
An Investigation of the Role of Common and Rare Variants in a Large Italian Multiplex Family of Multiple Sclerosis Patients

 , , , , , ,
, , , , , ,  , , , , and
, , , , and
Abstract
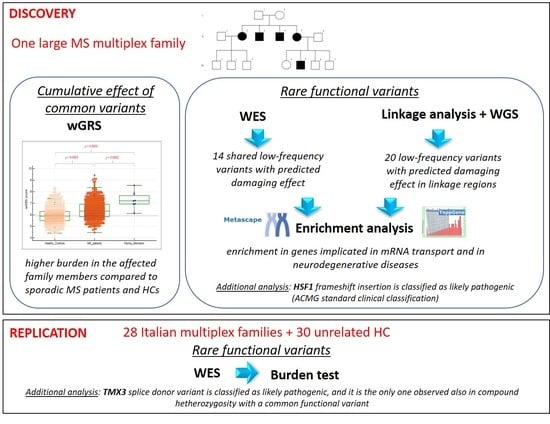
:
1. Introduction
2. Materials and Methods
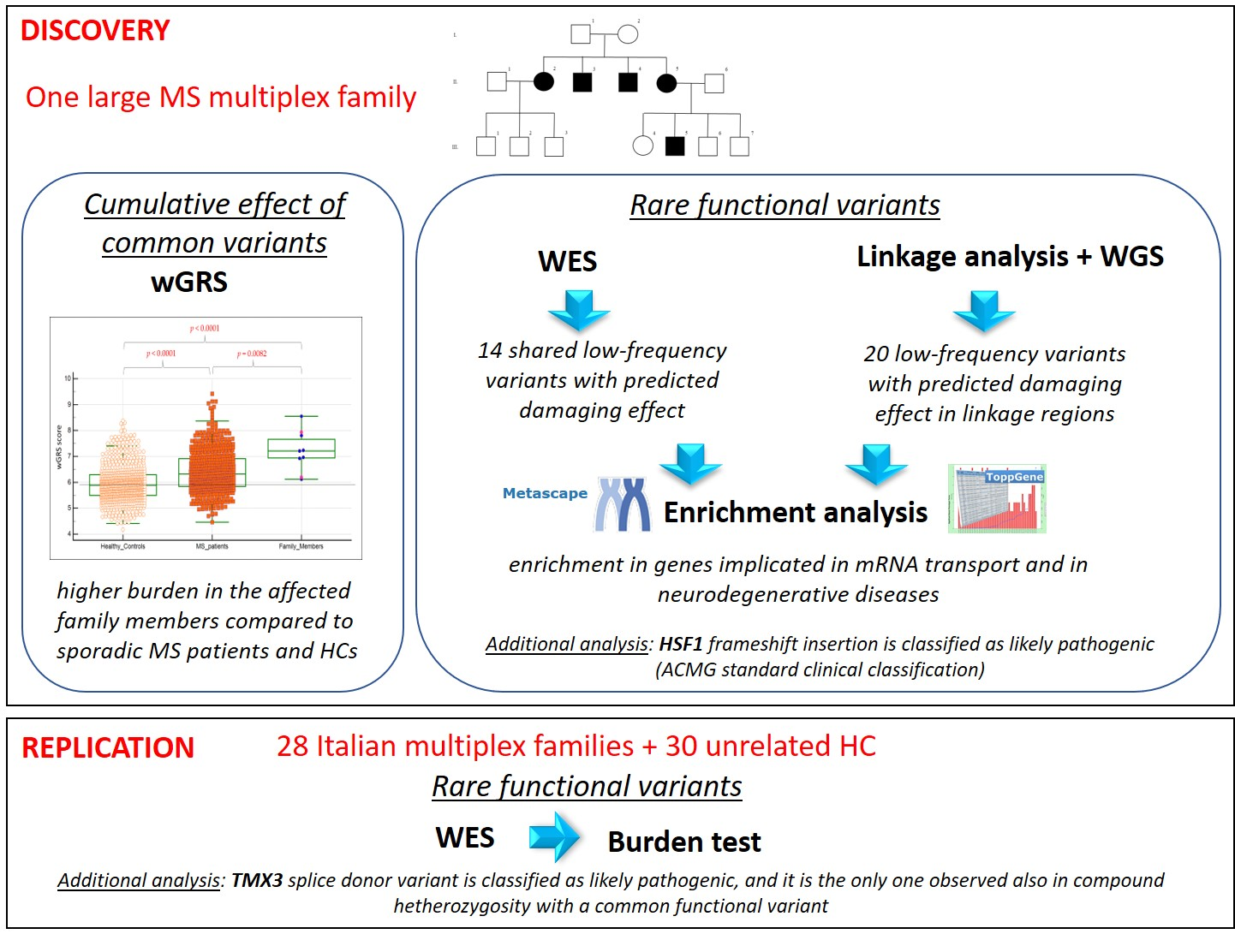
2.1. Index Family
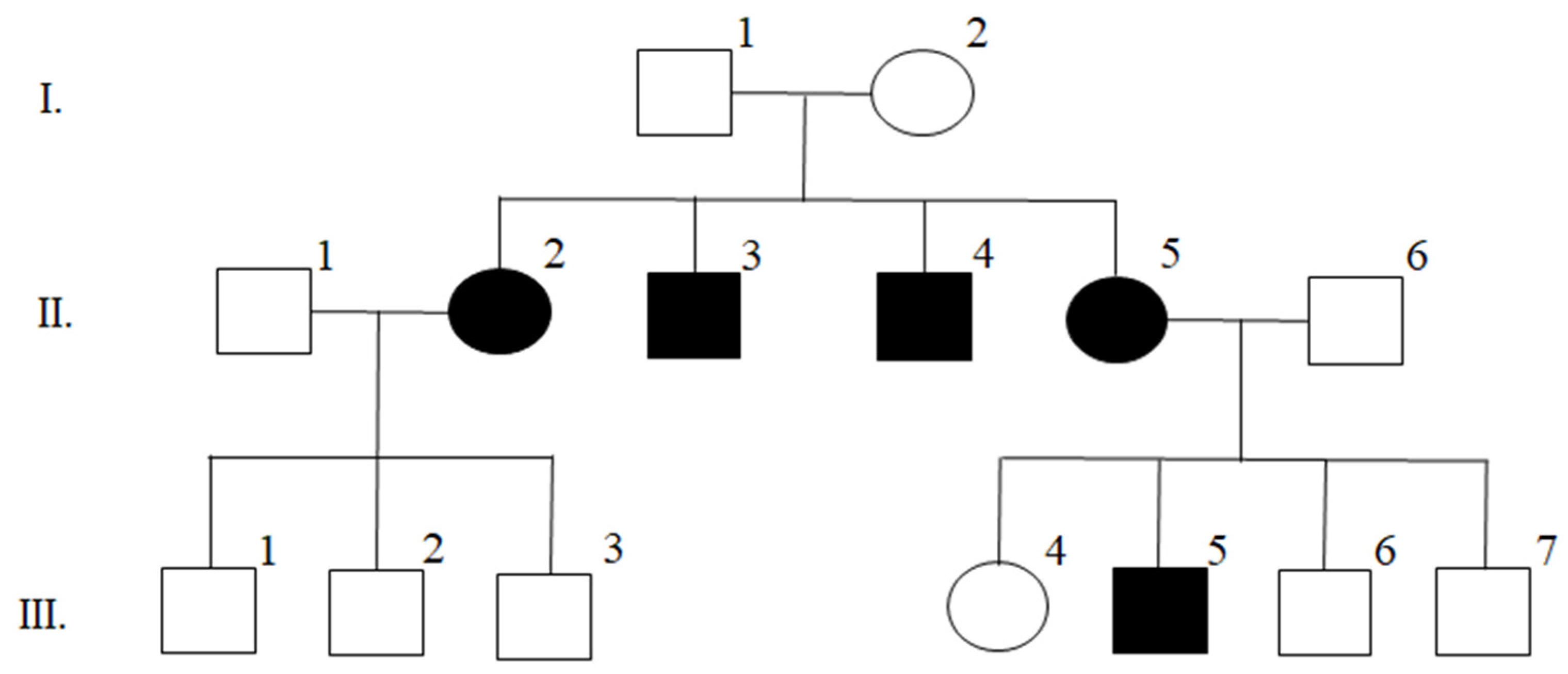
2.2. Weighted Genetic Risk Score
2.3. Whole Exome Sequencing (WES) and Filtering Pipeline
- -
- -
- -
- All shared low-frequency variants belonging to categories with an expected high functional impact (i.e., splicing, nonframe/frameshift indels, stopgain, stoploss variants) were selected.
- -
2.4. Linkage-Analysis
- (a)
- 42,764 variants whose genotypes were inferred from the WES data (for II.1,2,3,4; III.3,5 individuals); and
- (b)
- 23,443 variants previously obtained from a genotyping platform (exome chip, IMSGC, Cell, 2018) containing 247,871 SNPs, mainly derived from published exome sequencing studies also including rare variants. Standard quality controls were applied as previously described [5] (for I.2; II.1,2,3,4; III.3,5 individuals).
2.5. Whole Genome Sequencing (WGS) and Filtering Pipeline
- -
- for all the coding variants, we applied the same filtering pipeline described for WES.
- -
- for non-coding (ncRNA, UTR, intronic, upstream) variants, we selected those with a SpliceAI score > 0.4 [21]. SpliceAI is a deep learning-based tool that allows the identification of putative splice variants.
2.6. Association of Single Variants in Large International Datasets
2.7. Gene-Based Burden Analysis
2.8. Enrichment Analysis
3. Results
3.1. Description of the Index Family and Workflow of the Study
3.2. Role of Classical HLA Alleles
3.3. Burden of Common MS Susceptibility Variants
3.4. Identification of Shared Low-Frequency Functional Variants in the Index Family by Whole Exome Sequencing
3.5. Linkage Analysis on Index Family
3.6. Identification of Low-Frequency Functional Variants in Linkage Regions
3.7. Gene-Based Analysis of Identified Genes in an Independent Cohort
3.8. Enrichment Analysis of Genes Carrying Selected Low-Frequency Functional Variants Shared within the Index Multiplex Family
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Raine, C.S. Multiple sclerosis: Immune system molecule expression in the central nervous system. J. Neuropathol. Exp. Neurol. 1994, 53, 328–337. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium (IMSGC). The Multiple Sclerosis Genomic Map: Role of peripheral immune cells and resident microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodmer, W.; Bonilla, C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008, 40, 695–701. [Google Scholar] [CrossRef] [Green Version]
- Schork, N.J.; Murray, S.S.; Frazer, K.A.; Topol, E.J. Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 2009, 19, 212–219. [Google Scholar] [CrossRef] [Green Version]
- International Multiple Sclerosis Genetics Consortium (IMSGC). Low frequency and rare coding variation contributes to multiple sclerosis risk. Cell 2018, 175, 1679–1687.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Gorman, C.; Lin, R.; Stankovich, J.; Broadley, S.A. Modelling genetic susceptibility to multiple sclerosis with family data. Neuroepidemiology 2012, 40, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sawcer, S. The complex genetics of multiple sclerosis: Pitfalls and prospects. Brain 2008, 131, 3118–3131. [Google Scholar] [CrossRef] [Green Version]
- Harirchian, M.H.; Fatehi, F.; Sarraf, P.; Honarvar, N.M.; Bitarafan, S. Worldwide prevalence of familial multiple sclerosis: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2018, 20, 43–47. [Google Scholar] [CrossRef]
- Dyment, D.A.; Cader, M.Z.; Herrera, B.M.; Ramagopalan, S.V.; Orton, S.M.; Chao, M.; Willer, C.J.; Sadovnick, A.D.; Risch, N.; Ebers, G.C. A genome scan in a single pedigree with a high prevalence of multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 158–162. [Google Scholar] [CrossRef]
- Eraksoy, M.; Kurtuncu, M.; Akman-Demir, G.; Kilinc, M.; Gedizlioglu, M.; Mirza, M.; Anlar, O.; Kutlu, C.; Demirkiran, M.; Idrisoglu, H.A.; et al. A whole genome screen for linkage in Turkish multiple sclerosis. J. Neuroimmunol. 2003, 143, 17–24. [Google Scholar] [CrossRef]
- Broadly, S.; Sawcer, S.; D’Alfonso, S.; Hensiek, H.; Coraddu, F.; Gray, J.; Roxburgh, R.; Clayton, D.; Buttinelli, C.; Quattrone, A.; et al. A genome screen for multiple sclerosis in Italian families. Genes Immun. 2001, 2, 205–210. [Google Scholar] [CrossRef] [PubMed]
- The International Multiple Sclerosis Genetics Consortium (IMSGC). A High-Density Screen for Linkage in Multiple Sclerosis. Am. J. Hum. Genet. 2005, 77, 454–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mescheriakova, J.Y.; Verkerk, A.J.; Amin, N.; Uitterlinden, A.G.; van Duijn, C.M.; Hintzen, R.Q. Linkage analysis and whole exome sequencing identify a novel candidate gene in a Dutch multiple sclerosis family. Mult. Scler. J. 2019, 25, 909–917. [Google Scholar] [CrossRef]
- Sadovnick, A.D.; Gu, B.J.; Traboulsee, A.L.; Bernales, C.Q.; Encarnacion, M.; Yee, I.M.; Criscuoli, M.G.; Huang, X.; Ou, A.; Milligan, C.J.; et al. Purinergic receptors P2RX4 and P2RX7 in familial multiple sclerosis. Hum. Mutat. 2017, 38, 736–744. [Google Scholar] [CrossRef] [Green Version]
- Vidmar, L.; Maver, A.; Drulović, J.; Sepčić, J.; Novaković, I.; Ristič, S.; Šega, S.; Peterlin, B. Multiple Sclerosis patients carry an increased burden of exceedingly rare genetic variants in the inflammasome regulatory genes. Sci. Rep. 2019, 9, 9171. [Google Scholar] [CrossRef] [Green Version]
- De Jager, P.L.; Chibnik, L.B.; Cui, J.; Reischl, J.; Lehr, S.; Simon, K.C.; Aubin, C.; Bauer, D.; Heubach, J.F.; Sandbrink, R.; et al. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility: A weighted genetic risk score. Lancet Neurol. 2009, 8, 1111–1119. [Google Scholar] [CrossRef] [Green Version]
- International Multiple Sclerosis Genetics Consortium (IMSGC); Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [PubMed]
- Dilthey, A.T.; Moutsianas, L.; Leslie, S.; McVean, G. HLA*IMP—An integrated framework for imputing classical HLA alleles from SNP genotypes. Bioinformatics 2011, 27, 968–972. [Google Scholar] [CrossRef] [Green Version]
- Moutsianas, L.; Jostins, L.; Beecham, A.H.; Dilthey, A.T.; Xifara, D.K.; Ban, M.; Shah, T.S.; Patsopoulos, N.A.; Alfredsson, L.; Anderson, C.A.; et al. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat. Genet. 2015, 47, 1107–1113. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007, 8, R232. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the functional, molecular and phenotypic consequences of amino acid substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Ritchie, G.R.; Dunham, I.; Zeggini, E.; Flicek, P. Functional annotation of noncoding sequence variants. Nat. Methods 2014, 11, 294–296. [Google Scholar] [CrossRef]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; NISC Comparative Sequencing Program; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007, 449, 851–861. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.C.; Cardon, L.R. MERLIN—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide copy number detection and visualization from targeted sequencing. PLoS Comput. Biol. 2014, 12, e1004873. [Google Scholar] [CrossRef]
- Mascia, E.; Clarelli, F.; Zauli, A.; Guaschino, C.; Sorosina, M.; Barizzone, N.; Basagni, C.; Santoro, S.; Ferrè, L.; Bonfiglio, S.; et al. Burden of rare coding variants in an Italian cohort of familial multiple sclerosis. J. Neuroimmunol. 2021. Under Review. [Google Scholar]
- Shaid, D.J.; McDonnell, S.K.; Sinnwell, J.P.; Thibodeau, S.N. Multiple genetic variant association testing by collapsing and kernel methods with pedigree or population structured data. Genet. Epidemiol. 2013, 37, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Gazal, S.; Gosset, S.; Verdura, E.; Bergametti, F.; Guey, S.; Babron, M.C.; Tournier-Lasserve, E. Can whole-exome sequencing data be used for linkage analysis? Eur. J. Hum. Genet. 2016, 24, 581–586. [Google Scholar] [CrossRef] [Green Version]
- Toma, C.; Shaw, A.D.; Heath, A.; Pierce, K.D.; Mitchell, P.B.; Schofield, P.R.; Fullerton, J.M. A linkage exome study of multiplex families with bipolar disorder implicates rare coding variants of ANK3 and additional rare alleles at 10q11-q21. J. Psychiatry Neurosci. 2021, 46, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Wilson, J.E.; Wilson, A.F. Linkage Analysis in the Next-Generation Sequencing Era. Hum. Hered. 2011, 72, 228–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susgun, S.; Kasan, K.; Yucesan, E. Gene hunting approaches through the combination of linkage analysis with Whole-Exome Sequencing in mendelian diseases: From Darwin to the present day. Public Health Genom. 2021, 24, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Gökmen-Polar, Y. HSF1 as a Cancer Biomarker and Therapeutic Target. Curr. Cancer Drug Targets 2019, 19, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dello Russo, C.; Polak, P.E.; Mercado, P.R.; Spagnolo, A.; Sharp, A.; Murphy, P.; Kamal, A.; Burrows, F.J.; Fritz, L.C.; Feinstein, D. The heat-shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin suppresses glial inflammatory responses and ameliorates experimental autoimmune encephalomyelitis. J. Neurochem. 2006, 99, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Cwiklinska, H.; Mycko, M.P.; Szymanska, B.; Matysiak, M.; Selmaj, K.W. Aberrant stress-induced Hsp70 expression in immune cells in multiple sclerosis. J. Neurosci. Res. 2010, 88, 3102–3110. [Google Scholar] [CrossRef] [PubMed]
- Courtois, E.; Schmid, M.; Wajsbrot, O.; Barau, C.; Le Corvoisier, P.; Aouizerate, B.; Bellivier, F.; Belzeaux, R.; Dubertret, C.; Kahn, J.P.; et al. Contribution of common and rare damaging variants in familial forms of bipolar disorder and phenotypic outcome. Transl. Psychiatry 2020, 10, 124. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Chr | No. of Variants a | Burden b | p |
|---|---|---|---|---|
| MYO5C | 15 | 4 | −1.6295 | 0.103214 |
| NOTCH1 | 9 | 4 | −1.3859 | 0.165765 |
| PPRC1 | 10 | 5 | −1.3563 | 0.174993 |
| ABCA1 | 9 | 2 | 1.2021 | 0.229316 |
| DNAH8 | 6 | 7 | −1.0906 | 0.275457 |
| LOXHD1 | 18 | 10 | −0.781 | 0.434797 |
| HSF1 | 8 | 3 | −0.6585 | 0.510208 |
| ADAMTS5 | 21 | 6 | 0.4399 | 0.659993 |
| MYO1C | 17 | 5 | 0.3498 | 0.726526 |
| ASXL3 | 18 | 4 | −0.1488 | 0.88168 |
| KIF1A | 2 | 2 | 0.1461 | 0.883868 |
| TLN2 | 15 | 2 | 0.1461 | 0.883868 |
| ATN1 | 12 | 3 | −0.1299 | 0.896626 |
| KIF26B | 1 | 4 | 0.0279 | 0.977741 |
| Gene | Chr | rs ID | POS (hg19) | REF | ALT | n MS a | n CT a | Effect | Ann Impact b | AF EUR c | CADD Phred d |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CCNL2e | 1 | rs3831366; rs368050244 | 1334052 | CTAGAG | C | 6 f | 5 f | Splice acceptor variant | HIGH | 0.0487 | NA |
| TMX3 | 18 | chr18:66358530 | 66358530 | A | G | 2 | 0 | Splice donor variant | HIGH | −1 | 24.5 |
| ToppGene | |||||||
|---|---|---|---|---|---|---|---|
| Category | GO Term | Description | Genes | No. of Selected Genes | No. of Hits in Genome | p | p Adjusted |
| Biological processes a | GO:0051028 | mRNA transport | AGFG1,HSF1,MYO1C,SMG1,MAGOHB | 5 | 157 | 6.2 × 10−6 | 0.011 |
| Molecular function | GO:0003774 | motor activity | MYO1C,KIF1A,DNAH8,KIF26B,MYO5C | 5 | 134 | 3.44 × 10−6 | 9.50 × 10−4 |
| Molecular function | GO:0003713 | transcription coactivator activity b | MMS19,ATN1,PPRC1,NOTCH1,RNF20 | 5 | 285 | 1.29 × 10−4 | 3.57 × 10−2 |
| Disease | C0524851 | Neurodegenerative Disorders | AGFG1,PPP2R2B,ABCA1,GRIN2B,ATN1,TDP1,HSF1,KIF1A,DNAH8,LDLR,NOTCH1 | 11 | 1480 | 1.01 × 10−5 | 0.0156 |
| Disease | C0027765 | nervous system disorder c | AGFG1,SMG1,PPP2R2B,ABCA1,GRIN2B,TDP1,KIF1A,NOTCH1,RNF20 | 9 | 942 | 1.12 × 10−5 | 0.0173 |
| Transcription factors | RFX1_01 d | SMG1,PPP2R2B,HSF1,MYO1C | 4 | 194 | 7.82 × 10−5 | 0.0375 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barizzone, N.; Cagliani, R.; Basagni, C.; Clarelli, F.; Mendozzi, L.; Agliardi, C.; Forni, D.; Tosi, M.; Mascia, E.; Favero, F.; et al. An Investigation of the Role of Common and Rare Variants in a Large Italian Multiplex Family of Multiple Sclerosis Patients. Genes 2021, 12, 1607. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101607
Barizzone N, Cagliani R, Basagni C, Clarelli F, Mendozzi L, Agliardi C, Forni D, Tosi M, Mascia E, Favero F, et al. An Investigation of the Role of Common and Rare Variants in a Large Italian Multiplex Family of Multiple Sclerosis Patients. Genes. 2021; 12(10):1607. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101607
Chicago/Turabian StyleBarizzone, Nadia, Rachele Cagliani, Chiara Basagni, Ferdinando Clarelli, Laura Mendozzi, Cristina Agliardi, Diego Forni, Martina Tosi, Elisabetta Mascia, Francesco Favero, and et al. 2021. "An Investigation of the Role of Common and Rare Variants in a Large Italian Multiplex Family of Multiple Sclerosis Patients" Genes 12, no. 10: 1607. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12101607