
Evaluation of a Custom Design Gene Panel as a Diagnostic Tool for Human Non-Syndromic Infertility


1
IPPTS, Université de Strasbourg, 67000 Strasbourg, France
2
Laboratoire de Diagnostic Génétique UF de génétique de l’infertilité, Hôpitaux Universitaires de Strasbourg, 67000 Strasbourg, France
3
Laboratoires de Diagnostic Génétique, UF de Génétique Moléculaire, Hôpitaux Universitaires de Strasbourg, 67000 Strasbourg, France
4
Laboratoire de Génétique Médicale, INSERM, UMRS_1112, Institut de Génétique Médicale d’Alsace (IGMA), Université de Strasbourg Faculté de Médecine de Strasbourg, 67000 Strasbourg, France
5
Unité Fonctionnelle de Bioinformatique Médicale Appliquée au Diagnostic (UF7363), Hôpitaux Universitaires de Strasbourg, 67000 Strasbourg, France
*
Author to whom correspondence should be addressed.
Genes 2021, 12(3), 410; https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030410
Submission received: 15 February 2021
/
Revised: 4 March 2021
/
Accepted: 5 March 2021
/
Published: 12 March 2021
(This article belongs to the Special Issue Genetics and Genomics of Reproductive Medicine)
Abstract
:Infertility is a global healthcare problem, which affects men and women equally. With the advance of genome-wide analysis, an increasing list of human genes involved in infertility is now available. In order to evaluate the diagnostic interest to analyze these genes, we have designed a gene panel allowing the analysis of 51 genes involved in non-syndromic human infertility. In this initial evaluation study, a cohort of 94 non-syndromic infertility cases with a well-defined infertility phenotype was examined. Five patients with previously known mutations were used as positive controls. With a mean coverage of 457×, and 99.8% of target bases successfully sequenced with a depth coverage over 30×, we prove the robustness and the quality of our panel. In total, we identified pathogenic or likely pathogenic variations in eight patients (five male and three female). With a diagnostic yield of 8.5% and the identification of a variety of variants including substitution, insertion, deletion, and copy number variations, our results demonstrate the usefulness of such a strategy, as well as the efficiency and the quality of this diagnostic gene panel.
1. Introduction
The demonstration that infertility may in some instances have a genetic basis was provided soon after the first human karyotype was established. Indeed, in the late 1950s, numerical and structural chromosome abnormalities were identified in infertile patients [1]. For many years, karyotyping was the only available test to establish a genetic etiology of infertility. About twenty years later, Tiepolo and Zuffardi demonstrated a correlation between male infertility and Y chromosome deletions [2]. This locus was then defined as the “azoospermia factor” (AZF). However, the complexity of the AZF locus was only uncovered in the mid-1990s when it was subdivided into three sub-genomic regions, AZFa, AZFb, and AZFc, from proximal to distal Yq [3,4]. It was at about the same time that the identification of monogenic causes of male and female infertility began. Since then, the number of monogenic mutations discovered has increased very rapidly, with acceleration in the early 21st century due to the advent of whole-genome analyses, in particular high-throughput sequencing (HTS).
Mutations can be responsible for either syndromic or non-syndromic infertility. Syndromic cases associate infertility with other symptoms, which are usually the patient’s primary concern. With a few exceptions (for example, patients with cystic fibrosis or myotonic dystrophy) these patients, due to their health conditions, don’t have the opportunity to plan a parental project. On the other hand, non-syndromic cases involve patients with infertility not associated with any other symptoms. During the last two decades, large-scale genome-wide analyses of family cases of infertility or groups of infertile patients have identified a fast-growing number of “infertility genes”, solely responsible for non-syndromic infertility. This progress suggests it is now time to provide fertility practitioners with access to a new diagnostic tool for their patients. Unfortunately, only a limited amount of information is available so far from most studies on infertility genes, as nicely underlined by Oud et al. on male infertility [5]. In fact, a majority of the mutations described still await validation. This renders it tricky to choose a panel of genes to include in a diagnostic practice.
Until recently, the genetic diagnosis of infertility was mainly limited to karyotyping, CFTR mutation screening, Yq microdeletion testing for azoospermic or severe oligozoospermic patients, and, for women, to FMR1 gene screening to exclude Fragile X-associated primary ovarian insufficiency (FXPOI). However, the advent of HTS technologies has opened the field. Indeed, gene panel sequencing, allowing the simultaneous analysis of dozens to hundreds of genes, is now the common tool in human genetics, and some laboratories are already dedicating panels to infertility [6,7,8,9,10,11].
We present here the development, validation, and results of our activity based on a panel of 51 genes involved in different forms of non-syndromic male and female infertility.
2. Materials and Methods
2.1. Studied Population
Male and female patients showing different infertility phenotypes were recruited (Table 1). Germethèque biobank (BB-003-00081), site of Toulouse, (https://www.chu-toulouse.fr/-germetheque-centre-de-ressources-biologiques-, accessed on 3 March 2021), provided 40 male DNA samples extracted from whole blood and their associated data to realize this study [12]. Germethèque obtained consent form from each patient to use their samples (CPP 2.15.27). The Germethèque pilotage committee approved the study design on 1 December 2016. The biobank has a declaration DC-2014-2202 and an authorization AC-2015-2350. The number of the request made to Germethèque is 20161013, and its contract is referenced under the number 17 008 C. For the remaining set, saliva or blood samples were collected from collaborating clinics.
All patients were diagnosed through a complete diagnostic work-up for couple infertility. The minimum requirement was to follow the institutional directives already established for the diagnosis of a pathology being studied.
In the field of male infertility, World Health Organization (WHO) instructions were followed, and at least two detailed spermiograms performed at an interval of at least three months, in order to define the defect in terms of sperm number (azoospermia or oligozoospermia) and the motility of the sperm cells present (asthenozoospermia). If the study was focused on morphological defects (teratozoospermia), both a spermiogram and a spermocytogram were required. The patient may have had a combination of these defects, some of them (oligoasthenozoospermia, asthenoteratozoospermia, oligoteratozoospermia), or all (oligoasthenoteratozoospermia). In the case of non-obstructive azoospermia, it can be worthwhile, but not required, to have information on testis histology. Normal karyotype and absence of Y chromosome microdeletion were required for azoospermic and severe oligozoospermic cases. The workflow described by our group [13] was followed for the diagnosis of male infertility.
For female infertility, there is no defined workflow except for the premature ovarian insufficiency (POI) phenotype; however, a normal karyotype is required in all defined phenotypes. Diagnosis for POI was based on ESHRE recommendations, which defined POI as oligo/amenorrhea for at least 4 months and an elevated FSH level (>25 IU/L) on two occasions > 4 weeks apart (https://www.eshre.eu/Guidelines-and-Legal/Guidelines/Management-of-premature-ovarian-insufficiency.aspx, accessed on 10 February 2021). Additionally, FMR1 premutation testing was required for POI patients. At least two oocyte pick up (OPU) cycles with identified oocyte maturation defect and estradiol level at the day of OPU were decisive for the diagnosis of women with oocyte maturation defect.
Genomic DNA from patients was extracted from peripheral blood using a QIAamp® DNA Mini kit (Qiagen, Hilden, Germany) or from saliva using an Oragene DNA self-collection kit (DNA Genotek, Ottawa, Canada) according to the manufacturers’ instructions.
A positive control cohort was defined with 5 patients for whom a gene mutation was identified from previous whole-exome sequencing (WES) runs and confirmed by Sanger sequencing. The phenotype of control samples and their previously defined mutations are listed in Table 2.
2.2. Gene Panel Design
The gene panel was designed at the beginning of 2017. Gene selection was based on the following criteria:
- (i)
- Infertility genes: Genes defined by Online Mendelian Inheritance in Man (OMIM), at the time of design, as responsible for a non-syndromic male and/or female infertility phenotype; coded as spermatogenic failure (SPGF) for male infertility, and as premature ovarian failure (POF) and as oocyte maturation defect (OOMD) for female infertility.
- (ii)
- Candidate genes: Genes for which at least one variant potentially pathogenic for the related phenotype in humans has been identified by good-quality WES studies, but which need further confirmation.
- (iii)
- FMR1 sequencing: There is an association between pre-mutation of the FMR1 gene and increased susceptibility to idiopathic POI. We added FMR1 on the gene list in order to elucidate possible disease-causing variants for POI.
The Infertility Panel V2, HUS, Strasbourg, France (referred to as “infertility panel” in the rest of text), includes 51 genes in total; comprising 34 genes for male infertility, 15 genes for female infertility, and 2 genes shared by both. Table 3 lists the genes explored in this study; it comprises 33 Infertility genes (IG), 17 candidate genes (CG) and FMR1 for sequencing (S). In the meantime, among genes listed as “candidate genes”, NR0B1 (known also as DAX1), Wt1, and CCDC39 were validated as “infertility genes” with strong or definitive evidence [5].
The Infertility panel included also 6 common single-nucleotide polymorphisms (SNPs). The genotypes of 6 SNPs were used as identity-vigilance markers to verify and monitor the identity of patients by comparing the results obtained using an independent Taqman technology: PRSS12, chr4:119,237,348 (rs2292597); TRAPPC9, chr8:141,461,062 (rs3735803); AP4E1, chr15:51,217,361 (rs2306331); GRIN2B, chr12:14,018,777 (rs7301328); FTCD, chr21:47,558,473 (rs1047179); DOCK8, chr9: 286,593 (rs529208). Control SNPs were selected on the basis of allele frequency (∼0.5) and location on different chromosomes.
The design of the probes was carried out with the Agilent SureSelect application (www.agilent.com/genomics/suredesign, accessed on 4 January 2017). The list of genes and the genomic positions of control SNPs were submitted to SureDesign for the choice of capture probes according to the following specifications: cover the exons of the genes of interest as well as 25 bp of intronic sequence on either side of the exons and without the UTR regions, with 3 probes per nucleotide in the target regions (3× tiling). Coding regions of all the transcripts of the genes of interest were obtained by SureDesign via several sources (Refseq, ensembl, CCDS). The genomic coordinates of the proposed design were assessed in the UCSC genome browser [14] by using genome assembly Human GRCh37/hg19.
2.3. Gene Panel Sequencing
The method of detection of constitutional variants by capture and massive parallel sequencing on NextSeq550 has been validated and is used in our medical department (Genetic Diagnosis Laboratory, HUS, Strasbourg, France) in various diagnostic tests [15,16,17].
Libraries were prepared using SureSelectQXT Target Enrichment System (Agilent Technologies, Santa Clara, CA, USA), which uses RNA probes to capture known coding DNA. Briefly, 50 ng genomic DNA was fragmented enzymatically, and specific adaptor oligos were ligated to fragments of the DNA to be sequenced. The probes hybridized on the regions of interest were then captured by a magnetic system (beads coupled to streptavidin). After purification of the enriched DNA fragments, a PCR was carried out in order to increase the number of enriched libraries, which were then double-indexed by oligonucleotide barcodes (a unique barcode per patient within the same series) and pooled by 30 before sequencing on NextSeq 550 System (Illumina, San Diego, CA, USA) with 2 × 75 bp reads for a total 51 genes and 6 control SNPs.
2.4. Data Analysis
Sequencing data was analyzed by STARK (Stellar Tools from raw sequencing data Analysis to variant RanKing), our in-house bioinformatics pipeline. STARK adopts the Genome Analysis Toolkit (GATK) recommendations [18]. It performs reads demultiplexing, alignment to the reference human genome (GRCh37/hg19), indel (insertion or deletion) realignment, bam recalibration, variant detection (calling), and variant annotation steps. It takes as input the raw sequencing data (BCL, FASTQ, or unaligned BAM) and generates annotated results (VCF) as well as quality reports. SNV/indels were then annotated and ranked using VaRank [19]. VaRank incorporates the annotations retrieved by the Alamut Batch software (Interactive Biosoftware, France) as well as allele frequency from our internal exome database. Annotations include HGVS nomenclature (genomic, cDNA, and proteic), and population database frequencies from the 1000 Genomes Project ([20], http://www.1000genomes.org/, accessed on 15 February 2021) and gnomAD databases ([21], http://gnomad.broadinstitute.org/, accessed on 15 February 2021). Data processing and analysis were performed as described previously with minor changes [22].
Copy number variants (CNV) have been called using the CANOES program [23] and annotated using AnnotSV [24] with similar databases as SNV/indel, but also including DGV and dbVar.
Variant filtering has been carried out according to allele frequency in 1000 Genomes Project data and in GnomAD (filtering out allele frequency when >1%). Variant classification was performed following guidelines for the interpretation of sequence variants according to the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) [25]. We consider each transmission mode without prioritizing one. Pathogenicity of missense variants were predicted via available software like Provean/SIFT (http://provean.jcvi.org/, accessed on 15 February 2021 [26]) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/, accessed on 15 February 2021 [27]). Adapted ACMG/AMP guidelines for single-gene copy number variants were used for interpretation of CNV [28].
2.5. Identity Control and Confirmation of Mutations
The identity of samples was systematically checked via control SNPs by real-time PCR and then compared with the sequencing results. The predesign TaqMan PCR assay was performed independently on the extracted DNA. The amplification was executed on the patient series in simplex with a blank PCR and a control DNA with known genotype according to manufacturer’s protocol (FastStart TaqMan® Probe Master, Merck KGaA, Darmstadt, Germany).
Selected candidate variations have been confirmed via Sanger sequencing. Primers for Sanger have been designed using the Primer3Plus online program for the region harboring the identified variation. Primer pairs were checked via In-SilicoPCR (https://genome.ucsc.edu/cgi-bin/hgPcr, accessed on 8 February 2020) and primer BLAST (https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/tools/primer-blast/, accessed on 8 February 2020) for the specificity. The region of interest was amplified by using polymerase chain reaction (PCR). Amplification conditions and all primers are listed in Supplementary Table S1. Amplification and size of the PCR product were checked on 2% agarose gel. PCR products were purified, and double-strand sequencing of each DNA fragment was performed by GATC Services, Eurofins Genomics (Ebersberg, Germany). Sequences were aligned on a reference sequence (GrCH37, hg19) by using ApE plasmid editor in order to check variation(s).
3. Results
3.1. Cohort Description
A total of 94 patients were included in this study, 79 men and 15 women. The phenotype of each individual is presented in Table 1. Male patients were classified into four categories: sperm production defect (SPD) (50 cases), either azoospermia or oligozoospermia; teratozoospermia (7 cases); asthenozoospermia (1 case); and a mixed phenotype (21 cases). Female patients were classified into two categories, including oocyte maturation defect (OOMD; 4 cases) and premature ovarian insufficiency (POI; 11 cases).
3.2. Validation of the Infertility Panel and Identity Control
Among the five patients we used as controls, there were 2 non-obstructive azoospermia patients; C1 had a hemizygous stop-loss in MAGEB4 [29] and C2 had a homozygous stop-gain in TEX15 [22]. The remaining three samples were from globozoospermia patients, C3, C4, and C5, who carried either deletions and/or point mutations in DPY19L2 [30]. All control sample variants could be redetected by the panel analysis (Table 2, Supplementary Figures S1–S4).
The identity of all samples was checked via the included SNPs by TaqMan amplification. All results matched with sequencing results and confirmed the identity of the samples.
3.3. Sequencing Results and Identification of Variants
The total panel size was 187 kb with 57 loci comprising 883 distinct regions. Overall, the mean coverage was 457×, and 99.8% of target bases were successfully sequenced with a minimum depth of coverage of 30× (see Supplementary Tables S2 and S3, per gene and per patient, respectively). Genes were analyzed according to the patient’s phenotype. Targeted sequencing and variant analysis statistics are given in Supplementary Table S3. After filtering for allele frequency in the general population (filtered out >1%), predictions of the effect on protein function and a literature check were conducted. We retained causative mutations explaining the infertility phenotype for eight patients (8/94; 8.5%); five male patients (5/79; 6.3%) and three female patients (3/15; 20%). No pathogenic CNV was found related to studied phenotype. Detailed information of the mutations identified and relevant patient information are shown in Table 4.
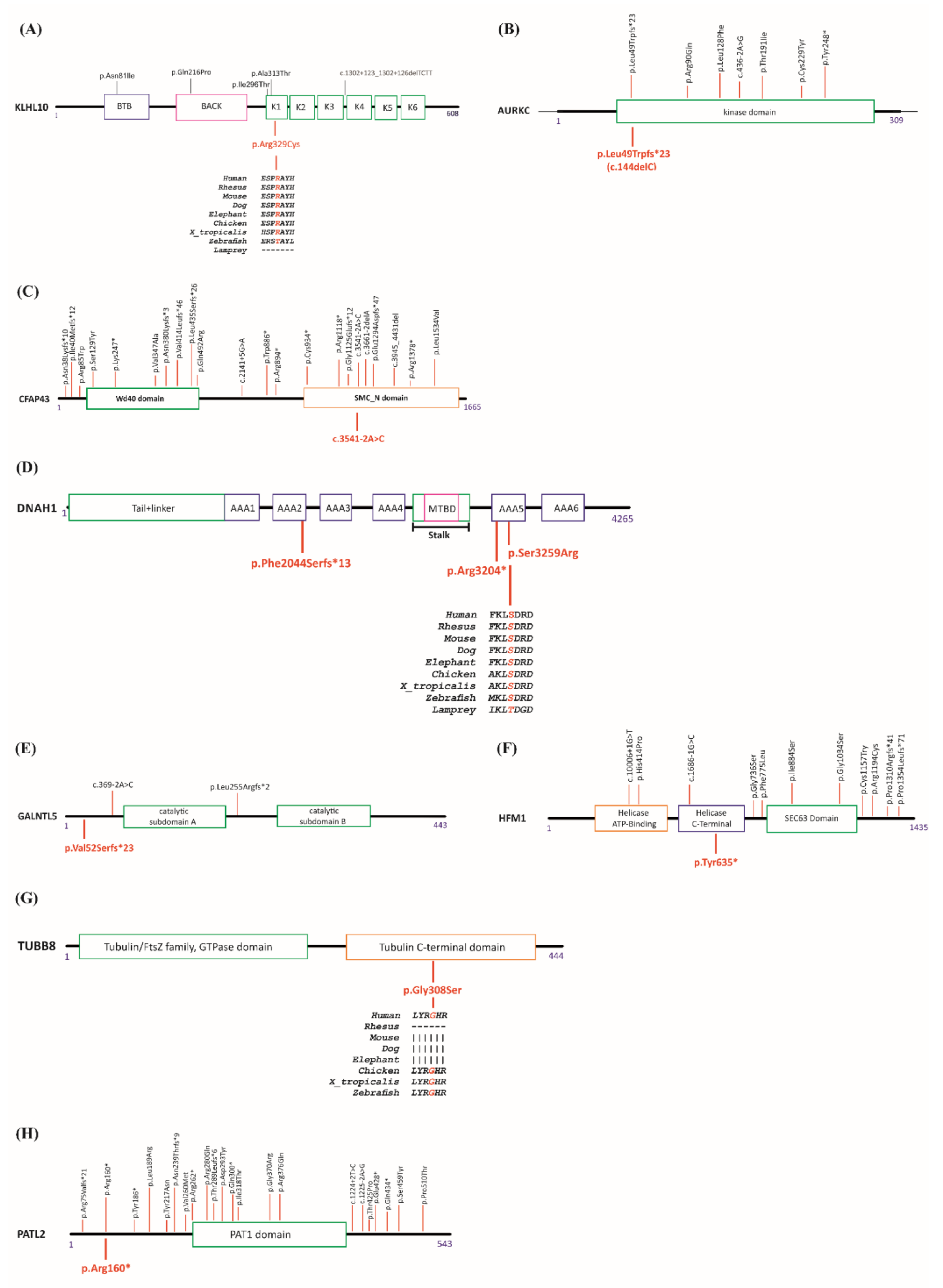
Among the identified mutations, three of them, affecting PATL2, AURKC, and CFAP43, were already described in at least one patient with a related phenotype. In addition, we identified five new mutations affecting HFM1, GALNTL5, KLHL10, DNAH1, and TUBB8. Schematic diagrams of the relevant genes and the known mutations are shown in Figure 1A–H and Supplementary Table S4.
Among male patients, patient Pt12 was diagnosed with azoospermia. We identified a heterozygous missense mutation (c.985C > T, p.Arg329Cys) in KLHL10. The encoded protein is involved in the ubiquitination process and subsequent proteasomal degradation of target proteins during spermatogenesis. The variation is in the conserved Kelch1 domain (Figure 1A), as is the missense mutation previously identified in one azoospermic patient [31]. Prediction tools indicate a possible damaging/deleterious effect (PolyPhen-2 and Provean/SIFT, respectively).
Patient Pt41 was a 30-year-old man of Algerian descent; his spermiogram showed oligo-astheno-teratozoospermia with total head abnormality. Panel analysis revealed the recurrent homozygous mutation (c.144delC, p.Leu49Trpfs*23) in AURKC. The gene product plays a role in meiosis and more particularly in spermatogenesis. With a lack of functional AURKC protein, chromosomal segregation would be perturbed. The identified mutation has been described as the most frequent one in the Maghrebian population [32]. A list of known AURKC mutations is given in Figure 1B.
Patient Pt55 was a 28-year-old Moroccan man suffering infertility due to a sperm flagellar problem. Analysis revealed a homozygous splice mutation (c.3541-2A > C, p.?) in CFAP43. This gene encodes a protein involved in sperm flagellum axoneme organization and function. The same mutation was reported before in two unrelated Tunisian patients homozygously for sperm flagella problems [33]. A list of known CFAP43 mutations is given in Figure 1C.
Patient Pt65 was a 33-year-old male of French origin, suffering from severe astheno-teratozoospermia, with 97% immotile sperm and 92% abnormal flagella. Three heterozygous variations in DNAH1 were identified, one frameshift (c.6131del, p.Phe2044Serfs*13) giving rise to a premature stop codon thirteen codons downstream from the start codon, a stop gain mutation (c.9610C > T, p.Arg3204*), and one missense (c.9777T > G, p.Ser3259Arg). The missense variation, which is located in a conserved region, is predicted as deleterious or possibly damaging using the prediction tools Provean/SIFT and PolyPhen-2 respectively (Figure 1D). DNAH1 gene product is required in spermatozoa for the formation of the inner dynein arms and biogenesis of the axoneme; it is an energy-generating protein needed for sperm motility. In order to establish the allelic distribution of these variants, we analyzed his parents by Sanger sequencing. He inherited two variations from his mother (c.6131del, p.Phe2044Serfs*13 and c.9777T > G, p.Ser3259Arg) and one heterozygous variation from his father (c.9610C > T, p.Arg3204*), ruling out the possible involvement of the missense in the patient’s phenotype. His brother was also tested; he carried the same mutations and suffered from the same infertility problems.
Patient Pt77 was a 29-year-old male; his spermiogram was diagnosed as oligo-astheno-teratozoospermia with 90% immotile sperm. Our analysis revealed a heterozygous frameshift mutation (c.153dup, p.Val52Serfs*23) in GALNTL5 (Figure 1E). The functional gene product is essential for mammalian sperm formation. The identified mutation theoretically leads to an early translational termination at the very beginning part of the protein.
Among female patients, patient Pt2 was a 30-year-old woman of Turkish descent. She had low anti-Müllerian hormone (AMH) (0.04 ng/mL) and a normal karyotype, and her parents were first degree cousins. The patient had normal FMR1 alleles with 23 and 30 CGG repeats. Our panel gene analysis revealed a homozygous stop gain mutation (c.1905T > A, p.Tyr635*) in HFM1. The list of known mutations is given in Figure 1F.
Patient Pt 38 was a 32-year-old woman from a Turkish consanguineous family. She had a history of 12 years of primary infertility. She underwent four failed in vitro fertilization (IVF) attempts; all retrieved oocytes were either degenerate or immature. We identified a homozygous missense mutation (c.922G > A p.Gly308Ser) in TUBB8. The mutation was located in the conserved C-terminal domain of the protein and was predicted as possibly damaging or deleterious by PolyPhen-2 and Provean/SIFT respectively (Figure 1G).
Patient Pt 71 was a 31-year-old woman of Tunisian descent with primary infertility and a history of consanguinity in her family line. She had undergone three failed IVF cycles. Seven to 30 oocytes were retrieved in each cycle; however, all of the oocytes were at a germinal vesicle stage or atretic. She carried a homozygous stop gain mutation, (c.478C > T, p.Arg160*) in PATL2. This mutation was previously listed as a causative mutation in patients with oocyte maturation arrest [34]. The list of previously known PATL2 mutations is given in Figure 1H.
4. Discussion
The recent improvement of whole genome analysis has allowed an exponential rate of infertility gene identification. Indeed, during the last two decades, the number of genes proven to be involved in male or female non-syndromic infertility has increased very quickly. As for many, if not all medical fields, the time has come to translate this research to the clinical setting.
The goal of the present study was to assess the clinical value of a diagnostic gene panel in the ART practice and to establish the prevalence of mutations in selected genes for the cohort of patients studied. Diagnostic yields of available infertility panels worldwide range between 0.4% to 25% (Table 5). It is worth noting that results provided by Canerella et al. are confusing, and their diagnostic yield of 25% seems to be overestimated by collapsing results with their previous study [10]. However, available reports are difficult to compare because the list of genes, the studied phenotypes, the number of patients analyzed, and the quality of HTS are different in each study. Most of them do not report the quality of their sequencing, or do so only partially, which should be: (1) high sequence coverage of 100× to guarantee sequence of whole gene, which is compensatory for the detection of all rare variants; (2) more than 95% of the region of interest covered by at least 30 reads for a good quality HTS study [35]. In addition, most of these studies do not provide patients’ clinical data, and none of them analyze CNVs. It is almost certain that the diagnostic yield will be improved nonetheless by the HTS quality, but also by the strict gene selection and the number of genes analyzed.
For this purpose, we set up a panel of 51 genes responsible for a non-syndromic infertility phenotype. Our control cohort was chosen in order to challenge as much as possible the ability of our panel to detect various types of variants. Indeed, we show that our strategy allows us to identify substitutions, indels, and CNVs. Subsequently, we analyzed 94 patients (79 men and 15 women) with precise infertility phenotypes. We have shown the efficiency and the quality of our panel with a mean coverage of 457× and 99.8% of target bases successfully sequenced with a depth coverage over 30×. In total, we identified causative mutations for eight of the tested patients (8.5%; 8/94), five for the male cohort (6.3%; 5/79) and three for the female patients (20%; 3/15). Mutations identified in three patients in the present study have already been reported and new mutations were identified in five patients.
Among male patients, a new heterozygous missense mutation in KLHL10 was identified in an azoospermic patient. OMIM gives an autosomal dominant inheritance for KLHL10 mutations. Initially, KLHL10 gene mutations were mainly identified in oligospermic patients [36]; however, a recent study described a mutation in KLH10 in an azoospermic patient [31]. Therefore, we cannot rule out that mutations in this gene may lead to an oligozoospermia evolving towards a complete azoospermia over time. Unfortunately, we have only recent spermiograms for this patient. Further investigations are critical to confirm this finding. Indeed, if mutations in KLHL10 imply an evolution from oligozoospermia to azoospermia, this could impact the care proposed to such patients and their relatives. It is important to offer patients, as soon as possible, sperm cryopreservation, and also important to test brothers and other male relatives in order to propose cryopreservation for the carriers.
A second patient was suffering from macrozoospermia, and he carried the recurrent AURKC mutation found in the North African population. Overall, with full AURKC gene sequencing, a positive mutation diagnosis is found in 83.7% of macrozoospermic patients [37]. Although different homozygous or heterozygous mutations have been identified, two mutations in AURKC are recurrent. The first one, c.144delC (p.Leu49Trpfs*23), is found in North African populations, and the other, c.744C > G (p.Y248*), is found in European populations. For the first one, it has been clearly demonstrated that in such a situation, the majority of spermatozoa are tetraploid, therefore the only ART option possible is sperm donation [38]. OMIM defines the pathology linked to AURKC as transmitted under an autosomal recessive mode.
For a third patient showing MMAF, we found a previously reported homozygote mutation in CFAP43. Autosomal recessive inheritance was reported in OMIM for CFAP43 mutations in men with MMAF, including absent, short, coiled, bent, and irregular-caliber flagella (Figure 1C). It seems that ICSI can be safely proposed, with a reasonable success, to these patients [39]. Next, we identified in a patient diagnosed with severe astheno-teratozoospermia three heterozygous variations in DNAH1, two transmitted by his mother and one by his father. So far, more than forty mutations have been identified in DNAH1 as possible causes of MMAF (Figure 1D and Supplementary Table S4. WES was the common technique in all published studies, and, except for two, all patients were homozygous or carried two heterozygous mutations. This supports a recessive mode of transmission as reported by OMIM. Here again ICSI can be safely proposed, with a reasonable success.
The last male patient, showing an oligo-astheno-teratozoospermia, had a new heterozygous frameshift variation, possibly disease-causing, in GALNTL5. Human GALNTL5 consists of nine exons and codes for 443 amino acid (aa) protein. The variation we identified is on exon 2 (aa52); it is a nucleotide duplication causing a frame shift, creating a premature stop codon and leading to an early translational termination of 23aa. A different heterozygous one-nucleotide deletion has been identified in exon 6 of GALNTL5 as being causative for male fertility due to immotile sperm [40]. These results have been confirmed in mice. Indeed, heterozygous mutations affected male mice fertility due to immotile sperm [40]. This strongly supports a dominant mode of transmission, although it is not listed in OMIM. GALNTL5 was classified as CG, though our results confirm and re-inforce genotype/phenotype relation in the asthenozoospermia phenotype in men, therefore contributing to the upgrade of GALNTL5 as IG.
Among the female patients, one presented a POI, and she carried a new homozygote mutation in HFM1. Initially, compound heterozygous mutations in HFM1 were identified in women with POI [41], and the inheritance mode was given as autosomal recessive by OMIM. Subsequently, it was postulated that heterozygous missense mutations might also be associated with POI [42,43]; however, this needs to be further investigated, since in the first study, using WES, the mother was a heterozygous carrier, and she was reported as clinically normal [41].
The second mutated woman produced, following ovarian stimulation, only degenerate or immature oocytes. She carries a new homozygous mutation in TUBB8. As of January 2021, 98 different heterozygous or homozygous mutations in TUBB8 have been identified as a cause for oocyte maturation arrest in females, and the list is still growing (Supplementary Table S4. Both an autosomal dominant and autosomal recessive mode of inheritance are indicated in OMIM.
The third woman diagnosed by our panel showed an oocyte maturation arrest at the germinal vesicle stage, with an already identified homozygous mutation in PATL2. Patients with mutations in PATL2 can present variable phenotypes, with some oocytes exhibiting maturation arrest at the germinal vesicle stage and others at the metaphase I stage, as well as fertilization failure or, in those that are fertilized, early embryonic arrest. So far, about twenty homozygous or compound heterozygous PATL2 mutations have been identified in women with oocyte maturation arrest, confirming a recessive mode of transmission as given in OMIM.
The diagnostic yield of our custom designed panel in the present study was 8.5%. Such a result is one of the highest reported so far in this field, but remains lower than for other medical specialties. For instance, a success rate of 25% was reported for the diagnosis of intellectual disability using targeted HTS [15]. These results can be, most probably, explained by the heterogeneity of infertility. This success rate will certainly be improved by including the latest genes identified, increasing the cohort of patients, and narrowing the inclusion criteria of infertility phenotypes.
We are at a transition period where basic research is translated into clinical practice. This will have many positive consequences for patients as well as for ART practitioners. First of all, for an increasing number, it will pinpoint the etiology of the infertility, which is a relief both for patients and their doctors. Precise diagnosis also opens the way to offer genetic counseling for patients, as well as their relatives. Moreover, targeted sequencing studies are valuable to re-classify reported CG genes related to infertility as IG based on the identification of new patients showing the same phenotype and sharing mutations in the same gene.
Having a diagnosis will improve patient care by adapting treatment to the patient’s situation. Being at the beginning of this genetic activity, there are, so far, only a few genes for which a specific action can be proposed. This is the case for DPY19L2, where artificial oocyte activation must be offered [44]; AURKC, for which sperm donation or renouncing parenthood are the only possibilities [32]; and TEX15, where sperm cryopreservation has to be proposed to the patient, but also to his affected brothers even if they do not yet have a parental project [22]. Similar to TEX15, KLHL10 mutations may correlate with a decrease in sperm count over time, and sperm cryopreservation might be proposed to the patient. Considering the low cost of the technique, it could be proposed even before acquiring proof that the sperm concentration will decrease with time.
In order to improve patient care through the genetic diagnosis of infertility, more studies have to be carried out. We are here at the frontier of two activities; research will enrich the diagnostic tools offered, and the diagnostic practice will allow a better definition of the genotype/phenotype correlations to enable personalized care. Indeed, the in-depth analysis of clinical data will allow us to better define the criteria for each genetic test and therefore improve the efficiency of the proposed diagnoses.
5. Conclusions
Our custom designed infertility panel is validated and proved to be able to detect various types of variants including substitutions, indels, and CNVs. In total, we identified causative mutations for eight of the tested patients (8.5%; 8/94). The quickest improvement of diagnosis based on panel analysis will come from increasing the number of genes analyzed.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4425/12/3/410/s1, Figure S1: Visualization of HTS results for control samples C1 and C2 by the Integrative Genomics Viewer (IGV), Figure S2: HTS data visualized with the IGV for control sample C3 showing heterozygous DPY19L2 gene deletion with point mutation in exon 8, Figure S3: Data visualization of HTS for control sample C4, Figure S4: A whole DPY19L2 deletion in control sample C5. Table S1: Primers used for amplification of ten candidate variants selected in 8 genes after panel analysis, Table S2: Sequencing quality for each gene from the infertility panel, Table S3: Sequencing quality and bioinformatics analysis information of the 94 patients, Table S4: Previously identified mutations described in the literature for the 8 genes that we determined as mutated in this study, with corresponding zygosity and relevant references.
Author Contributions
O.O. designed the panel, and carried out sequencing and analysis of the samples. J.T. helped with the design of the panel. J.M. supervised the bioinformatics analysis. S.V. supervised all the studies. O.O. and S.V. designed and wrote the first draft of the study, and all authors contributed to the revision process. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Agence de la Biomédicine “AMP, diagnostic prénatal et diagnostic génétique 2016”, grant number: ADB/CLV-DLE, and the APC was funded by Hôpital universitaire de Strasbourg (HUS).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Comité de Protection de la Personne (CPP) of Strasbourg University Hospital, France (CPP 09/40—WAC-2008-438 1W DC-2009-I 002).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data generated in this study are included in the following article and corresponding Supporting Information. The raw sequencing data generated in the course of this study are not publicly available due to the protocol and the corresponding consents used that did not include such information. All variants have been submitted to ClinVar (SCV001478453, SCV001478457, SCV001478463, SCV001478464, SCV001478466, SCV001478469, SCV001478470, SCV001478471) (https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar).
Acknowledgments
We thank Timur Gürgan and Senior Clinical Embryologist Halil Ruso from Gürgan Clinic Women’s Health and IVF Center, Ankara, Turkey, and the IVF specialist from CMCO, Strasbourg University Hospital, Strasbourg, France, for their assistance with the clinical data for patients with mutations. This study also involved samples from Germethéque Biobank (Toulouse, France). We would like also to thank Robert Drillien for his critical reading of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Jacobs, P.A.; Strong, J.A. A Case of Human Intersexuality Having a Possible XXY Sex-Determining Mechanism. Nature 1959, 183, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Tiepolo, L.; Zuffardi, O. Localization of Factors Controlling Spermatogenesis in the Nonfluorescent Portion of the Human Y Chromosome Long Arm. Hum. Genet. 1976, 34, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Reijo, R.; Lee, T.Y.; Salo, P.; Alagappan, R.; Brown, L.G.; Rosenberg, M.; Rozen, S.; Jaffe, T.; Straus, D.; Hovatta, O. Diverse Spermatogenic Defects in Humans Caused by Y Chromosome Deletions Encompassing a Novel RNA-Binding Protein Gene. Nat. Genet. 1995, 10, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.H.; Edelmann, A.; Kirsch, S.; Henegariu, O.; Hirschmann, P.; Kiesewetter, F.; Köhn, F.M.; Schill, W.B.; Farah, S.; Ramos, C.; et al. Human Y Chromosome Azoospermia Factors (AZF) Mapped to Different Subregions in Yq11. Hum. Mol. Genet. 1996, 5, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Oud, M.S.; Volozonoka, L.; Smits, R.M.; Vissers, L.E.L.M.; Ramos, L.; Veltman, J.A. A Systematic Review and Standardized Clinical Validity Assessment of Male Infertility Genes. Hum. Reprod. 2019, 34, 932–941. [Google Scholar] [CrossRef] [Green Version]
- França, M.M.; Funari, M.F.A.; Nishi, M.Y.; Narcizo, A.M.; Domenice, S.; Costa, E.M.F.; Lerario, A.M.; Mendonca, B.B. Identification of the First Homozygous 1-Bp Deletion in GDF9 Gene Leading to Primary Ovarian Insufficiency by Using Targeted Massively Parallel Sequencing. Clin. Genet. 2018, 93, 408–411. [Google Scholar] [CrossRef]
- Riera-Escamilla, A.; Enguita-Marruedo, A.; Moreno-Mendoza, D.; Chianese, C.; Sleddens-Linkels, E.; Contini, E.; Benelli, M.; Natali, A.; Colpi, G.M.; Ruiz-Castañé, E.; et al. Sequencing of a “mouse Azoospermia” Gene Panel in Azoospermic Men: Identification of RNF212 and STAG3 Mutations as Novel Genetic Causes of Meiotic Arrest. Hum. Reprod. 2019, 34, 978–988. [Google Scholar] [CrossRef]
- Lorenzi, D.; Fernández, C.; Bilinski, M.; Fabbro, M.; Galain, M.; Menazzi, S.; Miguens, M.; Perassi, P.N.; Fulco, M.F.; Kopelman, S.; et al. First Custom Next-Generation Sequencing Infertility Panel in Latin America: Design and First Results. JBRA Assist. Reprod. 2020, 24, 104–114. [Google Scholar] [CrossRef]
- Rocca, M.S.; Msaki, A.; Ghezzi, M.; Cosci, I.; Pilichou, K.; Celeghin, R.; Foresta, C.; Ferlin, A. Development of a Novel Next-Generation Sequencing Panel for Diagnosis of Quantitative Spermatogenic Impairment. J. Assist. Reprod. Genet. 2020, 37, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Cannarella, R.; Condorelli, R.A.; Paolacci, S.; Barbagallo, F.; Guerri, G.; Bertelli, M.; La Vignera, S.; Calogero, A.E. Next-Generation Sequencing: Toward an Increase in the Diagnostic Yield in Patients with Apparently Idiopathic Spermatogenic Failure. Asian J. Androl. 2021, 23, 24–29. [Google Scholar] [CrossRef]
- Cannarella, R.; Precone, V.; Guerri, G.; Busetto, G.M.; Di Renzo, G.C.; Gerli, S.; Manara, E.; Dautaj, A.; Bertelli, M.; Calogero, A.E. Clinical Evaluation of a Custom Gene Panel as a Tool for Precision Male Infertility Diagnosis by Next-Generation Sequencing. Life 2020, 10, 242. [Google Scholar] [CrossRef]
- BB-0033-00081; Contract ref. 17 008 C, Request no 20161013; CRB Germethèque: Toulouse, France, 2016.
- Okutman, O.; Rhouma, M.B.; Benkhalifa, M.; Muller, J.; Viville, S. Genetic Evaluation of Patients with Non-Syndromic Male Infertility. J. Assist. Reprod. Genet. 2018, 35, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Redin, C.; Gérard, B.; Lauer, J.; Herenger, Y.; Muller, J.; Quartier, A.; Masurel-Paulet, A.; Willems, M.; Lesca, G.; El-Chehadeh, S.; et al. Efficient Strategy for the Molecular Diagnosis of Intellectual Disability Using Targeted High-Throughput Sequencing. J. Med. Genet. 2014, 51, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Montaut, S.; Tranchant, C.; Drouot, N.; Rudolf, G.; Guissart, C.; Tarabeux, J.; Stemmelen, T.; Velt, A.; Fourrage, C.; Nitschké, P.; et al. Assessment of a Targeted Gene Panel for Identification of Genes Associated With Movement Disorders. JAMA Neurol. 2018, 75, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Rey, T.; Tarabeux, J.; Gerard, B.; Delbarre, M.; Le Béchec, A.; Stoetzel, C.; Prasad, M.; Laugel-Haushalter, V.; Kawczynski, M.; Muller, J.; et al. Protocol GenoDENT: Implementation of a New NGS Panel for Molecular Diagnosis of Genetic Disorders with Orodental Involvement. Methods Mol. Biol. 2019, 1922, 407–452. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Geoffroy, V.; Pizot, C.; Redin, C.; Piton, A.; Vasli, N.; Stoetzel, C.; Blavier, A.; Laporte, J.; Muller, J. VaRank: A Simple and Powerful Tool for Ranking Genetic Variants. PeerJ 2015, 3, e796. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Okutman, O.; Muller, J.; Baert, Y.; Serdarogullari, M.; Gultomruk, M.; Piton, A.; Rombaut, C.; Benkhalifa, M.; Teletin, M.; Skory, V.; et al. Exome Sequencing Reveals a Nonsense Mutation in TEX15 Causing Spermatogenic Failure in a Turkish Family. Hum. Mol. Genet. 2015, 24, 5581–5588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backenroth, D.; Homsy, J.; Murillo, L.R.; Glessner, J.; Lin, E.; Brueckner, M.; Lifton, R.; Goldmuntz, E.; Chung, W.K.; Shen, Y. CANOES: Detecting Rare Copy Number Variants from Whole Exome Sequencing Data. Nucleic Acids Res. 2014, 42, e97. [Google Scholar] [CrossRef]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An Integrated Tool for Structural Variations Annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN Web Server: A Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, T.; Sack, L.M.; Arjona, D.; Tan, D.; Mei, H.; Cui, H.; Gao, H.; Bean, L.J.H.; Ankala, A.; Del Gaudio, D.; et al. Adapting ACMG/AMP Sequence Variant Classification Guidelines for Single-Gene Copy Number Variants. Genet. Med. 2020, 22, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Okutman, O.; Muller, J.; Skory, V.; Garnier, J.M.; Gaucherot, A.; Baert, Y.; Lamour, V.; Serdarogullari, M.; Gultomruk, M.; Röpke, A.; et al. A No-Stop Mutation in MAGEB4 Is a Possible Cause of Rare X-Linked Azoospermia and Oligozoospermia in a Consanguineous Turkish Family. J. Assist. Reprod. Genet. 2017, 34, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Koscinski, I.; Elinati, E.; Fossard, C.; Redin, C.; Muller, J.; de la Calle, J.V.; Schmitt, F.; Ben Khelifa, M.; Ray, P.F.; Ray, P.; et al. DPY19L2 Deletion as a Major Cause of Globozoospermia. Am. J. Hum. Genet. 2011, 88, 344–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, T.F.; Friedrich, C.; Grangeiro, C.H.P.; Martelli, L.R.; Grzesiuk, J.D.; Emich, J.; Wyrwoll, M.J.; Kliesch, S.; Simões, A.L.; Tüttelmann, F. Sequence Analysis of 37 Candidate Genes for Male Infertility: Challenges in Variant Assessment and Validating Genes. Andrology 2020, 8, 434–441. [Google Scholar] [CrossRef]
- Dieterich, K.; Zouari, R.; Harbuz, R.; Vialard, F.; Martinez, D.; Bellayou, H.; Prisant, N.; Zoghmar, A.; Guichaoua, M.R.; Koscinski, I.; et al. The Aurora Kinase C c.144delC Mutation Causes Meiosis I Arrest in Men and Is Frequent in the North African Population. Hum. Mol. Genet. 2009, 18, 1301–1309. [Google Scholar] [CrossRef] [Green Version]
- Coutton, C.; Vargas, A.S.; Amiri-Yekta, A.; Kherraf, Z.-E.; Ben Mustapha, S.F.; Le Tanno, P.; Wambergue-Legrand, C.; Karaouzène, T.; Martinez, G.; Crouzy, S.; et al. Mutations in CFAP43 and CFAP44 Cause Male Infertility and Flagellum Defects in Trypanosoma and Human. Nat. Commun. 2018, 9, 686. [Google Scholar] [CrossRef]
- Maddirevula, S.; Coskun, S.; Alhassan, S.; Elnour, A.; Alsaif, H.S.; Ibrahim, N.; Abdulwahab, F.; Arold, S.T.; Alkuraya, F.S. Female Infertility Caused by Mutations in the Oocyte-Specific Translational Repressor PATL2. Am. J. Hum. Genet. 2017, 101, 603–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthijs, G.; Souche, E.; Alders, M.; Corveleyn, A.; Eck, S.; Feenstra, I.; Race, V.; Sistermans, E.; Sturm, M.; Weiss, M.; et al. Guidelines for Diagnostic Next-Generation Sequencing. Eur. J. Hum. Genet. 2016, 24, 2–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatsenko, A.N.; Roy, A.; Chen, R.; Ma, L.; Murthy, L.J.; Yan, W.; Lamb, D.J.; Matzuk, M.M. Non-Invasive Genetic Diagnosis of Male Infertility Using Spermatozoal RNA: KLHL10 Mutations in Oligozoospermic Patients Impair Homodimerization. Hum. Mol. Genet. 2006, 15, 3411–3419. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.F.; Toure, A.; Metzler-Guillemain, C.; Mitchell, M.J.; Arnoult, C.; Coutton, C. Genetic Abnormalities Leading to Qualitative Defects of Sperm Morphology or Function. Clin. Genet. 2017, 91, 217–232. [Google Scholar] [CrossRef]
- Dieterich, K.; Rifo, R.S.; Faure, A.K.; Hennebicq, S.; Ben Amar, B.; Zahi, M.; Perrin, J.; Martinez, D.; Sèle, B.; Jouk, P.S.; et al. Homozygous mutation of AURKC yields large-headed polyploid spermatozoa and causes male infertility. Nat. Genet. 2007, 39, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.-W.; Wang, X.; Su, Z.-Y.; Mei, L.-B.; Ji, Z.-Y.; Bao, H.; Li, P. Patients with Multiple Morphological Abnormalities of the Sperm Flagella Harbouring CFAP44 or CFAP43 Mutations Have a Good Pregnancy Outcome Following Intracytoplasmic Sperm Injection. Andrologia 2019, 51, e13151. [Google Scholar] [CrossRef]
- Takasaki, N.; Tachibana, K.; Ogasawara, S.; Matsuzaki, H.; Hagiuda, J.; Ishikawa, H.; Mochida, K.; Inoue, K.; Ogonuki, N.; Ogura, A.; et al. A Heterozygous Mutation of GALNTL5 Affects Male Infertility with Impairment of Sperm Motility. Proc. Natl. Acad. Sci. USA 2014, 111, 1120–1125. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, W.; Jiang, H.; Wu, B.-L.; Primary Ovarian Insufficiency Collaboration. Mutations in HFM1 in Recessive Primary Ovarian Insufficiency. N. Engl. J. Med. 2014, 370, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Pu, D.; Wang, C.; Cao, J.; Shen, Y.; Jiang, H.; Liu, J.; Wu, B.L.; Zhang, W.; Wu, J. Association Analysis between HFM1 Variation and Primary Ovarian Insufficiency in Chinese Women. Clin. Genet. 2016, 89, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Zhe, J.; Chen, S.; Chen, X.; Liu, Y.; Li, Y.; Zhou, X.; Zhang, J. A Novel Heterozygous Splice-Altering Mutation in HFM1 May Be a Cause of Premature Ovarian Insufficiency. J. Ovarian Res. 2019, 12, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuentz, P.; Vanden Meerschaut, F.; Elinati, E.; Nasr-Esfahani, M.H.; Gurgan, T.; Iqbal, N.; Carré-Pigeon, F.; Brugnon, F.; Gitlin, S.A.; Velez de la Calle, J.; et al. Assisted Oocyte Activation Overcomes Fertilization Failure in Globozoospermic Patients Regardless of the DPY19L2 Status. Hum. Reprod. 2013, 28, 1054–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic representation of (A) KLHL10, (B) AURKC, (C) CFAP43, (D) DNAH1, (E) GALNTL5, (F) HFM1, (G) TUBB8, and (H) PATL2 proteins and their published mutations. Published mutations are indicated on top, while mutations identified in this study are shown at the bottom. Since more than 40 mutations have been identified for TUBB8 and DNAH1, identified mutations are not shown on the figure. Details of mutations on relative genes are given in Supplementary Table S4. Protein sequence alignments for different species are shown for missense mutations identified in this study.
Figure 1.
Schematic representation of (A) KLHL10, (B) AURKC, (C) CFAP43, (D) DNAH1, (E) GALNTL5, (F) HFM1, (G) TUBB8, and (H) PATL2 proteins and their published mutations. Published mutations are indicated on top, while mutations identified in this study are shown at the bottom. Since more than 40 mutations have been identified for TUBB8 and DNAH1, identified mutations are not shown on the figure. Details of mutations on relative genes are given in Supplementary Table S4. Protein sequence alignments for different species are shown for missense mutations identified in this study.


{kind=link}
Table 1.
Infertility phenotype and sex of the recruited patients. The number of patients in each group is indicated.
Table 1.
Infertility phenotype and sex of the recruited patients. The number of patients in each group is indicated.
| Sex | Infertility Phenotype | #Patients | |
|---|---|---|---|
| Male | Teratozoopsermia | 7 | |
| Asthenozoospermia | 1 | ||
| Sperm production defect (SPD) | Azoospermia | 30 | |
| Oligozoospermia | 20 | ||
| Mixed phenotype | 21 | ||
| Female | Oocyte maturation defect (OOMD) | 4 | |
| Premature ovarian insufficiency (POI) | 11 | ||
#Patients: Number of patients.
Table 2.
Control samples: phenotype, zygosity, and mutation information. SPD: Sperm production defect, hom: homozygous, het: heterozygous, hemi: hemizygous.
Table 2.
Control samples: phenotype, zygosity, and mutation information. SPD: Sperm production defect, hom: homozygous, het: heterozygous, hemi: hemizygous.
| Sample | Phenotype | Mutated Gene | Zygosity | Defined Mutation |
|---|---|---|---|---|
| C1 | SPD | MAGEB4 | hemi | p.*347Cysext*24 (c.1041A > T) |
| C2 | SPD | TEX15 | hom | p.Tyr710* (c.2130T > G) |
| C3 | Teratozoospermia | DPY19L2 | Het gene del and point mutation | Heterozygous DPY19L2 deletion with p.Arg290His (c.869G > A) |
| C4 | Teratozoospermia | DPY19L2 | Hom | exon 5-exon 6 deletion in DPY19L2 |
| C5 | Teratozoospermia | DPY19L2 | Hom | del DPY19L2 |
Table 3.
Selected genes for infertility panel: 34 genes related to non-syndromic male infertility, 15 genes related to female infertility, [13 genes for premature ovarian insufficiency (POI), and 2 genes for oocyte maturation defect (OOMD)]; 2 shared genes for non-syndromic male and female infertility have been included in the panel.
Table 3.
Selected genes for infertility panel: 34 genes related to non-syndromic male infertility, 15 genes related to female infertility, [13 genes for premature ovarian insufficiency (POI), and 2 genes for oocyte maturation defect (OOMD)]; 2 shared genes for non-syndromic male and female infertility have been included in the panel.
| MALE INFERTILITY | Phenotype | Gene Name | OMIM # | RefSeq | IG/CG |
| Teratozoospermia | AKAP4 | 300185 | NM_003886.2 | CG | |
| AURKC | 603495 | NM_001015878.1 | IG | ||
| BRDT | 602144 | NM_001242806.2 | IG | ||
| CFAP43 | 617558 | NM_025145.6 | IG | ||
| CFAP44 | 617559 | NM_001164496.1 | IG | ||
| DNAH1 | 603332 | NM_015512.4 | IG | ||
| DPY19L2 | 613893 | NM_173812.4 | IG | ||
| MTUS1 ** | 609589 | NM_001001924.2 | CG | ||
| PICK1 | 605926 | NM_001039583.1 | CG | ||
| SEPT12 | 611562 | NM_144605.4 | IG | ||
| SPATA16 | 609856 | NM_031955.5 | IG | ||
| Asthenozoospermia | CATSPER1 | 606389 | NM_053054.3 | IG | |
| GALNTL5 | 615133 | NM_145292.3 | CG | ||
| SLC26A8 | 608480 | NM_001193476.1 | IG | ||
| SPAG17 | 616554 | NM_206996.3 | CG | ||
| Sperm production defect (SPD) | CCDC39 | 613798 | NM_181426.1 | CG * | |
| DNAH6 | 603336 | NM_001370.1 | CG | ||
| HIWI (PIWIL1) | 605571 | NM_004764.4 | CG | ||
| HSF2 | 140581 | NM_004506.3 | CG | ||
| KLHL10 | 608778 | NM_152467.4 | IG | ||
| MAGEB4 | 300153 | NM_002367.3 | CG | ||
| MEIOB | 617670 | NM_001163560.2 | IG | ||
| NANOS1 | 608226 | NM_199461.3 | IG | ||
| NPAS2 | 603347 | NM_002518.3 | IG | ||
| NROB1 (DAX1) | 300473 | NM_000475.4 | CG * | ||
| SOHLH1 | 610224 | NM_001012415.2 | IG | ||
| SPINK2 | 605753 | NM_001271722.1 | IG | ||
| TAF4B | 601689 | NM_001293725.1 | IG | ||
| TEX11 | 300311 | NM_001003811.1 | IG | ||
| TEX14 | 605792 | NM_001201457.1 | IG | ||
| TEX15 | 605795 | NM_031271.3 | IG | ||
| Wt1 | 607102 | NM_024426.3 | CG * | ||
| ZMYND15 | 614312 | NM_001267822.1 | IG | ||
| Total fertilization problem | PLCZ1 | 608075 | NM_033123.3 | IG | |
| Phenotype | Gene Name | OMIM | RefSeq | IG/CG | |
| FEMALE INFERTILITY | Primary Ovarian Insufficiency (POI) | BMP15 | 300247 | NM_005448.2 | IG |
| FIGLA | 608697 | NM_001004311.3 | IG | ||
| FMR1 | 309550 | NM_002024.5 | S | ||
| FSHR | 136435 | NM_000145.3 | CG | ||
| GDF9 | 601918 | NM_005260.5 | IG | ||
| HFM1 | 615684 | NM_001017975.4 | IG | ||
| MCM8 | 608187 | NM_001281521.1 | IG | ||
| MCM9 | 610098 | NM_017696.2 | CG | ||
| MSH4 | 602105 | NM_002440.3 | CG | ||
| NANOS3 | 608229 | NM_001098622.2 | CG | ||
| NOBOX | 610934 | NM_001080413.3 | IG | ||
| PGRMC1 | 300435 | NM_006667.4 | CG | ||
| STAG3 | 608489 | NM_001282717.1 | IG | ||
| Oocyte Maturation Defect (OOMD) | TUBB8 | 616768 | NM_177987.2 | IG | |
| PATL2 | 614661 | NM_001145112.1 | IG | ||
| Gene Name | OMIM | RefSeq | IG/CG | ||
| Male/Female infertility (SPD/POI) | NR5A1 | 184757 | NM_004959.4 | IG | |
| SYCE1 | 611486 | NM_001143764.1 | IG | ||
IG: infertility genes, CG: candidate genes, S: gene to sequence, (*): Candidate genes that were validated since the beginning of this study as strongly or definitely linked to male infertility, (**): genes identified through in-house screening project, not published.OMIM #: OMIM number.
Table 4.
Identified mutations according to phenotype and sex of the patients.
| Sex | Patient Code | Phenotype | Gene Name (Refseq Id) | Coding Effect | Zygosity | Consanguinity | cNomen | pNomen | Allele Frequency (gnomAD) | ART Option |
|---|---|---|---|---|---|---|---|---|---|---|
| M | Pt12 | SPD (Azoospermia) | KLHL10 (NM_001329595.1) | Missense | Het | NP | c.985C > T | p.Arg329Cys | 0.00001202 | Cryo- preservation * |
| Pt41 | Teratozoopermia | AURKC (NM_001015878) | Frameshift | Hom | No | c.144delC | p.Leu49TrpfsTer23 | 0.00008749 | Sperm donation | |
| Pt55 | Teratozoopermia (MMAF) | CFAP43 (NM_025145.5) | Splice site | Hom | NP | c.3541-2A > C | p.? | Not listed | ICSI | |
| Pt65 | AT | DNAH1 (NM_015512.4) | Stop-gain Frameshift Missense | Comp. Het | No | c.9610C > T c.6131del c.9777T > G | p.Arg3204 * p.Phe2044Serfs *13 p.Ser3259Arg | Not listed Not listed 0.000008037 | ICSI | |
| Pt77 | SPD (OAT) | GALNTL5 (NM_145292.3) | Frameshift | Het | NP | c.153dup | p.Val52Serfs*23 | Not listed | * | |
| F | Pt2 | POI | HFM1 (NM_001017975.4) | Stop-gain | Hom | Yes | c.1905T > A | p.Tyr635* | Not listed | Oocyte Donation ** |
| Pt38 | OOMD | TUBB8 (NM_177987.2) | Missense | Hom | Yes | c.922G > A | p.Gly308Ser | Not listed | Oocyte donation | |
| Pt71 | OOMD | PATL2 (NM_001145112.1) | Stop-gain | Hom | Yes | c.478C > T | p.Arg160* | 0.00003245 | Oocyte donation |
M: male, F: female, SPD: sperm production defect, MMAF: multiple morphological abnormalities of the sperm flagella, AT: astheno-teratozoospermia, OAT: oligo-astheno-teratozoospermia, POI: premature ovarian insufficiency, OOMD: oocyte maturation defect, Het: heterozygous, Hom: homozygous, Comp. Het: compound heterozygous, NP: not provided, ART option: options for assisted reproductive techniques, ICSI: intra-cytoplasmic sperm injection, *: further investigation needed, **: if ovarian reserve too low.
Table 5.
Available gene panel publications in the infertility field.
| Infertility Type | #Genes | #Cases (Male/ Female- Phenotype) | HTS Quality | Filtered-Out Frequency | Diagnostic Yield | References | ||
|---|---|---|---|---|---|---|---|---|
| Mean Coverage | Depth of Coverage | % Targetted Bases | ||||||
| Syndromic/ non-syndromic | 284 | 48 idiopathic POI females | 145X | 10X | 99.38% | > 0.1% | 2% | [6] |
| Male infertility (genes based on mouse model) | 175 | 33 idiopathic NOA | 300X | NP | NP | > 5% | 6.3% | [7] |
| Syndromic/ non-syndromic | 75 | 17 female, 6 male with different infertility phenotype | 180 | 20X | 98% | > 5% | 8.7% | [8] |
| Syndromic/ non-syndromic | 9 | 241 idiopathic male infertility cases | 351X | 10X | 93.5% | > 1% | 0.4% | [9] |
| Syndromic/ non-syndromic | 15 | 25 idiopathic male infertility with SPD | ND | ND | ND | ND | 12% | [10] |
| Syndromic/ non-syndromic | 110 | 22 male infertility cases | 286–539X * | 10X * | 91.3–98% * | ND | 25% | [11] |
| Non-syndromic | 51 | 15 female, 79 male with different infertility phenotype | 457X | 30X | 99.8% | >1% | 8.5% | Present study |
HTS: high-throughput sequencing. POI: primary ovarian insufficiency, NOA: non-obstructive azoospermia. Mean coverage: average number of reads that align to known reference bases. Depth of coverage: the number of unique reads that include a given nucleotide in the reconstructed sequence, also known as on-target read depth. % targeted bases: percentage of target bases that are successfully sequenced with a given depth of coverage. SPD: sperm production failure, oligozoospermia and non-obstructive azoospermia. NP: not provided. (*): quality data is given only for 5 positive cases.#: number.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Okutman, O.; Tarabeux, J.; Muller, J.; Viville, S. Evaluation of a Custom Design Gene Panel as a Diagnostic Tool for Human Non-Syndromic Infertility. Genes 2021, 12, 410. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030410
AMA Style
Okutman O, Tarabeux J, Muller J, Viville S. Evaluation of a Custom Design Gene Panel as a Diagnostic Tool for Human Non-Syndromic Infertility. Genes. 2021; 12(3):410. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030410
Chicago/Turabian StyleOkutman, Ozlem, Julien Tarabeux, Jean Muller, and Stéphane Viville. 2021. "Evaluation of a Custom Design Gene Panel as a Diagnostic Tool for Human Non-Syndromic Infertility" Genes 12, no. 3: 410. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12030410
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.