
Biochemical Studies in Fibroblasts to Interpret Variants of Unknown Significance in the ABCD1 Gene

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Patient Selection (AMC, The Netherlands)
2.2. Clinical Assessments of the Dutch ALD Cohort
2.3. ABCD1 VUS Fibroblasts
2.4. Biochemical and Functional Tests of VLCFA Metabolism
2.5. Statistical Analysis
3. Results
3.1. Patient Characteristics
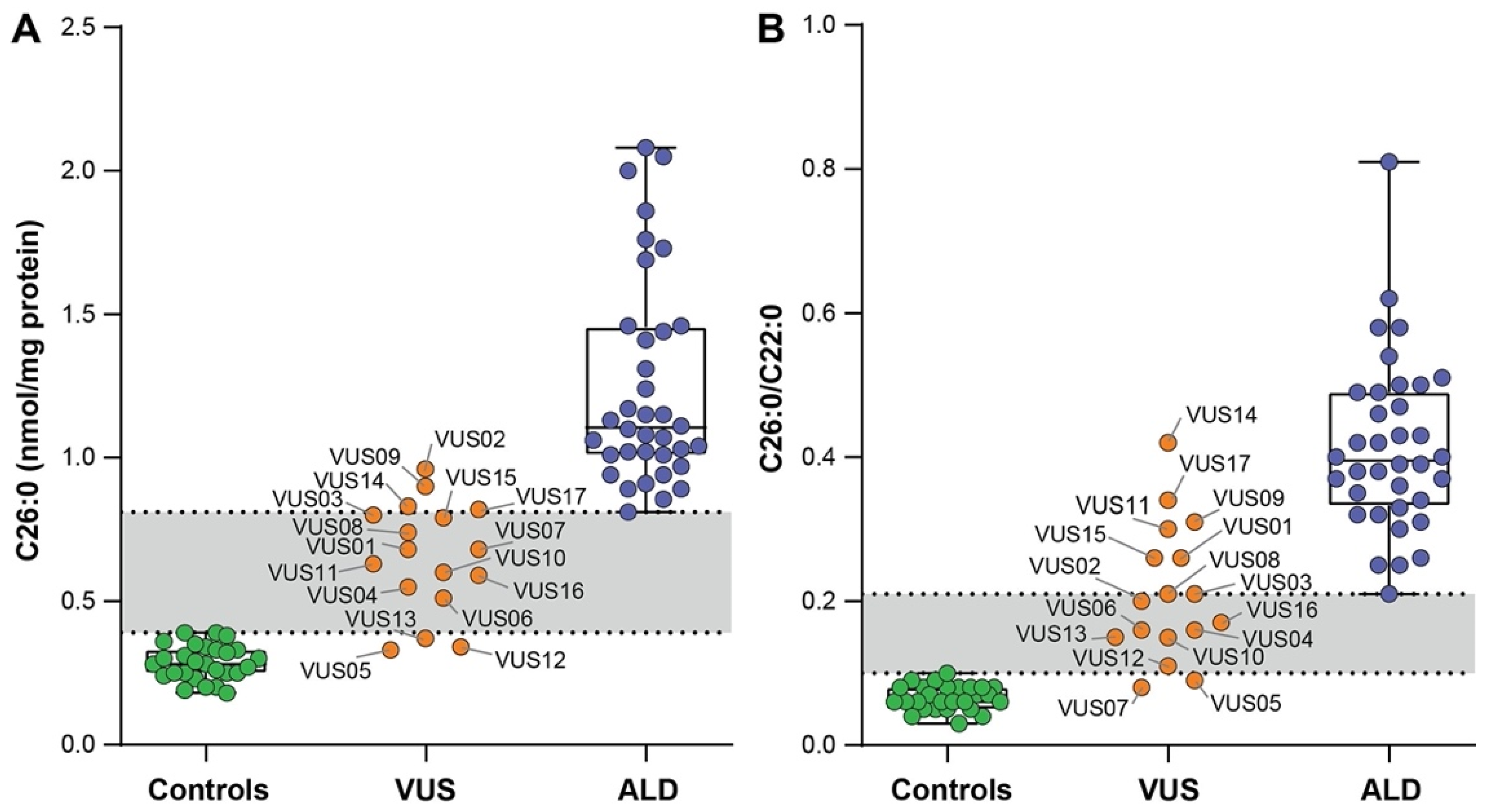
3.2. VLCFA Levels in Fibroblasts
3.3. D3-C22:0 Loading Test
3.4. ALDP Immunoblot
3.5. Clinical Correlations
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mosser, J.; Douar, A.-M.; Sarde, C.-O.; Kioschis, P.; Feil, R.; Moser, H.; Poustka, A.-M.; Mandel, J.-L.; Aubourg, P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 1993, 361, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Moser, H.W.; Smith, K.D.; Watkins, P.; Powers, J.; Moser, A.B. X-linked adrenoleukodystrophy. In The Metabolic and Molecular Bases of Inherited Disease; McGraw Hill: New York, NY, USA, 2001; Volume 8, pp. 3257–3301. [Google Scholar]
- Mosser, J.; Lutz, Y.; Stoeckel, M.E.; Sarde, C.O.; Kretz, C.; Douar, A.M.; Lopez, J.; Aubourg, P.; Mandel, J.L. The gene responsible for adrenoleukodystrophy encodes a peroxisomal membrane protein. Hum. Mol. Genet. 1994, 3, 265–271. [Google Scholar] [CrossRef]
- Singh, I.; Moser, A.B.; Moser, H.W.; Kishimoto, Y. Adrenoleukodystrophy: Impaired oxidation of very long chain fatty acids in white blood cells, cultured skin fibroblasts, and amniocytes. Pediatr. Res. 1984, 18, 286–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, M.; Schaumburg, H.H.; Powers, J.; Kishimoto, Y.; Koilodny, E.; Suzuki, K. Fatty acid abnormality in adrenoleukodystrophy. J. Neurochem. 1976, 26, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.; Huffnagel, I.C.; Linthorst, G.E.; Wanders, R.J.A.; Engelen, M. Adrenoleukodystrophy—Neuroendocrine pathogenesis and redefinition of natural history. Nat. Rev. Endocrinol. 2016, 12, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Huffnagel, I.C.; van Ballegoij, W.J.C.C.; Van Geel, B.M.; Vos, J.M.B.W.; Kemp, S.; Engelen, M. Progression of myelopathy in males with adrenoleukodystrophy: Towards clinical trial readiness. Brain 2019, 142, 334–343. [Google Scholar] [CrossRef]
- Huffnagel, I.C.; Dijkgraaf, M.G.W.W.; Janssens, G.E.; van Weeghel, M.; Van Geel, B.M.; Poll-The, B.T.; Kemp, S.; Engelen, M. Disease progression in women with X-linked adrenoleukodystrophy is slow. Orphanet J. Rare Dis. 2019, 14, 30. [Google Scholar] [CrossRef] [Green Version]
- Engelen, M.; Barbier, M.; Dijkstra, I.M.E.; Schür, R.; de Bie, R.M.A.; Verhamme, C.; Dijkgraaf, M.G.W.; Aubourg, P.A.; Wanders, R.J.A.; van Geel, B.M.; et al. X-linked adrenoleukodystrophy in women: A cross-sectional cohort study. Brain 2014, 137, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.; Aubourg, P.; Poll, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- De Beer, M.; Engelen, M.; Van Geel, B.M. Frequent occurrence of cerebral demyelination in adrenomyeloneuropathy. Neurology 2014, 83, 2227–2231. [Google Scholar] [CrossRef] [PubMed]
- Huffnagel, I.C.; Laheji, F.K.; Aziz-Bose, R.; Tritos, N.A.; Marino, R.; Linthorst, G.E.; Kemp, S.; Engelen, M.; Eichler, F. The Natural History of Adrenal Insufficiency in X-Linked Adrenoleukodystrophy: An International Collaboration. J. Clin. Endocrinol. Metab. 2019, 104, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, P.; Raymond, G.V.; Moser, A.B.; Kharkar, S.; Bezman, L.; Moser, H.W. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. J. Pediatr. 2005, 146, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.P.; Rothman, S.M.; Nascene, D.; Kivisto, T.; DeFor, T.E.; Ziegler, R.S.; Eisengart, J.; Leiser, K.; Raymond, G.; Lund, T.C.; et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: The largest single-institution cohort report. Blood 2011, 118, 1971–1978. [Google Scholar] [CrossRef]
- Mallack, E.J.; Turk, B.R.; Yan, H.; Price, C.; Demetres, M.; Moser, A.B.; Becker, C.; Hollandsworth, K.; Adang, L.; Vanderver, A.; et al. MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: Meta-analysis and consensus guidelines. J. Inherit. Metab. Dis. 2020, 44, 728–739. [Google Scholar] [CrossRef]
- Moser, A.B.; Jones, R.O.; Hubbard, W.C.; Tortorelli, S.; Orsini, J.J.; Caggana, M.; Vogel, B.H.; Raymond, G. V Newborn Screening for X-Linked Adrenoleukodystrophy. Int. J. Neonatal Screen. 2016, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Barendsen, R.W.; Dijkstra, I.M.E.; Visser, W.F.; Alders, M.; Bliek, J.; Boelen, A.; Bouva, M.J.; van der Crabben, S.N.; Elsinghorst, E.; van Gorp, A.G.M.; et al. Adrenoleukodystrophy Newborn Screening in the Netherlands (SCAN Study): The X-Factor. Front. Cell Dev. Biol. 2020, 8, 499. [Google Scholar] [CrossRef]
- Straus, S.; Paul, G.; Richardson, S.W.; Haynes, B.R. Evidence-Based Medicine, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- van de Beek, M.-C.; Dijkstra, I.M.E.; Kemp, S. Method for Measurement of Peroxisomal Very Long-Chain Fatty Acid β-Oxidation and De Novo C26:0 Synthesis Activity in Living Cells Using Stable-Isotope Labeled Docosanoic Acid. Methods Mol. Biol. 2017, 1595, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Kurtzke, J.F. Rating neurologic impairment in multiple sclerosis. Neurology 1983, 33, 1444–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castilhos, R.M.; Blank, D.; Netto, C.B.O.; Souza, C.F.M.; Fernandes, L.N.T.; Schwartz, I.V.D.; Giugliani, R.; Jardim, L.B. Severity score system for progressive myelopathy: Development and validation of a new clinical scale. Braz. J. Med. Biol. Res. 2012, 45, 565–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podsiadlo, D.; Richardson, S. The Timed “Up & Go”: A Test of Basic Functional Mobility for Frail Elderly Persons. J. Am. Geriatr. Soc. 1991, 39, 142–148. [Google Scholar] [CrossRef]
- Wiens, K.; Berry, S.A.; Choi, H.; Gaviglio, A.; Gupta, A.; Hietala, A.; Kenney-Jung, D.; Lund, T.; Miller, W.; Pierpont, E.I.; et al. A report on state-wide implementation of newborn screening for X-linked Adrenoleukodystrophy. Am. J. Med. Genet. A 2019, 179, 1205–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, P.; Gupta, N.; Gulati, S.; Ghosh, M.; Vasisht, S.; Sharma, R.; Gupta, A.K.; Kalra, V.; Kabra, M. Molecular analysis of ABCD1 gene in Indian patients with X-linked Adrenoleukodystrophy. Clin. Chim. Acta 2011, 412, 2289–2295. [Google Scholar] [CrossRef]
- Vreken, P.; van Lint, A.E.; Bootsma, A.H.; Overmars, H.; Wanders, R.J.; van Gennip, A.H. Rapid stable isotope dilution analysis of very-long-chain fatty acids, pristanic acid and phytanic acid using gas chromatography-electron impact mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1998, 713, 281–287. [Google Scholar] [CrossRef]
- Dacremont, G.; Cocquyt, G.; Vincent, G. Measurement of very long-chain fatty acids, phytanic and pristanic acid in plasma and cultured fibroblasts by gas chromatography. J. Inherit. Metab. Dis. 1995, 18, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Valianpour, F.; Selhorst, J.J.M.; Van Lint, L.E.M.; Van Gennip, A.H.; Wanders, R.J.A.; Kemp, S. Analysis of very long-chain fatty acids using electrospray ionization mass spectrometry. Mol. Genet. Metab. 2003, 79, 189–196. [Google Scholar] [CrossRef]
- Kemp, S.; Mooyer, P.A.; Bolhuis, P.A.; van Geel, B.M.; Mandel, J.L.; Barth, P.G.; Aubourg, P.; Wanders, R.J. ALDP expression in fibroblasts of patients with X-linked adrenoleukodystrophy. J. Inherit. Metab. Dis. 1996, 19, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Taneja, K.K.; Kalra, V.; Behari, M.; Aneja, S.; Bansal, S.K. Genomic Profiling Identifies Novel Mutations and SNPs in ABCD1 Gene: A Molecular, Biochemical and Clinical Analysis of X-ALD Cases in India. PLoS ONE 2011, 6, e25094. [Google Scholar] [CrossRef]
- Simhadri, V.L.; Hamasaki-Katagiri, N.; Lin, B.C.; Hunt, R.; Jha, S.; Tseng, S.C.; Wu, A.; Bentley, A.A.; Zichel, R.; Lu, Q.; et al. Single synonymous mutation in factor IX alters protein properties and underlies haemophilia B. J. Med. Genet. 2017, 54, 338–345. [Google Scholar] [CrossRef]
- Moser, A.B.; Kreiter, N.; Bezman, L.; Lu, S.; Raymond, G.V.; Naidu, S.; Moser, H.W. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann. Neurol. 1999, 45, 100–110. [Google Scholar] [CrossRef]
- Boles, D.J.; Craft, D.A.; Padgett, D.A.; Loria, R.M.; Rizzo, W.B. Clinical variation in X-linked adrenoleukodystrophy: Fatty acid and lipid metabolism in cultured fibroblasts. Biochem. Med. Metab. Biol. 1991, 45, 74–91. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ID | ABCD1 Variant | Consequence | ALDP Expression | C26:0 | C26:0/C22:0 |
|---|---|---|---|---|---|
| ALD022 | c.1A > G | p.Met1Val | Not detectable | 0.89 | 0.37 |
| ALD030 | c.1A > G | p.Met1Val | Not detectable | 1.06 | 0.42 |
| ALD043 | c.1A > G | p.Met1Val | Not detectable | 1.15 | 0.31 |
| ALD062 | c.1A > G | p.Met1Val | Not detectable | 1.10 | 0.62 |
| ALD063 | c.1A > G | p.Met1Val | Not detectable | 1.03 | 0.58 |
| ALD064 | c.1A > G | p.Met1Val | Not detectable | 0.89 | 0.21 |
| ALD031 | c.220C > T | p.Arg74Trp | Not detectable | 0.81 | 0.30 |
| ALD056 | c.411G > T | p.Trp137Cys | Not detectable | 0.94 | 0.40 |
| ALD034 | c.446G > A | p.Ser149Asn | Detectable | 1.08 | 0.37 |
| ALD222 | c.446G > A | p.Ser149Asn | Detectable | 1.04 | 0.49 |
| ALD003 | c.529C > T | p.Gln177 * | Not detectable | 1.15 | 0.25 |
| ALD055 | c.543C > A | p.Tyr181 * | Not detectable | 1.01 | 0.39 |
| ALD020 | c.659T > C | p.Leu220Pro | Reduced | 1.41 | 0.32 |
| ALD045 | c.734C > A | p.Ala245Asp | Detectable | 1.02 | 0.49 |
| ALD017 | c.832G > T | p.Glu278 * | Not detectable | 2.00 | 0.34 |
| ALD228 | c.874_76del | p.Glu292del | Not detectable | 1.07 | 0.50 |
| ALD058 | c.892G > A | p.Gly298Ser | Detectable | 1.17 | 0.43 |
| ALD059 | c.892G > A | p.Gly298Ser | Detectable | 1.11 | 0.26 |
| ALD081 | c.892G > A | p.Gly298Ser | Detectable | 0.91 | 0.81 |
| ALD001 | c.901-5C > A | p.Val301fs *? | Not detectable | 1.86 | 0.43 |
| ALD024 | c.1166G > A | p.Arg389His | Reduced | 0.97 | 0.38 |
| ALD202 | c.1166G > A | p.Arg389His | Reduced | 1.24 | 0.33 |
| ALD006 | c.1322insA | p.Asp442Glyfs * 114 | Not detectable | 1.31 | 0.46 |
| ALD011 | c.1390C > T | p.Arg464 * | Not detectable | 1.44 | 0.25 |
| ALD899 | c.1390C > T | p.Arg464 * | Not detectable | 1.46 | 0.54 |
| ALD009 | c.1415_16delAG | p.Gln472Argfs * 83 | Not detectable | 2.08 | 0.35 |
| ALD068 | c.1488+3A > G | p.Val497fs *? | Not detectable | 1.01 | 0.39 |
| ALD069 | c.1488+3A > G | p.Val497fs *? | Not detectable | 1.01 | 0.39 |
| ALD079 | c.1846G > A | p.Ala616Thr | Not detectable | 1.46 | 0.42 |
| ALD025 | c.1866-10G > A | p.Pro623fs *? | Not detectable | 1.02 | 0.32 |
| ALD048 | c.1866-2A > T | p.Pro623fs *? | Not detectable | 2.05 | 0.47 |
| ALD049 | c.1866-2A > T | p.Pro623fs *? | Not detectable | 1.69 | 0.50 |
| ALD027 | c.1899delC | p.Ser633Argfs * 3 | Not detectable | 1.73 | 0.58 |
| ALD008 | c.1961T > C | p.Leu654Pro | Not detectable | 1.76 | 0.51 |
| ALD015 | c.1961T > C | p.Leu654Pro | Not detectable | 0.94 | 0.40 |
| ALD019 | c.1970delTCA | p.Ile657del | Not detectable | 1.13 | 0.38 |
| Clinical Characteristics | ALD Patients (n = 36) |
|---|---|
| Age (year) | 41.8 ± 17.2 |
| Body weight (kg) | 80.4 ± 12.2 |
| Adrenal insufficiency, n (%) | 23 (64) |
| EDSS | 3.0 (0.0–6.5) |
| SSPROM | 89 (61.5–100) |
| TUG (s) | 6.1 (2.9–18.6) |
| ID | ABCD1 Variant | Consequence | Method of Detection | Age | ALD Symptoms |
|---|---|---|---|---|---|
| VUS01 | c.1597A > C | p.Lys533Gln b | NBS | 11 months | None |
| VUS02 | c.1448C > T | p.Thr483Met | NBS | 12 months | None |
| VUS03 a | c.1828A > G | p.Lys610Glu | NBS | 24 months | None |
| VUS04 | c.1696A > G | p.Met566Val | NBS | 5 months | None |
| VUS05 | c.2134C > T | p.Arg712Cys | NBS | 6 months | None |
| VUS06 | c.566G > A | p.Arg189Gln | NBS | 5 months | None |
| VUS07 | c.895C > T | p.His299Tyr | NBS | 2 years | None |
| VUS08 a | c.1828A > G | p.Lys610Glu | Extended family screening | 4 years | None |
| VUS09 | c.1597A > C | p.Lys533Gln b | NBS | 2 months | None |
| VUS10 | c.1000C > T | p.Leu334Phe | NBS | 3 years | None |
| VUS11 | c.1979G > A | p.Arg660Gln b | WES | 5 years | None |
| VUS12 | c.896A > G | p.His299Arg | Decline in cognitive function | 13 years | None |
| VUS13 | c.1966_1967dup | p.Ile657ProfsX35 c | NBS | 2 months | None |
| VUS14 | c.970C > T | p.Arg324Cys | NBS | 2 months | None |
| VUS15 | c.1900G > A | p.Ala643Thr | Family screening | 34 years | None |
| VUS16 | c.1438C > A | p.Pro480Thr | Family screening | 9 years | None |
| VUS17 | c.574A > G | p.Asn192Asp | NBS | 2 months | None |
| Test | Reference Range | Gray Zone | ALD Range | VUS 17 | VUS 14 | VUS 02 | VUS 03 | VUS 01 | VUS 04 | VUS 15 | VUS 09 | VUS 08 | VUS 05 | VUS 06 | VUS 07 | VUS 10 | VUS 11 | VUS 12 | VUS 13 | VUS 16 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C26:0 | 0.18–0.39 | 0.40–0.80 | 0.81–2.08 | 0.82 | 0.83 | 0.96 | 0.80 | 0.68 | 0.55 | 0.79 | 0.9 | 0.74 | 0.33 | 0.51 | 0.68 | 0.60 | 0.63 | 0.34 | 0.37 | 0.56 |
| C26:0/C22:0 | 0.03–0.10 | 0.11–0.20 | 0.21–0.81 | 0.34 | 0.42 | 0.20 | 0.21 | 0.26 | 0.16 | 0.26 | 0.31 | 0.21 | 0.09 | 0.17 | 0.08 | 0.16 | 0.30 | 0.11 | 0.15 | 0.18 |
| D3-C16:0 | 15.9–31.7 | 12.6–15.8 | 5.9–12.5 | 5.5 | 7.1 | 6.2 | 5.6 | 4.8 | 3.2 | 7.4 | 12.8 | 9.5 | 9.8 | 9.3 | 14.3 | 17.0 | 17.0 | 20.8 | 23.0 | 19.1 |
| D3-C26:0 | 0.15–0.51 | 0.52–0.99 | 1.00–3.69 | 2.51 | 1.94 | 2.11 | 2.79 | 1.38 | 2.62 | 1.86 | 1.39 | 1.88 | 1.41 | 1.04 | 0.66 | 0.85 | 0.86 | 0.76 | 0.52 | 0.50 |
| D3-C16:0/D3-C22:0 | 0.88–2.00 | 0.47–0.87 | 0.15–0.46 | 0.29 | 0.49 | 0.23 | 0.30 | 0.27 | 0.17 | 0.27 | 0.63 | 0.48 | 0.43 | 0.53 | 1.15 | 1.14 | 1.12 | 1.07 | 1.28 | 1.18 |
| % of control cells | >57 | 27–56 | 12–26 | 13 | 25 | 17 | 19 | 23 | 14 | 18 | 34 | 34 | 32 | 43 | 67 | 57 | 59 | 107 | 72 | 61 |
| Immunoblot | present | present | reduced | present | absent | reduced | present | absent | present | present | present | present | present | present | present | present | ND | |||
| VLCFA elevated at follow-up | Yes | Yes | Yes | Yes | ND | Yes | Yes | Yes | No | No | No | No | No | No | No | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van de Stadt, S.I.W.; Mooyer, P.A.W.; Dijkstra, I.M.E.; Dekker, C.J.M.; Vats, D.; Vera, M.; Ruzhnikov, M.R.Z.; van Haren, K.; Tang, N.; Koop, K.; et al. Biochemical Studies in Fibroblasts to Interpret Variants of Unknown Significance in the ABCD1 Gene. Genes 2021, 12, 1930. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12121930
van de Stadt SIW, Mooyer PAW, Dijkstra IME, Dekker CJM, Vats D, Vera M, Ruzhnikov MRZ, van Haren K, Tang N, Koop K, et al. Biochemical Studies in Fibroblasts to Interpret Variants of Unknown Significance in the ABCD1 Gene. Genes. 2021; 12(12):1930. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12121930
Chicago/Turabian Stylevan de Stadt, Stephanie I. W., Petra A. W. Mooyer, Inge M. E. Dijkstra, Conny J. M. Dekker, Divya Vats, Moin Vera, Maura R. Z. Ruzhnikov, Keith van Haren, Nelson Tang, Klaas Koop, and et al. 2021. "Biochemical Studies in Fibroblasts to Interpret Variants of Unknown Significance in the ABCD1 Gene" Genes 12, no. 12: 1930. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12121930