
Illegitimate Recombination between Duplicated Genes Generated from Recursive Polyploidizations Accelerated the Divergence of the Genus Arachis

 , , , , , , , , , , , , , , , , , and
, , , , , , , , , , , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
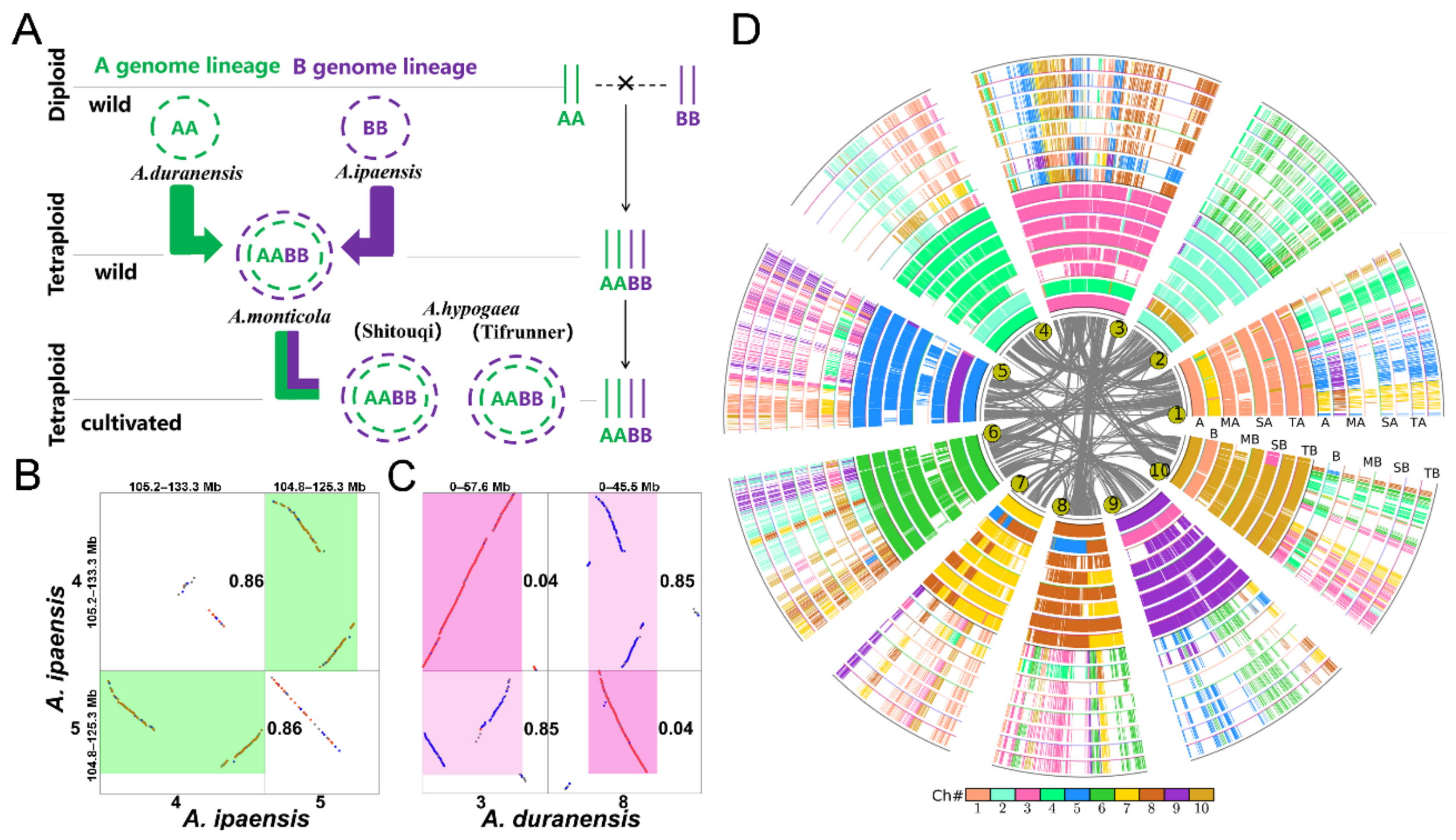
2.1. Genome Data
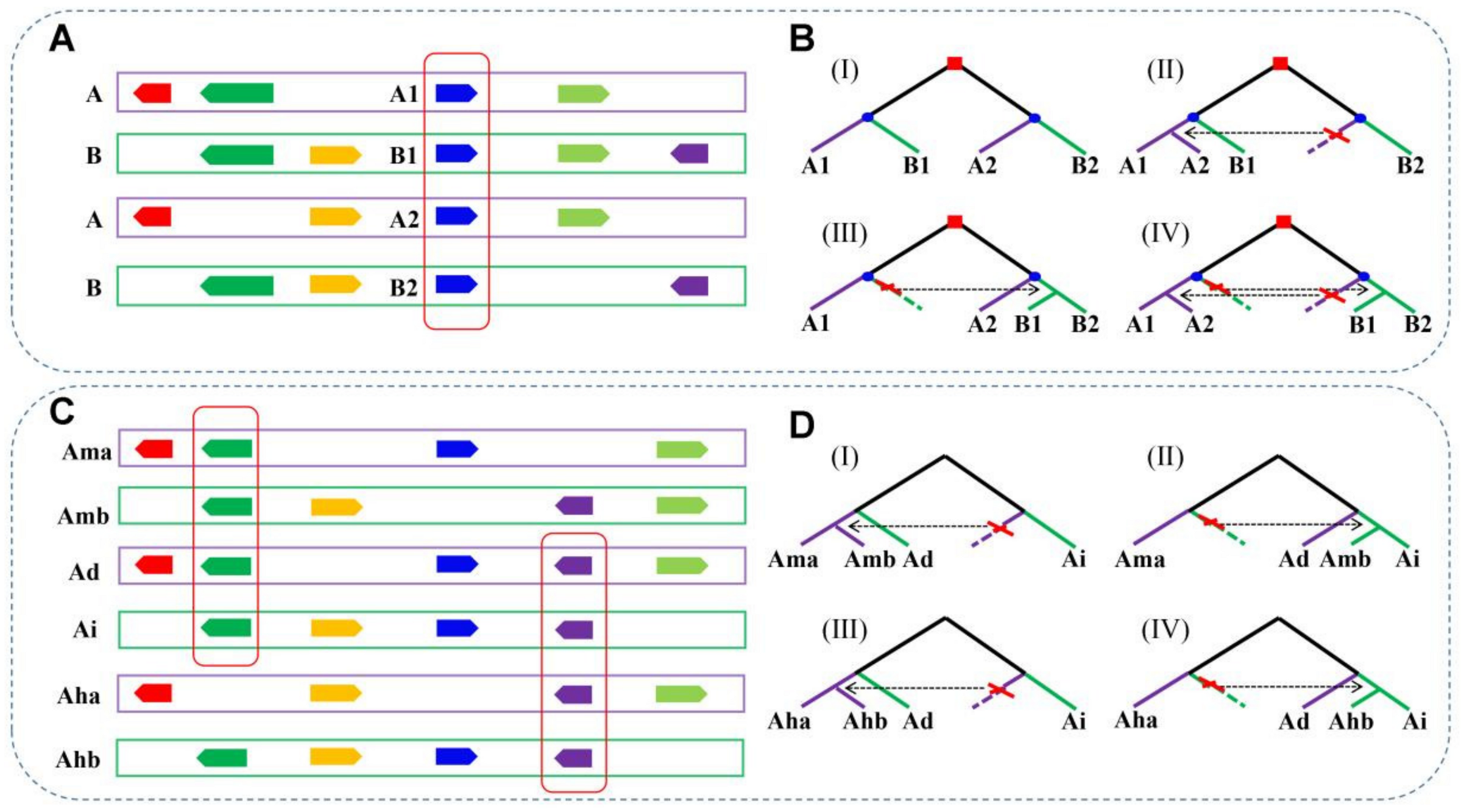
2.2. Detection of Duplicated Genes
2.3. Construction of Homologous Gene Quartets
2.4. Calculation of Ks and Ka
2.5. Gene Conversion Inference
2.6. Statistical Analysis of the Correlation between Conversion and Physical Location
2.7. Gene Ontology Analysis
3. Results
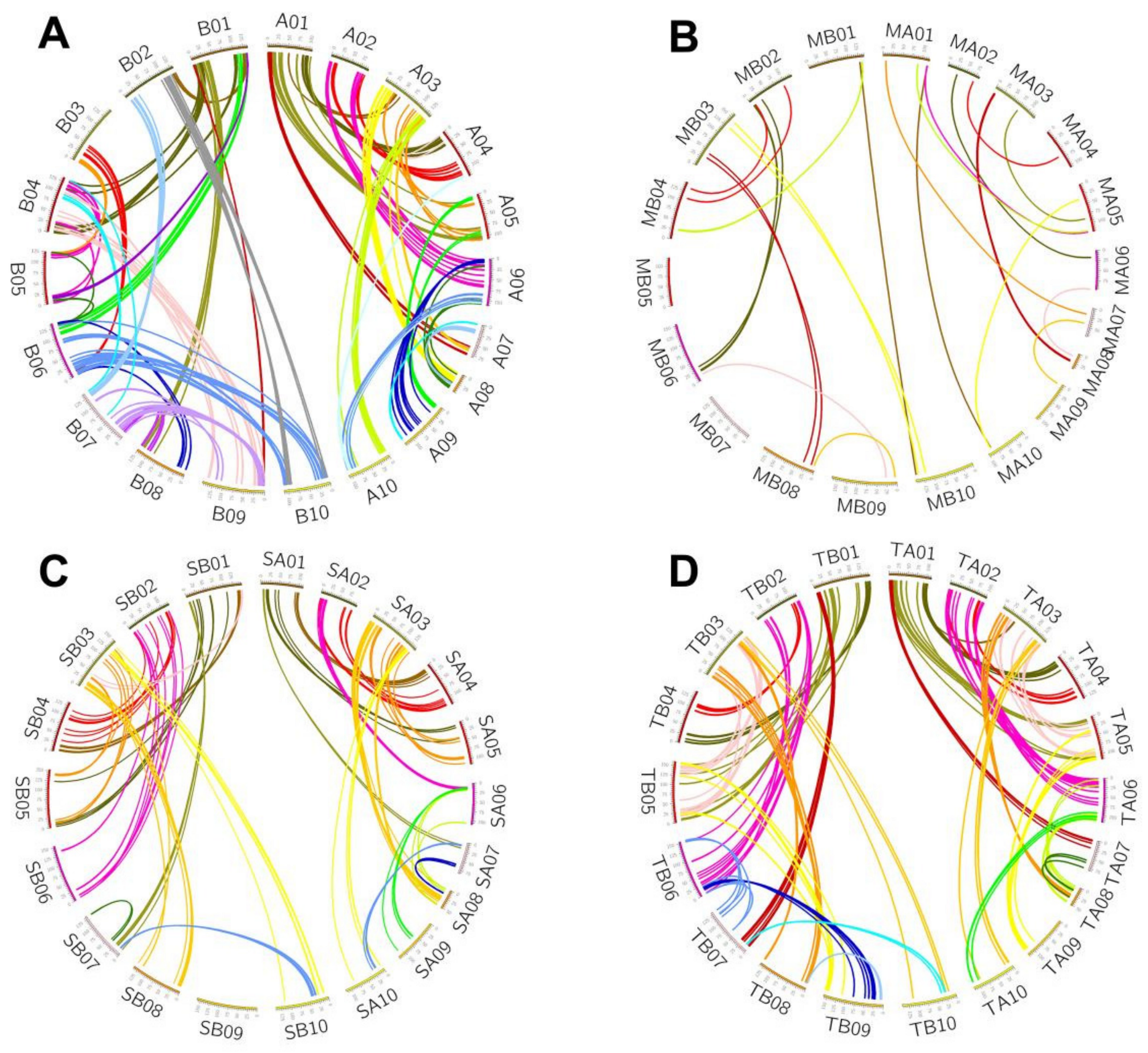
3.1. Genomic Homology
3.2. Homologous Gene Quartets
3.3. Gene Conversion between LCT-Related Duplicated Genes
3.4. Gene Conversion between PRT-Related Duplicated Genes
3.5. Conversion and Evolution
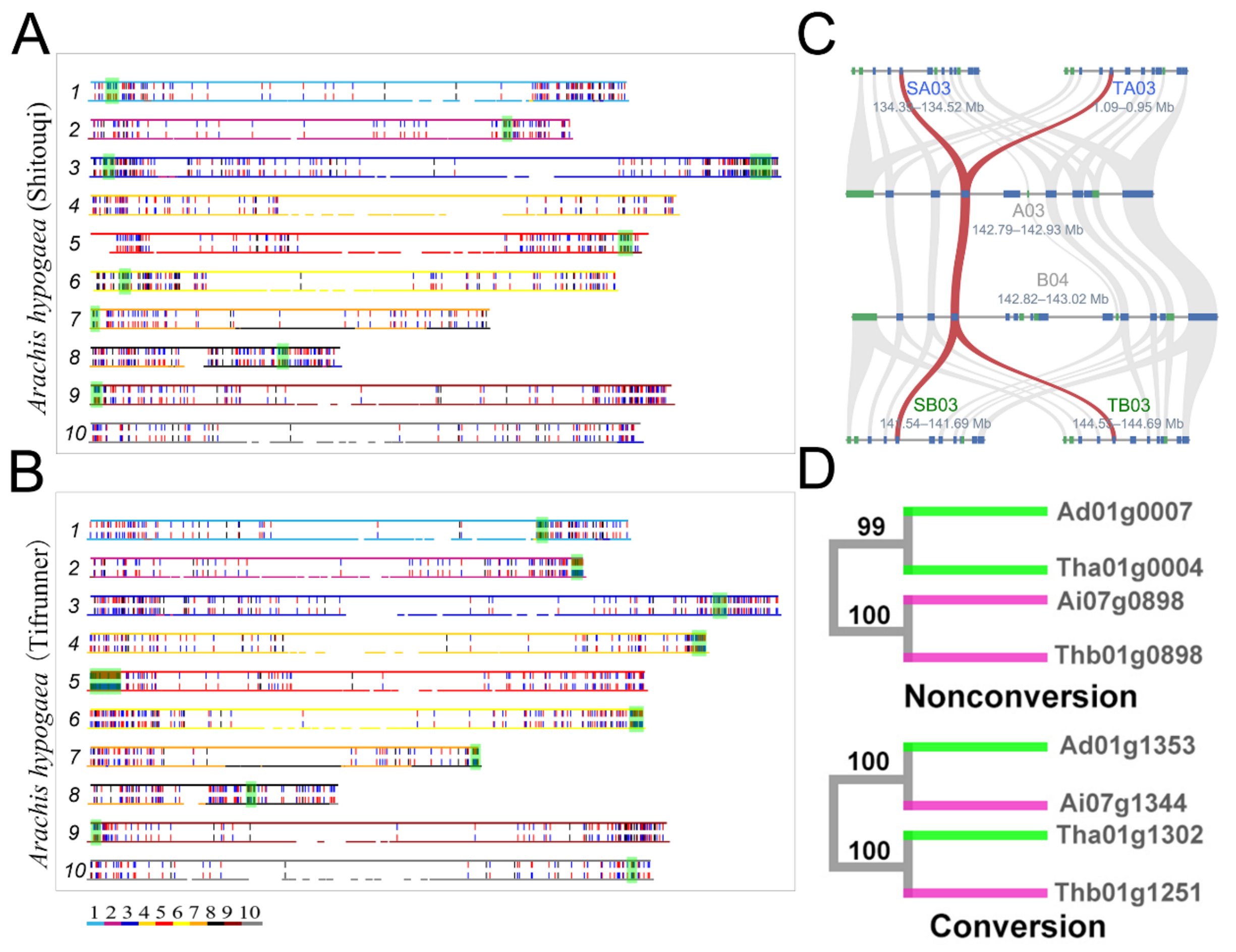
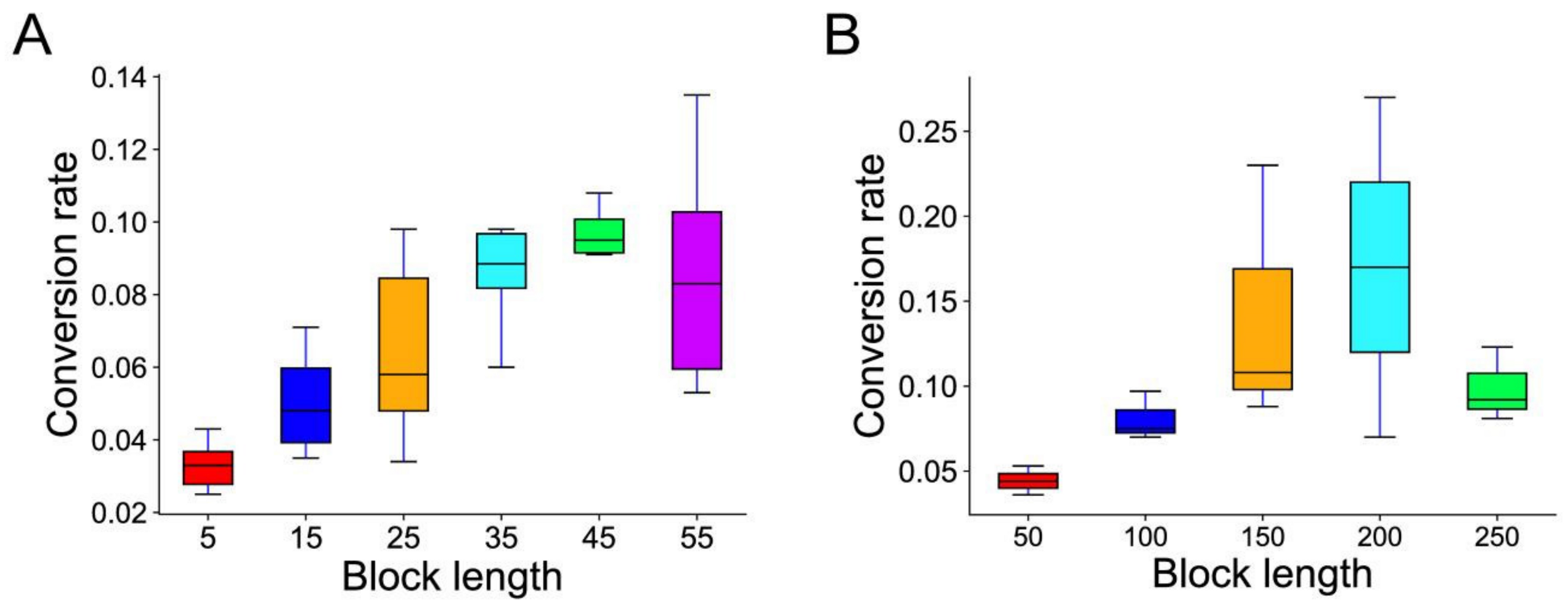
3.6. Conversion and Physical Position
3.7. Chromosome Rearrangements and Conversion
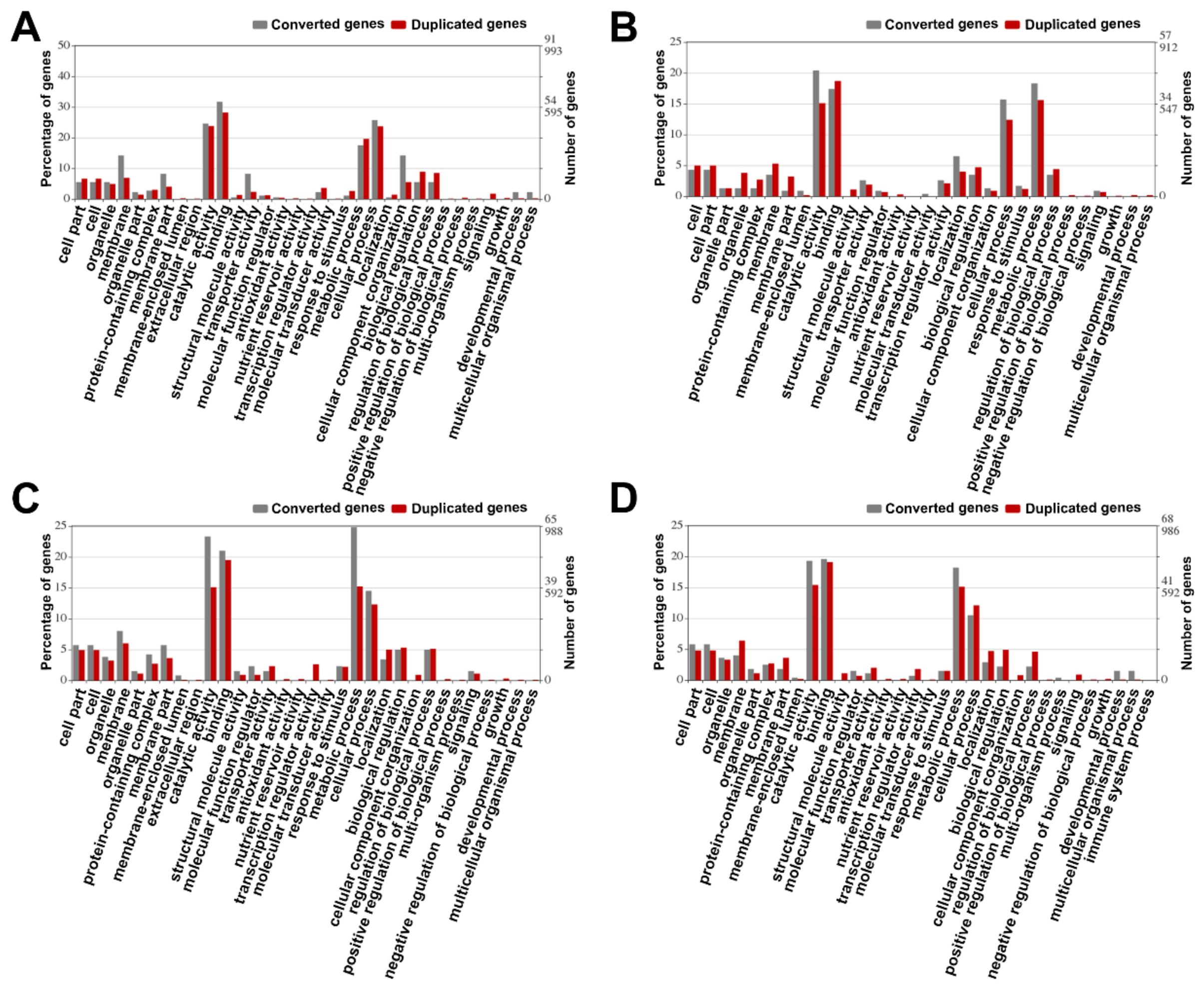
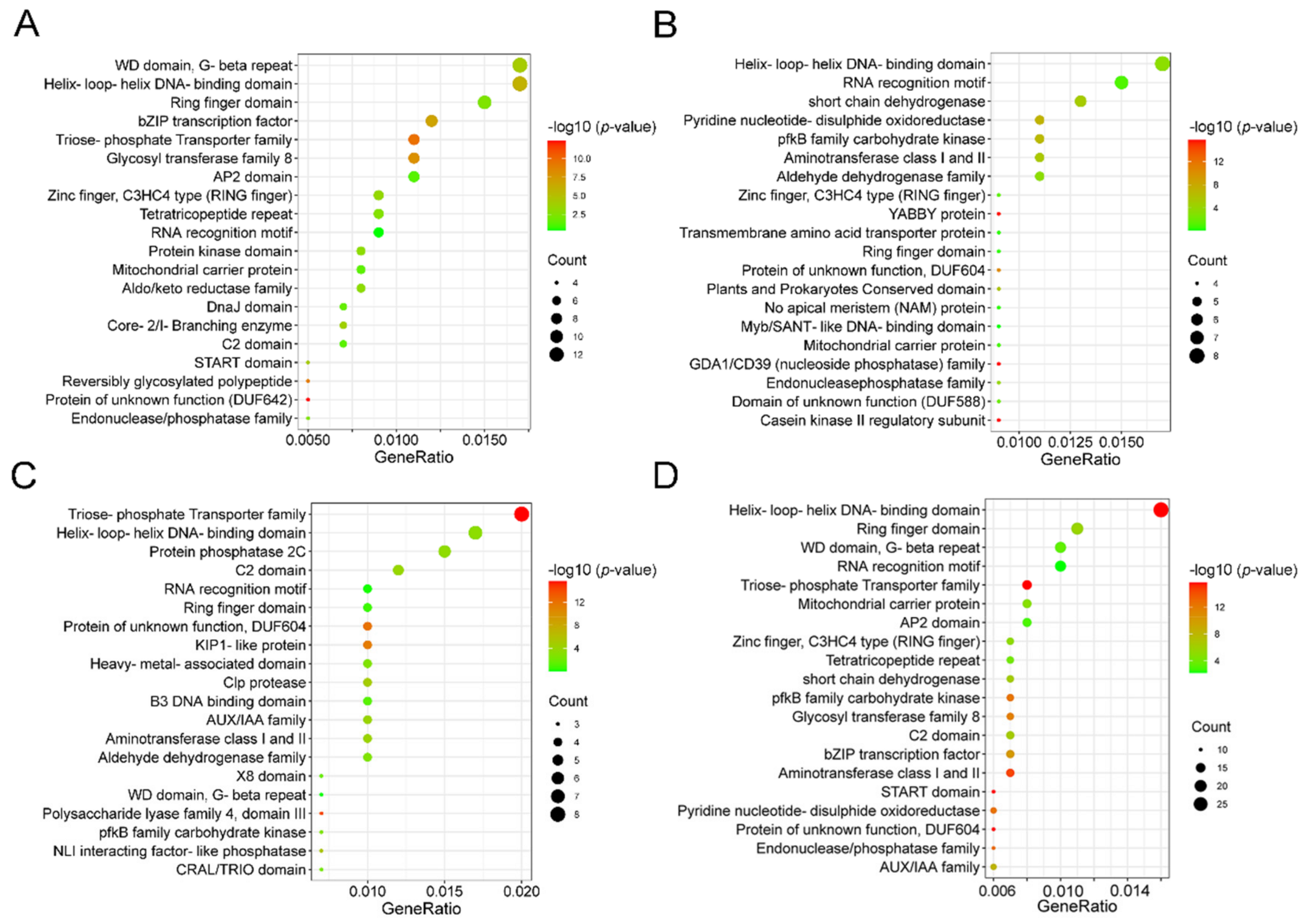
3.8. Gene Function Analysis
4. Discussion
4.1. Long-Lasting Extensive Conversions Affected the Evolution of Duplicated Genes in Peanut Genomes
4.2. Conversion Contributes to the Divergence of Genus Arachis Genomes
4.3. Donor Genes Are Preferred as Donors
4.4. Conversion and Genomic Rearrangements
4.5. Conversion and Function
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, J.K.; Bruce, V.M.; McDonald, B.E. Dietary α-linolenic acid is as effective as oleic acid and linoleic acid in lowering blood cholesterol in normolipidemic men. Am. J. Clin. Nutr. 1991, 53, 1230–1234. [Google Scholar] [CrossRef]
- Zhuang, W.; Chen, H.; Yang, M.; Wang, J.; Pandey, M.K.; Zhang, C.; Chang, W.-C.; Zhang, L.; Zhang, X.; Tang, R.; et al. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat. Genet. 2019, 51, 865–876. [Google Scholar] [CrossRef]
- Chen, X.; Li, H.; Pandey, M.; Yang, Q.; Wang, X.; Garg, V.; Li, H.; Chi, X.; Doddamani, D.; Hong, Y.; et al. Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc. Natl. Acad. Sci. USA 2016, 113, 6785–6790. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Lu, Q.; Liu, H.; Zhang, J.; Hong, Y.; Lan, H.; Li, H.; Wang, J.; Liu, H.; Li, S.; et al. Sequencing of Cultivated Peanut, Arachis hypogaea, Yields Insights into Genome Evolution and Oil Improvement. Mol. Plant 2019, 12, 920–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, D.; Ji, C.; Song, Q.; Zhang, W.; Zhang, X.; Zhao, K.; Chen, C.Y.; Wang, C.; He, G.; Liang, Z.; et al. Comparison of Arachis monticola with Diploid and Cultivated Tetraploid Genomes Reveals Asymmetric Subgenome Evolution and Improvement of Peanut. Adv. Sci. 2020, 7, 1901672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertioli, S.C.M.L.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S.; et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 2016, 48, 438–446. [Google Scholar] [CrossRef]
- Hilu, K.W.; Stalker, H.T. Genetic relationships between peanut and wild species of Arachis sect. Arachis (Fabaceae): Evidence from RAPDs. Plant Syst. Evol. 1995, 198, 167–178. [Google Scholar] [CrossRef]
- Favero, A.P.; Simpson, C.E.; Valls, M.; Vello, N.A. Study of the Evolution of Cultivated Peanut through Crossability Studies among Arachis ipaensis, A. duranensis, and A. hypogaea. Plant Genet. Resour. 2006, 46, 1546–1552. [Google Scholar] [CrossRef]
- Puchta, H.; Dujon, B.; Hohn, B. Two different but related mechanisms are used in plants for the repair of genomic double-strand breaks by homologous recombination. Proc. Natl. Acad. Sci. USA 1996, 93, 5055–5060. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Barker, M.S.; Husband, B.C.; Pires, J.C. Spreading Winge and flying high: The evolutionary importance of polyploidy after a century of study. Am. J. Bot. 2016, 103, 1139–1145. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Han, B.; Jiao, Y. Genetic Contribution of Paleopolyploidy to Adaptive Evolution in Angiosperms. Mol. Plant 2020, 13, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.; Chapman, B.; Rong, J.; Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 2003, 422, 433–438. [Google Scholar] [CrossRef]
- Wang, X.; Shi, X.; Hao, B.; Ge, S.; Luo, J. Duplication and DNA segmental loss in the rice genome: Implications for diploidization. New Phytol. 2005, 165, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Bowers, J.; Chapman, B.; Peterson, D.; Rong, J.; Wicker, T.M. Comparative genome analysis of monocots and dicots, toward characterization of angiosperm diversity. Curr. Opin. Biotechnol. 2004, 15, 120–125. [Google Scholar] [CrossRef]
- Simillion, C.; Vandepoele, K.; Van Montagu, M.C.E.; Zabeau, M.; Van de Peer, Y. The hidden duplication past of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2002, 99, 13627–13632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Gaut, B.S.; Vision, T.J. Gene duplication and evolution. Science 2001, 293, 1551. [Google Scholar] [PubMed]
- He, X.; Zhang, J. Rapid Subfunctionalization Accompanied by Prolonged and Substantial Neofunctionalization in Duplicate Gene Evolution. Genetics 2005, 169, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Innan, H.; Kondrashov, F. The evolution of gene duplications: Classifying and distinguishing between models. Nat. Rev. Genet. 2010, 11, 97–108. [Google Scholar] [CrossRef]
- Wang, X.Y.; Paterson, A.H. Gene conversion in angiosperm genomes with an emphasis on genes duplicated by polyploidization. Genes 2011, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Wang, Z.; Wang, J.; Teng, J.; Shen, S.; Xiao, Q.; Bao, S.; Feng, Y.; Zhang, Y.; Li, Y.; et al. Conversion between 100-million-year-old duplicated genes contributes to rice subspecies divergence. BMC Genom. 2021, 22, 460. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tang, H.; Bowers, J.E.; Feltus, F.A.; Paterson, A.H. Extensive concerted evolution of rice paralogs and the road to regaining independence. Genetics 2007, 177, 1753–1763. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Leebens-Mack, J.; Ayyampalayam, S.; Bowers, J.E.; McKain, M.R.; McNeal, J.; Rolf, M.; Ruzicka, D.R.; Wafula, E.; Wickett, N.J.; et al. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 2012, 13, R3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sun, P.; Li, Y.; Liu, Y.; Yu, J.; Ma, X.; Sun, S.; Yang, N.; Xia, R.; Lei, T.; et al. Hierarchically Aligning 10 Legume Genomes Establishes a Family-Level Genomics Platform. Plant Physiol. 2017, 174, 284–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, D.; Ji, C.; Ma, X.; Li, H.; Zhang, W.; Li, S.; Liu, F.; Zhao, K.; Li, F.; Li, K.; et al. Genome of an allotetraploid wild peanut Arachis monticola: A de novo assembly. GigaScience 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Bertioli, D.J.; Jenkins, J.; Clevenger, J.; Dudchenko, O.; Gao, D.; Seijo, G.; Leal-Bertioli, S.C.M.; Ren, L.; Farmer, A.D.; Pandey, M.K.; et al. The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 2019, 51, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, W.; Wang, X.; Paterson, A.H.; Chen, H.; Yang, M.; Zhang, C.; Sun, P.; Zheng, Y.; Wang, L.; Xie, W.; et al. Reply to: Evaluating two different models of peanut’s origin. Nat. Genet. 2020, 52, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Robledo, G.; Lavia, G.I.; Seijo, G. Species relations among wild Arachis species with the A genome as revealed by FISH mapping of rDNA loci and heterochromatin detection. Theor. Appl. Genet. 2009, 118, 1295–1307. [Google Scholar] [CrossRef]
- Grabiele, M.; Chalup, L.; Robledo, G.; Seijo, G. Evolution: Genetic and geographic origin of domesticated peanut as evidenced by 5S rDNA and chloroplast DNA sequences. Plant. Syst. Evol. 2012, 298, 1151–1165. [Google Scholar] [CrossRef]
- Kochert, G.; Stalker, H.T.; Gimenes, M.; Galgaro, L.; Lopes, C.R.; Moore, K. Rflp and Cytogenetic Evidence on the Origin and Evolution of Allotetraploid Domesticated Peanut, Arachis hypogaea (Leguminosae). Am. J. Bot. 1996, 83, 1282–1291. [Google Scholar] [CrossRef]
- Simpson, C.E.; Krapovickas, A.; Valls, J.F.M. History of Arachis Including Evidence of A. hypogaea L. Progenitors. Peanut Sci. 2001, 28, 78–80. [Google Scholar] [CrossRef]
- Moretzsohn, M.C.; Gouvea, E.G.; Inglis, P.W.; Leal-Bertioli, S.C.M.; Valls, J.; Bertioli, D.J. A study of the relationships of cultivated peanut (Arachis hypogaea) and its most closely related wild species using intron sequences and microsatellite markers. Ann. Bot. 2013, 111, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Kochert, G.; Halward, T.; Branch, W.D.; Simpson, C.E. RFLP variability in peanut (Arachis hypogaea L.) cultivars and wild species. Theor. Appl. Genet. 1991, 81, 565–570. [Google Scholar] [CrossRef]
- Kohei, K.; Kunihiro, O. Genetic Diversification by Somatic Gene Conversion. Genes 2011, 2, 48–58. [Google Scholar]
- Wang, X.; Tang, H.; Bowers, J.E.; Paterson, A.H. Comparative inference of illegitimate recombination between rice and sorghum duplicated genes produced by polyploidization. Genome Res. 2009, 19, 1026–1032. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, L.J.; Wingen, L.U.; Bailey, P.; Joynson, R.; Brabbs, T.; Wright, J.; Higgins, J.D.; Hall, N.; Griffiths, S.; Clavijo, B.J.; et al. Analysis of the recombination landscape of hexaploid bread wheat reveals genes controlling recombination and gene conversion frequency. Genome Biol. 2019, 20, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Hendrix, M.; Lipsitch, M.; Jinks-Robertson, S. Dual roles for DNA sequence identity and the mismatch repair system in the regulation of mitotic crossing-over in yeast. Proc. Natl. Acad. Sci. USA 1997, 94, 9757–9762. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.Z.; Innan, H. Very low gene duplication rate in the yeast genome. Science 2004, 306, 1367–1370. [Google Scholar] [CrossRef] [Green Version]
- Slightom, J.L.; Blechl, A.E.; Smithies, O. Human fetal G gamma- and A gamma-globin genes: Complete nucleotide sequences suggest that DNA can be exchanged between these duplicated genes. Cell 1980, 21, 627–638. [Google Scholar] [CrossRef]
- Liu, C.; Wang, J.; Sun, P.; Yu, J.; Meng, F.; Zhang, Z.; Guo, H.; Wei, C.; Li, X.; Shen, S.; et al. Illegitimate Recombination Between Homeologous Genes in Wheat Genome. Front. Plant Sci. 2020, 11, 1076. [Google Scholar] [CrossRef]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhao, M.; Wang, X.; Tong, C.; Huang, S.; Tehrim, S.; Liu, Y.; Hua, W.; Liu, S. Bolbase: A comprehensive genomics database for Brassica oleracea. BMC Genom. 2013, 14, 664. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tang, H.; Paterson, A.H. Seventy Million Years of Concerted Evolution of a Homoeologous Chromosome Pair, in Parallel, in Major Poaceae Lineages. Plant Cell 2011, 23, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Li, H.; Hong, Y.; Zhang, G.; Wen, S.; Li, X.; Zhou, G.; Li, S.; Liu, H.; Liu, H.; et al. Corrigendum: Genome Sequencing and Analysis of the Peanut B-Genome Progenitor (Arachis ipaensis). Front. Plant Sci. 2018, 9, 1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sun, P.; Li, Y.; Liu, Y.; Yang, N.; Yu, J.; Ma, X.; Sun, S.; Xia, R.; Liu, X.; et al. An Overlooked Paleotetraploidization in Cucur-bitaceae. Mol. Biol. Evol. 2018, 35, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yuan, J.; Yu, J.; Meng, F.; Sun, P.; Li, Y.; Yang, N.; Wang, Z.; Pan, Y.; Ge, W.; et al. Recursive Paleohexaploidization Shaped the Durian Genome. Plant Physiol. 2019, 179, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitu-tions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar] [PubMed]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Som, A. Theoretical foundation to estimate the relative efficiencies of the Jukes–Cantor + gamma model and the Jukes–Cantor model in obtaining the correct phylogenetic tree. Gene 2006, 385, 103–110. [Google Scholar] [CrossRef]
- Edgar, R.C.; Batzoglou, S. Multiple sequence alignments. Curr. Opin. Struct. Biol. 2006, 16, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Audano, P.A.; Sulovari, A.; Graves-Lindsay, T.A.; Cantsilieris, S.; Sorensen, M.; Welch, A.E.; Dougherty, M.L.; Nelson, B.J.; Shah, A.; Dutcher, S.K.; et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 2019, 176, 663–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef]
- Chen, J.-M.; Cooper, D.N.; Chuzhanova, N.; Férec, C.; Patrinos, G.P. Gene conversion: Mechanisms, evolution and human disease. Nat. Rev. Genet. 2007, 8, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Hollister, J.D. Polyploidy: Adaptation to the genomic environment. New Phytol. 2015, 205, 1034–1039. [Google Scholar] [CrossRef]
- Peer, V.D.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- One Thousand Plant. Transcriptomes I: One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Soltis, P.S.; Soltis, D.E. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 2016, 30, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Fox, D.T.; Soltis, D.E.; Soltis, P.S.; Ashman, T.-L.; Van de Peer, Y. Polyploidy: A Biological Force from Cells to Ecosystems. Trends Cell Biol. 2020, 30, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.A.; Maere, S.; Van de Peer, Y. Plants with double genomes might have had a better chance to survive the Creta-ceous-Tertiary extinction event. Proc. Natl. Acad. Sci. USA 2009, 14, 5737–5742. [Google Scholar] [CrossRef] [Green Version]
- Koszul, R.; Fischer, G. A prominent role for segmental duplications in modeling eukaryotic genomes. Comptes R. Biol. 2009, 332, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, J.; Chaparro, C.; Laudie, M.; Berger, A.; Gavory, F.; Goicoechea, J.L.; Wing, R.A.; Cooke, R. Long-Range and Targeted Ectopic Recombination between the Two Homeologous Chromosomes 11 and 12 in Oryza Species. Mol. Biol. Evol. 2011, 28, 3139–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harpak, A.; Lan, X.; Gao, Z.; Pritchard, J.K. Frequent nonallelic gene conversion on the human lineage and its effect on the divergence of gene duplicates. Proc. Natl. Acad. Sci. USA 2017, 114, 12779–12784. [Google Scholar] [CrossRef] [Green Version]
- Bowers, J.; Arias, M.A.; Asher, R.; Avise, J.A.; Ball, R.T.; Brewer, G.A.; Buss, R.W.; Chen, A.H.; Edwards, T.M.; Estill, J.; et al. Comparative physical mapping links conservation of microsynteny to chromosome structure and recombination in grasses. Proc. Natl. Acad. Sci. USA 2005, 102, 13206–13211. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Wang, J.; Lin, W.; Li, S.; Li, H.; Zhou, J.; Ni, P.; Dong, W.; Hu, S.; Zeng, C.; et al. The Genomes of Oryza sativa: A History of Duplications. PLOS Biol. 2005, 3, e38. [Google Scholar] [CrossRef] [Green Version]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florent, M.; Alix, A.; Caroline, P.; Christophe, K.; Jérôme, S. Reconstructing the genome of the most recent common ancestor of flowering plants. Nat. Genet. 2017, 49, 490–496. [Google Scholar]
- Wang, X.; Jin, D.; Wang, Z.; Guo, H.; Zhang, L.; Wang, L.; Li, J.; Paterson, A.H. Telomere-centric genome repatterning determines recurring chromosome number reductions during the evolution of eukaryotes. New Phytol. 2015, 205, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chang, L.; Zhang, T.; Chen, H.; Zhang, L.; Lin, R.; Liang, J.; Wu, J.; Freeling, M.; Wang, X.; et al. Impacts of allopolyploidization and structural variation on intraspecific diversification in Brassica rapa. Genome Biol. 2021, 22, 166. [Google Scholar] [CrossRef]
- Daugherty, M.D.; Zanders, S.E. Gene conversion generates evolutionary novelty that fuels genetic conflicts. Curr. Opin. Genet. Dev. 2019, 58–59, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T. Population genetics theory of concerted evolution and its application to the immunoglobulin V gene tree. J. Mol. Evol. 1984, 20, 274–280. [Google Scholar] [CrossRef]
- Noonan, J.P.; Grimwood, J.; Schmutz, J.; Dickson, M.; Myers, R.M. Gene Conversion and the Evolution of Protocadherin Gene Cluster Diversity. Genome Res. 2004, 14, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Liao, D. Gene Conversion Drives Within Genic Sequences: Concerted Evolution of Ribosomal RNA Genes in Bacteria and Archaea. J. Mol. Evol. 2000, 51, 305–317. [Google Scholar] [CrossRef]
- Nei, M.; Rogozin, I.; Piontkivska, H. Purifying selection and birth-and-death evolution in the ubiquitin gene family. Proc. Natl. Acad. Sci. USA 2000, 97, 10866–10871. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Escalona, N.; Romero, J.; Espejo, R.T. Polymorphism and gene conversion of the 16S rRNA genes in the multiple rRNA operons ofVibrio parahaemolyticus. FEMS Microbiol. Lett. 2005, 246, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Rooney, A.P. Mechanisms Underlying the Evolution and Maintenance of Functionally Heterogeneous 18S rRNA Genes in Apicomplexans. Mol. Biol. Evol. 2004, 21, 1704–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, A.P.; Ward, T.J. Evolution of a large ribosomal RNA multigene family in filamentous fungi: Birth and death of a concerted evolution paradigm. Proc. Natl. Acad. Sci. USA 2005, 102, 5084–5089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godiard, L.; Grant, M.R.; Dietrich, R.A.; Kiedrowski, S.; Dangl, J.L. Perception and response in plant disease resistance. Curr. Opin. Genet. Dev. 1994, 4, 662–671. [Google Scholar] [CrossRef]
- Eickbush, M.T.; Young, J.M.; Zanders, S.E. Killer Meiotic Drive and Dynamic Evolution of the wtf Gene Family. Mol. Biol. Evol. 2019, 36, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Vogan, A.A.; Ament-Velásquez, S.L.; Granger-Farbos, A.; Svedberg, J.; Bastiaans, E.; Debets, A.J.; Coustou, V.; Yvanne, H.; Clavé, C.; Saupe, S.J.; et al. Combinations of Spok genes create multiple meiotic drivers in Podospora. eLife 2019, 8, e46454. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Quartet Patterns | Paralogues in Quartets | WCV-I a | WCV-II b | PCV c | Total | Conversion Rate (%) |
|---|---|---|---|---|---|---|---|
| A. duranensis | A1-B1-A2-B2 | 1871 | - | 4 | 216 | 220 | 11.8% |
| A. ipaensis | - | 2 | 241 | 242 | 13.0% | ||
| A. monticola A | Ama1-Amb1-Ama2-Amb2 | 99 | - | 2 | 11 | 13 | 13.1% |
| A. monticola B | - | - | 14 | 14 | 14.1% | ||
| A. hypogaea A (Shitouqi) | Aha1-Ahb1-Aha2-Ahb2 | 1314 | 3 | 3 | 121 | 126 | 9.6% |
| A. hypogaea B (Shitouqi) | 3 | 2 | 112 | 115 | 8.8% | ||
| A. hypogaea A (Tifrunner) | Aha1-Ahb1-Aha2-Ahb2 | 953 | 2 | 2 | 129 | 132 | 6.8% |
| A. hypogaea B (Tifrunner) | 3 | 3 | 134 | 139 | 7.1% |
| Orthologues | Converted Genes | Nonconverted Genes | p-Value (t-Test) | |
|---|---|---|---|---|
| A. duranensis–A. ipaensis | Pn | 0.055 | 0.023 | 9.48 × 10−14 |
| Ps | 0.109 | 0.064 | 7.05 × 10−7 | |
| Pn/Ps | 0.505 | 0.359 | 3.68 × 10−9 | |
| A. monticola A–A. monticola B | Pn | 0.114 | 0.0380 | 1.40 × 10−3 |
| Ps | 0.220 | 0.0846 | 3.60 × 10−3 | |
| Pn/Ps | 0.523 | 0.449 | 1.30 × 10−3 | |
| A. hypogaea A (Shitouqi)–A. hypogaea B (Shitouqi) | Pn | 0.0620 | 0.0483 | 2.10 × 10−3 |
| Ps | 0.116 | 0.0900 | 9.00 × 10−3 | |
| Pn/Ps | 0.534 | 0.537 | 4.98 × 10−5 | |
| A. hypogaea A (Tifrunner)–A. hypogaea B (Tifrunner) | Pn | 0.0704 | 0.0340 | 4.01 × 10−5 |
| Ps | 0.133 | 0.0753 | 4.10 × 10−5 | |
| Pn/Ps | 0.529 | 0.452 | 9.33 × 10−7 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, S.; Li, Y.; Wang, J.; Wei, C.; Wang, Z.; Ge, W.; Yuan, M.; Zhang, L.; Wang, L.; Sun, S.; et al. Illegitimate Recombination between Duplicated Genes Generated from Recursive Polyploidizations Accelerated the Divergence of the Genus Arachis. Genes 2021, 12, 1944. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12121944
Shen S, Li Y, Wang J, Wei C, Wang Z, Ge W, Yuan M, Zhang L, Wang L, Sun S, et al. Illegitimate Recombination between Duplicated Genes Generated from Recursive Polyploidizations Accelerated the Divergence of the Genus Arachis. Genes. 2021; 12(12):1944. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12121944
Chicago/Turabian StyleShen, Shaoqi, Yuxian Li, Jianyu Wang, Chendan Wei, Zhenyi Wang, Weina Ge, Min Yuan, Lan Zhang, Li Wang, Sangrong Sun, and et al. 2021. "Illegitimate Recombination between Duplicated Genes Generated from Recursive Polyploidizations Accelerated the Divergence of the Genus Arachis" Genes 12, no. 12: 1944. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12121944