.Y.; Wang, Y.L.; Wei, L.X.; Dong, J.H. Prognostic significance of mast cell count following curative resection for pancreatic ductal adenocarcinoma. Surgery 2011, 149, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xu, X.; Guo, S.; Zhang, C.; Tang, Y.; Tian, Y.; Ni, B.; Lu, B.; Wang, H. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS ONE 2014, 9, e91551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, N.B.; Mohamed, M.; Oien, K.A.; Foulis, A.K.; Dickson, E.J.; Imrie, C.W.; Carter, C.R.; McKay, C.J.; McMillan, D.C. The relationship between tumor inflammatory cell infiltrate and outcome in patients with pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2012, 19, 3581–3590. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Mitsunaga, S.; Kinoshita, T.; Konishi, M.; Takahashi, S.; Gotohda, N.; Kato, Y.; Aizawa, M.; Ochiai, A. Impact of tumor-associated macrophages on invasive ductal carcinoma of the pancreas head. Cancer Sci. 2012, 103, 2012–2020. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Carstens, J.L.; Correa de Sampaio, P.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef]
- Tahkola, K.; Mecklin, J.P.; Wirta, E.V.; Ahtiainen, M.; Helminen, O.; Bohm, J.; Kellokumpu, I. High immune cell score predicts improved survival in pancreatic cancer. Virchows Arch. 2018, 472, 653–665. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Liao, Q.; Zhao, Y. Chemotherapy and tumor microenvironment of pancreatic cancer. Cancer Cell Int. 2017, 17, 68. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology 2013, 144, 1230–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.A., 3rd; Yarchoan, M.; Lee, V.; Laheru, D.A.; Jaffee, E.M. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin. Cancer Res. 2017, 23, 1656–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Velez-Delgado, A.; Mathew, E.; Li, D.; Mendez, F.M.; Flannagan, K.; Rhim, A.D.; Simeone, D.M.; Beatty, G.L.; Pasca di Magliano, M. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017, 66, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Kamatham, S.; Shahjehan, F.; Kasi, P.M. Circulating Tumor DNA-Based Detection of Microsatellite Instability and Response to Immunotherapy in Pancreatic Cancer. Front. Pharmacol. 2020, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhang, H.; Chen, N.; Hao, J.; Jin, H.; Ma, X. Diagnostic value of various liquid biopsy methods for pancreatic cancer: A systematic review and meta-analysis. Medicine 2020, 99, e18581. [Google Scholar] [CrossRef]
- Carethers, J.M.; Smith, E.J.; Behling, C.A.; Nguyen, L.; Tajima, A.; Doctolero, R.T.; Cabrera, B.L.; Goel, A.; Arnold, C.A.; Miyai, K.; et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology 2004, 126, 394–401. [Google Scholar] [CrossRef]
- Benatti, P.; Gafa, R.; Barana, D.; Marino, M.; Scarselli, A.; Pedroni, M.; Maestri, I.; Guerzoni, L.; Roncucci, L.; Menigatti, M.; et al. Microsatellite instability and colorectal cancer prognosis. Clin. Cancer Res. 2005, 11, 8332–8340. [Google Scholar] [CrossRef] [Green Version]
- De Vos tot Nederveen Cappel, W.H.; Meulenbeld, H.J.; Kleibeuker, J.H.; Nagengast, F.M.; Menko, F.H.; Griffioen, G.; Cats, A.; Morreau, H.; Gelderblom, H.; Vasen, H.F. Survival after adjuvant 5-FU treatment for stage III colon cancer in hereditary nonpolyposis colorectal cancer. Int. J. Cancer 2004, 109, 468–471. [Google Scholar] [CrossRef]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Pinol, V.; Xicola, R.M.; Bujanda, L.; Rene, J.M.; et al. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut 2006, 55, 848–855. [Google Scholar] [CrossRef] [Green Version]
- Jacob, S.; Aguado, M.; Fallik, D.; Praz, F. The role of the DNA mismatch repair system in the cytotoxicity of the topoisomerase inhibitors camptothecin and etoposide to human colorectal cancer cells. Cancer Res. 2001, 61, 6555–6562. [Google Scholar] [PubMed]
- Fallik, D.; Borrini, F.; Boige, V.; Viguier, J.; Jacob, S.; Miquel, C.; Sabourin, J.C.; Ducreux, M.; Praz, F. Microsatellite instability is a predictive factor of the tumor response to irinotecan in patients with advanced colorectal cancer. Cancer Res. 2003, 63, 5738–5744. [Google Scholar]
- Duval, A.; Hamelin, R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: Toward a new concept of target genes for instability. Cancer Res. 2002, 62, 2447–2454. [Google Scholar] [PubMed]
- Sinicrope, F.A.; Sargent, D.J. Molecular pathways: Microsatellite instability in colorectal cancer: Prognostic, predictive, and therapeutic implications. Clin. Cancer Res. 2012, 18, 1506–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, D.; Aebi, S.; Howell, S.B. The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 1998, 4, 1–6. [Google Scholar]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Bergman, A.M.; Pinedo, H.M.; Peters, G.J. Determinants of resistance to 2′,2′-difluorodeoxycytidine (gemcitabine). Drug Resist. Updat. 2002, 5, 19–33. [Google Scholar] [CrossRef]
- Cloyd, J.M.; Katz, M.H.G.; Wang, H.; Cuddy, A.; You, Y.N. Clinical and Genetic Implications of DNA Mismatch Repair Deficiency in Patients with Pancreatic Ductal Adenocarcinoma. JAMA Surg. 2017, 152, 1086–1088. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, A.; Mahalingam, D. Immunotherapy in pancreatic adenocarcinoma-overcoming barriers to response. J. Gastrointest Oncol. 2018, 9, 143–159. [Google Scholar] [CrossRef] [Green Version]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Atkins, M.B. PD-1 as a potential target in cancer therapy. Cancer Med. 2013, 2, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475, Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schutz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. N. Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef]
- Plate, J.M.; Plate, A.E.; Shott, S.; Bograd, S.; Harris, J.E. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol. Immunother. 2005, 54, 915–925. [Google Scholar] [CrossRef]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [Green Version]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef] [Green Version]
- Nowak, A.K.; Robinson, B.W.; Lake, R.A. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res. 2003, 63, 4490–4496. [Google Scholar]
- Wainberg, Z.A.; Hochster, H.S.; Kim, E.J.; George, B.; Kaylan, A.; Chiorean, E.G.; Waterhouse, D.M.; Guiterrez, M.; Parikh, A.; Jain, R.; et al. Open-label, Phase I Study of Nivolumab Combined with nab-Paclitaxel Plus Gemcitabine in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4814–4822. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, T.; Crocenzi, T.; Durham, J.N.; Sugar, E.A.; Wu, A.A.; Onners, B.; Nauroth, J.M.; Anders, R.A.; Fertig, E.J.; Laheru, D.A.; et al. Evaluation of Cyclophosphamide/GVAX Pancreas Followed by Listeria-Mesothelin (CRS-207) with or without Nivolumab in Patients with Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 3578–3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borazanci, E.; Jameson, G.S.; Borad, M.; Ramanathan, R.K.; Korn, R.L.; Caldwell, L.; Ansaldo, K.; Hendrickson, K.; Marceau, K.; Von Hoff, D.D. A phase II pilot trial of nivolumab (N) + albumin bound paclitaxel (AP) + paricalcitol (P) + cisplatin (C) + gemcitabine (G) (NAPPCG) in patients with previously untreated metastatic pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Piha-Paul, S.A.; Luke, J.J. First-inhuman phase 1 dose escalation and expansion of a novel combination, anti–CSF-1 receptor (cabiralizumab) plus anti–PD-1 (nivolumab), in patients with advanced solid tumors. In Proceedings of the 32nd SITC Annual Meeting, National Harbor, MD, USA, 8–12 November 2017. [Google Scholar]
- Calvo, A.; Joensuu, H.; Sebastian, M. Phase Ib/II study of lacnotuzumab (MCS110) combined with spartalizumab (PDR001) in patients (pts) with advanced tumors. J. Clin. Oncol. 2018, 36, S3014. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; McWilliams, R.; Lockhart, A.C.; Tan, B.R.; Suresh, R.; Lim, K.-H.; Pedersen, K.S.; Trikalinos, N.; Aranha, O.; Park, H.S.; et al. Phase I study of defactinib combined with pembrolizumab and gemcitabine in patients with advanced cancer: Experiences of pancreatic ductal adenocarcinoma (PDAC) patients. Cancer Res. 2020, 80, CT118. [Google Scholar] [CrossRef]
- Hong, D.; Rasco, D.; Veeder, M.; Luke, J.J.; Chandler, J.; Balmanoukian, A.; George, T.J.; Munster, P.; Berlin, J.D.; Gutierrez, M.; et al. A Phase 1b/2 Study of the Bruton Tyrosine Kinase Inhibitor Ibrutinib and the PD-L1 Inhibitor Durvalumab in Patients with Pretreated Solid Tumors. Oncology 2019, 97, 102–111. [Google Scholar] [CrossRef]
- Overman, M.J.; Lorusso, P.; Strickler, J.H.; Patel, S.P.; Clarke, S.J.; Noonan, A.M.; Prasanna, T.; Amin, M.A.; Nemunaitis, J.J.; Desai, J.; et al. Safety, efficacy and pharmacodynamics (PD) of MEDI9447 (oleclumab) alone or in combination with durvalumab in advanced colorectal cancer (CRC) or pancreatic cancer (panc). J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Whiting, C.; Lutz, E.; Nair, N.; Chang, S.C.; Lemmens, V.E.; Chen, S.-J.; Solt, S.; Ferber, S.; Maecker, H.; Murphy, A.; et al. Phase II, randomized study of GVAX pancreas and CRS-207 immunotherapy in patients with metastatic pancreatic cancer: Clinical update on long term survival and biomarker correlates to overall survival. J. Clin. Oncol. 2015, 33 (Suppl. 261). [Google Scholar] [CrossRef]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.; Morse, M.A.; Zeh, H.J., 3rd; Weekes, C.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef]
- Perez, K.; Cleary, J.M.; Karasic, T.B.; Raghavan, S.; Rahma, O.E.; Nowak, J.; Borazanci, E.; Downes, M.; Drebin, J.A.; Tuveson, D.A.; et al. Vitamin D receptor agonist paricalcitol plus gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Steins, A.; van Mackelenbergh, M.G.; van der Zalm, A.P.; Klaassen, R.; Serrels, B.; Goris, S.G.; Kocher, H.M.; Waasdorp, C.; de Jong, J.H.; Tekin, C.; et al. High-grade mesenchymal pancreatic ductal adenocarcinoma drives stromal deactivation through CSF-1. EMBO Rep. 2020, 21, e48780. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Berlin, J.; Cardin, D. Harnessing the Immune System in Pancreatic Cancer. Curr. Treat. Options Oncol. 2018, 19, 48. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef]
- Tempero, M.A.; Oh, D.Y.; Macarulla, T.; Reni, M.; Van Cutsem, E.; Hendifar, A.; Waldschmidt, D.; Starling, N.; Bachet, J.; Chang, H.; et al. Ibrutinib in combination with nab-paclitaxel and gemcitabine as first-line treatment for patients with metastatic pancreatic adenocarcinoma: Results from the phase 3 RESOLVE study. Ann. Oncol. 2019. [Google Scholar] [CrossRef]
- Hecht, J.R.; Naing, A.; Falchook, G.; Patel, M.R.; Infante, J.R.; Aljumaily, R.; Lee Wong, D.J.; Autio, K.A.; Wainberg, Z.A.; Javle, M.; et al. Phase 1b study with PEGylated human IL-10 (AM0010) with 5-FU and oxaliplatin (FOLFOX) in metastatic pancreatic adenocarcinoma (PDAC). J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Hecht, J.R.; Lonardi, S.; Bendell, J.C.; Sim, H.-W.; Macarulla, T.; Lopez, C.D.; Van Cutsem, E.; Munoz Martin, A.J.; Park, J.O.; Grell, R.; et al. Randomized Phase III Study of FOLFOX Alone and with Pegilodecakin as Second-line Therapy in Patients with Metastatic Pancreatic Cancer (SEQUOIA). J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- Jin, D.; Fan, J.; Wang, L.; Thompson, L.F.; Liu, A.; Daniel, B.J.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 on tumor cells impairs antitumor T-cell responses: A novel mechanism of tumor-induced immune suppression. Cancer Res. 2010, 70, 2245–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Tan, X.L.; Xu, Y.; Wang, Z.Z.; Xiao, C.H.; Liu, R. Expression and Prognostic Value of Indoleamine 2,3-dioxygenase in Pancreatic Cancer. Chin. Med. J. 2017, 130, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahary, N.; Garrido-Laguna, I.; Wang-Gillam, A.; Nyak-Kapoor, A.; Kennedy, E.; Vahanian, N.N.; Link, C.J. Results of the phase Ib portion of a phase I/II trial of the indoleamine 2,3-dioxygenase pathway (IDO) inhibitor indoximod plus gemcitabine/nab-paclitaxel for the treatment of metastatic pancreatic cancer. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; O’Reilly, E.M.; Bendell, J.C.; Wainberg, Z.A.; Borazanci, E.H.; Bahary, N.; O’Hara, M.H.; Beatty, G.L.; Pant, S.; Cohen, D.J. A randomized phase II study of cabiralizumab (cabira) + nivolumab (nivo) ± chemotherapy (chemo) in advanced pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Beatty, G.L.; Li, Y.; Long, K.B. Cancer immunotherapy: Activating innate and adaptive immunity through CD40 agonists. Expert Rev. Anticancer Ther. 2017, 17, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.M.; Epelbaum, R.; Semenisty, V.; Geva, R.; Golan, T.; Borazanci, E.H.; Stemmer, S.M.; Borad, M.J.; Park, J.O.; Pedersen, K. Evaluation of pharmacodynamic (PD) biomarkers in patients with metastatic pancreatic cancer treated with BL-8040, a novel CXCR4 antagonist. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Chumsri, S.N.B.M.; Ordentlich, P.; Advani, P.; Moreno-Aspitia, A.; McLaughlin, S.A.; Geiger, X.; McDonough, M.; Vallow, L.A.; Perez, E.A.; Thompson, E.A. Immunomodulatory effects of entinostat on PD-L1 and MHC class I and II in different subtypes of breast cancer. Cancer Res. 2016. [Google Scholar] [CrossRef]
- Macherla, S.; Laks, S.; Naqash, A.R.; Bulumulle, A.; Zervos, E.; Muzaffar, M. Emerging Role of Immune Checkpoint Blockade in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 3505. [Google Scholar] [CrossRef] [Green Version]
- Vanderwalde, A.; Spetzler, D.; Xiao, N.; Gatalica, Z.; Marshall, J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018, 7, 746–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]


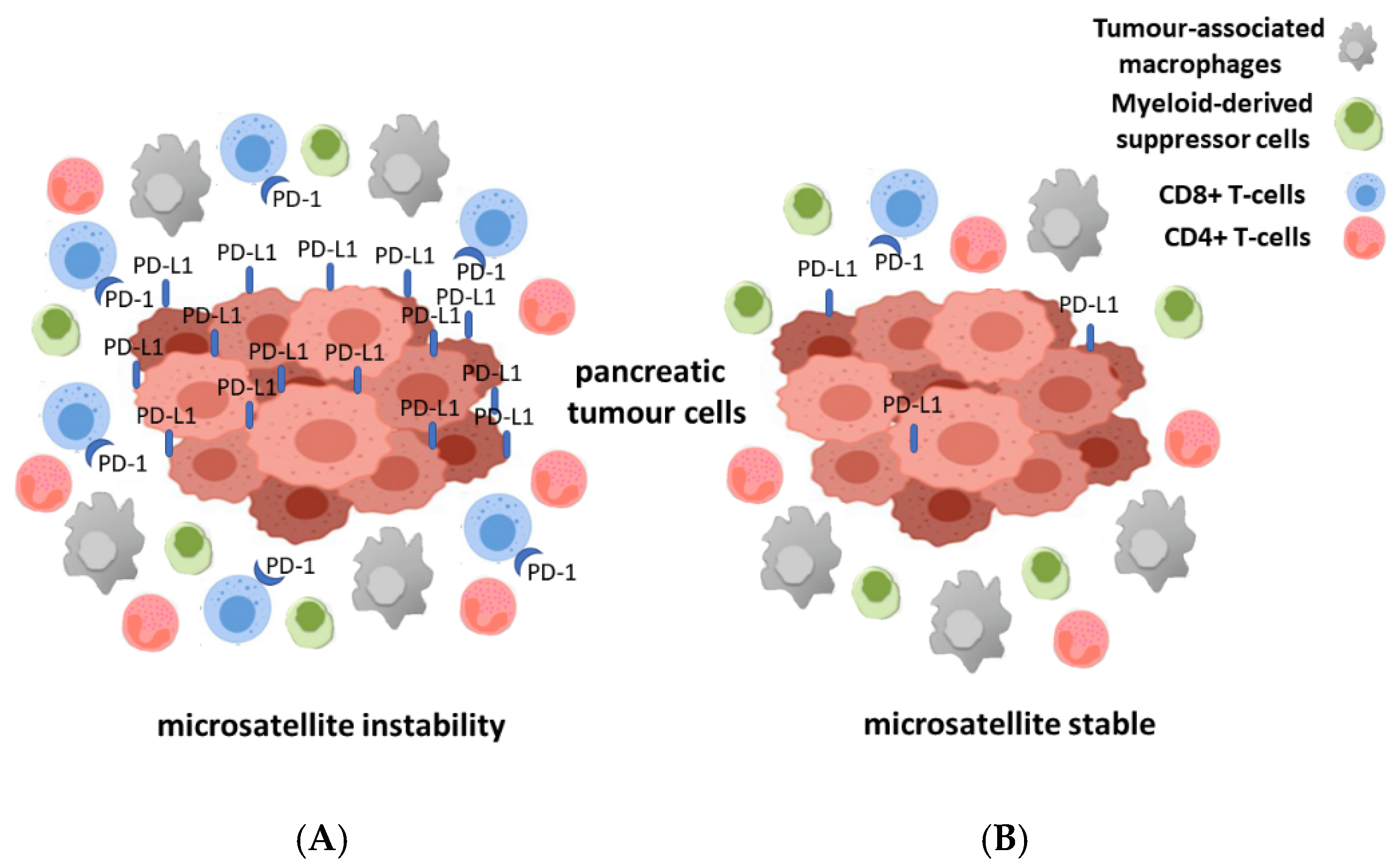{kind=link}
| Author/Year | Study Population | Methodology | MSI in % |
|---|---|---|---|
| Luipinacci/2018 [30] | 445 | IHC on resected samples from consecutive patients at multiple centers | 1.6 |
| Hu/2018 [31] | 833 | NGS, PCR-based and IHC on resected samples from consecutive patients | 0.8 |
| Liu/2014 [49] | 36 | IHC on resected and metastatic selected patients with acinar cell carcinoma | 13.8 |
| Abe/1996 [52] | 44 | PCR based on resected samples | 15.9 |
| Yamamoto/2001 [53] | 103 | PCR-based and IHC on resected samples from partially selected patients (3 Lynch Syndrome patients added to a series of 100 patients from multiple centers) | 15.5 |
| Abraham/2002 [54] | 21 | PCR based on resected samples (17 patients) and core biopsies (4 patients) from selected patients with acinar cell carcinoma | 7.7 |
| Tomaszewska/2003 [55] | 30 | IHC on resected samples from consecutive patients | 0.0 |
| Luttges/2003 [56] | 23 | PCR-based and IHC on resected samples from selected patients (extensive invasive mucinous component) | 4.3 |
| Maple/2005 [57] | 35 | PCR-based and IHC on selected patients (long-term survivors; ≥3 years) | 8.6 |
| Nakata/2002 [58] | 46 | PCR based on resected samples from consecutive patients | 17.4 |
| Fujii/2009 [59] | 21 | PCR based on resected samples | 0.0 |
| Laghi/2012 [60] | 338 | PCR-based and IHC on samples from consecutive patients at multiple centers | 0.3 |
| Han/1993 [61] | 9 | PCR based on resected samples | 67.0 |
| Seymour/1994 [62] | 33 | PCR based on resected samples | 21.2 |
| Brentnall/1995 [63] | 17 | PCR based on pancreatic juice | 75.0 |
| Venkatasubbarao/1998 [64] | 14 | PCR based on resected samples | 57.0 |
| Ouyang/1997 [65] | 51 | PCR based on resected samples | 14.0 |
| Ouyang/1998 [66] | 60 | PCR based on resected samples | 15.0 |
| Goggins/1998 [67] | 82 | PCR based on samples from consecutive patients | 3.7 |
| Ghimenti/1999 [68] | 21 | PCR based on samples from consecutive patients | 67.0 |
| Wilentz/2000 [69] | 18 | IHC and PCR based on samples from selected patients with medullary histology | 22.2 |
| Ueki/2000 [70] | 36 | PCR based on resected samples | 11.1 |
| Nakata/2003 [71] | 55 | IHC on resected samples | 9.2 |
| Ottenhof/2012 [72] | 78 | IHC on resected samples from patients at multiple centers | 12.8 |
| Mitsuhaski/2015 [73] | 283 | Methodology not specified | 0.0 |
| Riazy/2015 [74] | 265 | IHC on resected samples from consecutive patients | 15.4 |
| Grant/2015 [75] | 38 | NGS on blood samples from patients | 2.6 |
| Connor/2017 [76] | 255 | NGS, PCR-based and IHC on resected samples (243 primary tumors and 12 metastases) | 1.7 |
| Lucchini/2020 [78] | 8323 | Systematic review of 34 studies | 2.0 |
| Author/Year | Study Phase | N° pts | N° MSI-H/PD-L1-H (%) | Anti-PD-1/PD-L1 Agent | Combination Agent | Outcomes |
|---|---|---|---|---|---|---|
| Weiss/2018 [116] | I/II | 17 | 9 (53) MSI-H | pembro | gemcitabine/nab-paclitaxel | DCR 100%, mPFS 9.1 mo, mOS 15 mo |
| Wainberg/2020 [121] | I | 50 | 12 (24) PD-L1 ≥ 1 | nivo | gemcitabine/nab-paclitaxel | mPFS 5.5 mo, mOS 9.9 mo |
| O’Reilly/2019 [122] | II | 65 | 8 (12) PD-L1 ≥ 25 | durva | tremelimumab | DCR 9.4%, PR 3.1% |
| Tsujikawa/2020 [123] | II | 93 | NA | nivo | Cy/GVAX/CRS-207 | mOS 5.9 mo |
| Borazanci/2018 [124] | II | 25 | NA | nivo | paricalcitol, gemcitabine/nab-paclitaxel/cisplatin | PR 80%, DCR 100%, mPFS 8.2 mo, mOS NR |
| Wainberg/2017 [125] | I | 31 | NA | nivo | cabiralizumab | ORR 13% in MSS |
| Calvo/2018 [126] | I/II | 50 | NA | sparta | lacnotuzumab | DCR 46% |
| Wang-Gillam/2020 [127] | I | 20 | NA | pembro | defactinib/ gemcitabine | mPFS 2.9 mo, mOS 7.6 mo |
| Hong/2019 [128] | I/II | 49 | NA | durva | ibrutinib | mOS 4 mo |
| Overman/2018 [129] | I | 20 | NA | durva | oleclumab | PR 10%, DCR 25% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghidini, M.; Lampis, A.; Mirchev, M.B.; Okuducu, A.F.; Ratti, M.; Valeri, N.; Hahne, J.C. Immune-Based Therapies and the Role of Microsatellite Instability in Pancreatic Cancer. Genes 2021, 12, 33. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010033
Ghidini M, Lampis A, Mirchev MB, Okuducu AF, Ratti M, Valeri N, Hahne JC. Immune-Based Therapies and the Role of Microsatellite Instability in Pancreatic Cancer. Genes. 2021; 12(1):33. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010033
Chicago/Turabian StyleGhidini, Michele, Andrea Lampis, Milko B. Mirchev, Ali Fuat Okuducu, Margherita Ratti, Nicola Valeri, and Jens C. Hahne. 2021. "Immune-Based Therapies and the Role of Microsatellite Instability in Pancreatic Cancer" Genes 12, no. 1: 33. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12010033