
8p23.2-pter Microdeletions: Seven New Cases Narrowing the Candidate Region and Review of the Literature

 , , , , , , ,
, , , , , , ,  , , and
, , and
Abstract
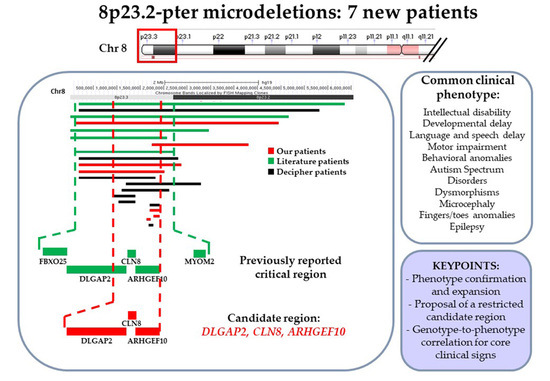
:
1. Introduction
2. Materials and Methods
3. Results
Cytogenomic and Phenotypic Characterization of Seven Novel 8p23-Pter Microdeletion Patients: Cross-Comparison to Previously Reported Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lo Bianco, M.; Vecchio, D.; Timpanaro, T.A.; Arena, A.; Macchiaiolo, M.; Bartuli, A.; Sciuto, L.; Presti, S.; Sciuto, S.; Sapuppo, A.; et al. Deciphering the Invdupdel(8p) Genotype-Phenotype Correlation: Our Opinion. Brain Sci. 2020, 10, 451. [Google Scholar] [CrossRef] [PubMed]
- Hollox, E.J.; Barber, J.C.; Brookes, A.J.; Armour, J.A. Defensins and the dynamic genome: What we can learn from structural variation at human chromosome band 8p23.1. Genome Res. 2008, 18, 1686–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, L.R.; Lee, J.Y.; Rector, L.; Kaminsky, E.B.; Brothman, A.R.; Martin, C.L.; South, S.T. U-type exchange is the most frequent mechanism for inverted duplication with terminal deletion rearrangements. J. Med. Genet. 2009, 46, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Fiedler, S.; Stegner, A.; Graf, W.D. Genomic profile of copy number variants on the short arm of human chromosome 8. Eur. J. Hum. Genet. 2010, 18, 1114–1120. [Google Scholar] [CrossRef]
- Ballarati, L.; Cereda, A.; Caselli, R.; Selicorni, A.; Recalcati, M.P.; Maitz, S.; Finelli, P.; Larizza, L.; Giardino, D. Genotype-phenotype correlations in a new case of 8p23.1 deletion and review of the literature. Eur. J. Med. Genet. 2011, 54, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Inagaki, H.; Miyai, S.; Suzuki, F.; Naru, Y.; Shinkai, Y.; Kato, A.; Kanyama, K.; Mizuno, S.; Muramatsu, Y.; et al. The involvement of U-type dicentric chromosomes in the formation of terminal deletions with or without adjacent inverted duplications. Hum. Genet. 2020, 139, 1417–1427. [Google Scholar] [CrossRef]
- Shi, S.; Lin, S.; Chen, B.; Zhou, Y. Isolated chromosome 8p23.2pter deletion: Novel evidence for developmental delay, intellectual disability, microcephaly and neurobehavioral disorders. Mol. Med. Rep. 2017, 16, 6837–6845. [Google Scholar] [CrossRef] [Green Version]
- DGV. Database of Genomic Variants. Available online: http://dgv.tcag.ca/dgv/app/home (accessed on 6 February 2021).
- Burnside, R.D.; Pappas, J.G.; Sacharow, S.; Applegate, C.; Hamosh, A.; Gadi, I.K.; Jaswaney, V.; Keitges, E.; Phillips, K.K.; Potluri, V.R.; et al. Three cases of isolated terminal deletion of chromosome 8p without heart defects presenting with a mild phenotype. Am. J. Med. Genet. Part A 2013, 161, 822–828. [Google Scholar] [CrossRef]
- Chien, W.H.; Gau, S.S.; Wu, Y.Y.; Huang, Y.S.; Fang, J.S.; Chen, Y.J.; Soong, W.T.; Chiu, Y.N.; Chen, C.H. Identification and molecular characterization of two novel chromosomal deletions associated with autism. Clin. Genet. 2010, 78, 449–456. [Google Scholar] [CrossRef]
- Wu, Y.; Ji, T.; Wang, J.; Xiao, J.; Wang, H.; Li, J.; Gao, Z.; Yang, Y.; Cai, B.; Wang, L.; et al. Submicroscopic subtelomeric aberrations in Chinese patients with unexplained developmental delay/mental retardation. BMC Med. Genet. 2010, 11, 72. [Google Scholar] [CrossRef] [Green Version]
- Kraus, D.M.; Elliott, G.S.; Chute, H.; Horan, T.; Pfenninger, K.H.; Sanford, S.D.; Foster, S.; Scully, S.; Welcher, A.A.; Holers, V.M. CSMD1 is a novel multiple domain complement-regulatory protein highly expressed in the central nervous system and epithelial tissues. J. Immunol. 2006, 176, 4419–4430. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, S.; Kawanai, T.; Hara, T.; Yamamoto, A.; Chaya, T.; Tokuhara, Y.; Tsuji, C.; Sakai, M.; Tachibana, T.; Inagaki, S. ARHGEF10 directs the localization of Rab8 to Rab6-positive executive vesicles. J. Cell Sci. 2016, 129, 3620–3634. [Google Scholar] [CrossRef] [Green Version]
- Shibata, S.; Teshima, Y.; Niimi, K.; Inagaki, S. Involvement of ARHGEF10, GEF for RhoA, in Rab6/Rab8-mediating membrane traffic. Small Gtpases 2019, 10, 169–177. [Google Scholar] [CrossRef]
- Rasmussen, A.H.; Rasmussen, H.B.; Silahtaroglu, A. The DLGAP family: Neuronal expression, function and role in brain disorders. Mol. Brain 2017, 10, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OMIM. Online Mendelian Inheritance in Man. Available online: https://www.omim.org/ (accessed on 6 February 2021).
- Lichter, P.; Cremer, T.; Borden, J.; Manuelidis, L.; Ward, D.C. Delineation of individual human chromosomes in metaphase and interphase cells by in situ suppression hybridization using recombinant DNA libraries. Hum. Genet. 1988, 80, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catusi, I.; Recalcati, M.P.; Bestetti, I.; Garzo, M.; Valtorta, C.; Alfonsi, M.; Alghisi, A.; Cappellani, S.; Casalone, R.; Caselli, R.; et al. Testing single/combined clinical categories on 5110 Italian patients with developmental phenotypes to improve array-based detection rate. Mol. Genet. Genom. Med. 2020, 8, e1056. [Google Scholar] [CrossRef] [Green Version]
- UCSC Genome Browser. Available online: https://genome.ucsc.edu/ (accessed on 6 February 2021).
- GnomAD. Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org/ (accessed on 6 February 2021).
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Di Ronza, A.; Bajaj, L.; Sharma, J.; Sanagasetti, D.; Lotfi, P.; Adamski, C.J.; Collette, J.; Palmieri, M.; Amawi, A.; Popp, L.; et al. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat. Cell Biol. 2018, 20, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.R.; Manfiolli, A.O.; Vieira, N.A.; Medeiros, A.C.; Coelho, P.O.; Santiago Guimaraes, D.; Schechtman, D.; Gomes, M.D. FBXO25 regulates MAPK signaling pathway through inhibition of ERK1/2 phosphorylation. Arch. Biochem. Biophys. 2017, 621, 38–45. [Google Scholar] [CrossRef]
- Watanabe, K.; Yokota, K.; Yoshida, K.; Matsumoto, A.; Iwamoto, S. Kbtbd11 contributes to adipocyte homeostasis through the activation of upstream stimulatory factor 1. Heliyon 2019, 5, e02777. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, H.; Zhou, W.; Zhang, Z.; Yang, Z.; Lu, Y.; Lu, B.; Wang, X.; Ding, Q.; Hu, R. Molecular cloning of a novel nuclear factor, TDRP1, in spermatogenic cells of testis and its relationship with spermatogenesis. Biochem. Biophys. Res. Commun. 2010, 394, 29–35. [Google Scholar] [CrossRef]
- Van der Ven, P.F.; Speel, E.J.; Albrechts, J.C.; Ramaekers, F.C.; Hopman, A.H.; Furst, D.O. Assignment of the human gene for endosarcomeric cytoskeletal M-protein (MYOM2) to 8p23.3. Genomics 1999, 55, 253–255. [Google Scholar] [CrossRef] [Green Version]
- GTEx. Genotype-Tissue Expression. Available online: https://www.gtexportal.org/home/ (accessed on 17 March 2021).
- DECIPHER. DatabasE of GenomiC VarIation and Phenotype in Humans Using Ensembl Resources. Available online: https://decipher.sanger.ac.uk/ (accessed on 6 February 2021).
- Li, L.; Huang, L.; Luo, Y.; Huang, X.; Lin, S.; Fang, Q. Differing Microdeletion Sizes and Breakpoints in Chromosome 7q11.23 in Williams-Beuren Syndrome Detected by Chromosomal Microarray Analysis. Mol. Syndr. 2016, 6, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Bonati, M.T.; Castronovo, C.; Sironi, A.; Zimbalatti, D.; Bestetti, I.; Crippa, M.; Novelli, A.; Loddo, S.; Dentici, M.L.; Taylor, J.; et al. 9q34.3 microduplications lead to neurodevelopmental disorders through EHMT1 overexpression. Neurogenetics 2019, 20, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Venturin, M.; Gervasini, C.; Orzan, F.; Bentivegna, A.; Corrado, L.; Colapietro, P.; Friso, A.; Tenconi, R.; Upadhyaya, M.; Larizza, L.; et al. Evidence for non-homologous end joining and non-allelic homologous recombination in atypical NF1 microdeletions. Hum. Genet. 2004, 115, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaya, T.; Shibata, S.; Tokuhara, Y.; Yamaguchi, W.; Matsumoto, H.; Kawahara, I.; Kogo, M.; Ohoka, Y.; Inagaki, S. Identification of a negative regulatory region for the exchange activity and characterization of T332I mutant of Rho guanine nucleotide exchange factor 10 (ARHGEF10). J. Biol. Chem. 2011, 286, 29511–29520. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, K.; De Jonghe, P.; Van de Putte, T.; Nelis, E.; Zwijsen, A.; Verpoorten, N.; De Vriendt, E.; Jacobs, A.; Van Gerwen, V.; Francis, A.; et al. Slowed conduction and thin myelination of peripheral nerves associated with mutant rho Guanine-nucleotide exchange factor 10. Am. J. Hum. Genet. 2003, 73, 926–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungerius, B.J.; Hoogendoorn, M.L.; Bakker, S.C.; Van’t Slot, R.; Bardoel, A.F.; Ophoff, R.A.; Wijmenga, C.; Kahn, R.S.; Sinke, R.J. An association screen of myelin-related genes implicates the chromosome 22q11 PIK4CA gene in schizophrenia. Mol. Psychiatry 2008, 13, 1060–1068. [Google Scholar] [CrossRef] [Green Version]
- Tabares-Seisdedos, R.; Rubenstein, J.L. Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: Implications for schizophrenia, autism and cancer. Mol. Psychiatry 2009, 14, 563–589. [Google Scholar] [CrossRef]
- Noto, Y.; Shiga, K.; Tsuji, Y.; Mizuta, I.; Higuchi, Y.; Hashiguchi, A.; Takashima, H.; Nakagawa, M.; Mizuno, T. Nerve ultrasound depicts peripheral nerve enlargement in patients with genetically distinct Charcot-Marie-Tooth disease. J. Neurol. Neurosurg. Psychiatry 2015, 86, 378–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boora, G.K.; Kulkarni, A.A.; Kanwar, R.; Beyerlein, P.; Qin, R.; Banck, M.S.; Ruddy, K.J.; Pleticha, J.; Lynch, C.A.; Behrens, R.J.; et al. Association of the Charcot-Marie-Tooth disease gene ARHGEF10 with paclitaxel induced peripheral neuropathy in NCCTG N08CA (Alliance). J. Neurol. Sci. 2015, 357, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SFARI Gene Database. Available online: https://gene.sfari.org/ (accessed on 6 February 2021).
- Deneault, E.; White, S.H.; Rodrigues, D.C.; Ross, P.J.; Faheem, M.; Zaslavsky, K.; Wang, Z.; Alexandrova, R.; Pellecchia, G.; Wei, W.; et al. Complete Disruption of Autism-Susceptibility Genes by Gene Editing Predominantly Reduces Functional Connectivity of Isogenic Human Neurons. Stem Cell Rep. 2019, 12, 427–429. [Google Scholar] [CrossRef] [Green Version]
- Gazzellone, M.J.; Zarrei, M.; Burton, C.L.; Walker, S.; Uddin, M.; Shaheen, S.M.; Coste, J.; Rajendram, R.; Schachter, R.J.; Colasanto, M.; et al. Uncovering obsessive-compulsive disorder risk genes in a pediatric cohort by high-resolution analysis of copy number variation. J. Neurodev. Disord. 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Kimura, H.; Wang, C.; Ishizuka, K.; Kushima, I.; Arioka, Y.; Yoshimi, A.; Nakamura, Y.; Shiino, T.; Oya-Ito, T.; et al. Resequencing and Association Analysis of Six PSD-95-Related Genes as Possible Susceptibility Genes for Schizophrenia and Autism Spectrum Disorders. Sci. Rep. 2016, 6, 27491. [Google Scholar] [CrossRef]
- Li, Y.; Wang, K.; Zhang, P.; Huang, J.; An, H.; Wang, N.; De Yang, F.; Wang, Z.; Tan, S.; Chen, S.; et al. Quantitative DNA Methylation Analysis of DLGAP2 Gene using Pyrosequencing in Schizophrenia with Tardive Dyskinesia: A Linear Mixed Model Approach. Sci. Rep. 2018, 8, 17466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodbury-Smith, M.; Zarrei, M.; Wei, J.; Thiruvahindrapuram, B.; O’Connor, I.; Paterson, A.D.; Yuen, R.K.C.; Dastan, J.; Stavropoulos, D.J.; Howe, J.L.; et al. Segregating patterns of copy number variations in extended autism spectrum disorder (ASD) pedigrees. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2020, 183, 268–276. [Google Scholar] [CrossRef]
- Wu, K.; Hanna, G.L.; Easter, P.; Kennedy, J.L.; Rosenberg, D.R.; Arnold, P.D. Glutamate system genes and brain volume alterations in pediatric obsessive-compulsive disorder: A preliminary study. Psychiatry Res. 2013, 211, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Curran, S.; Ahn, J.W.; Grayton, H.; Collier, D.A.; Ogilvie, C.M. NRXN1 deletions identified by array comparative genome hybridisation in a clinical case series—Further understanding of the relevance of NRXN1 to neurodevelopmental disorders. J. Mol. Psychiatry 2013, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Xiao, X.; Zhang, Z.; Li, M. Genetic insights and neurobiological implications from NRXN1 in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1400–1414. [Google Scholar] [CrossRef] [PubMed]
- Castronovo, P.; Baccarin, M.; Ricciardello, A.; Picinelli, C.; Tomaiuolo, P.; Cucinotta, F.; Frittoli, M.; Lintas, C.; Sacco, R.; Persico, A.M. Phenotypic spectrum of NRXN1 mono- and bi-allelic deficiency: A systematic review. Clin. Genet. 2020, 97, 125–137. [Google Scholar] [CrossRef] [Green Version]
- LaConte, L.E.W.; Chavan, V.; Elias, A.F.; Hudson, C.; Schwanke, C.; Styren, K.; Shoof, J.; Kok, F.; Srivastava, S.; Mukherjee, K. Two microcephaly-associated novel missense mutations in CASK specifically disrupt the CASK-neurexin interaction. Hum. Genet. 2018, 137, 231–246. [Google Scholar] [CrossRef]
- Pirozzi, F.; Nelson, B.; Mirzaa, G. From microcephaly to megalencephaly: Determinants of brain size. Dialogues Clin. Neurosci. 2018, 20, 267–282. [Google Scholar] [CrossRef] [Green Version]
- Jean, F.; Stuart, A.; Tarailo-Graovac, M. Dissecting the Genetic and Etiological Causes of Primary Microcephaly. Front. Neurol. 2020, 11, 570830. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Morrison, C.G.; Gergely, F. Molecular causes of primary microcephaly and related diseases: A report from the UNIA Workshop. Chromosoma 2020, 129, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Jiang-Xie, L.F.; Liao, H.M.; Chen, C.H.; Chen, Y.T.; Ho, S.Y.; Lu, D.H.; Lee, L.J.; Liou, H.H.; Fu, W.M.; Gau, S.S. Autism-associated gene Dlgap2 mutant mice demonstrate exacerbated aggressive behaviors and orbitofrontal cortex deficits. Mol. Autism 2014, 5, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.H.; Liao, H.M.; Chen, C.H.; Tu, H.J.; Liou, H.C.; Gau, S.S.; Fu, W.M. Impairment of social behaviors in Arhgef10 knockout mice. Mol. Autism 2018, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horner, A.E.; Norris, R.H.; McLaren-Jones, R.; Alexander, L.; Komiyama, N.H.; Grant, S.G.N.; Nithianantharajah, J.; Kopanitsa, M.V. Learning and reaction times in mouse touchscreen tests are differentially impacted by mutations in genes encoding postsynaptic interacting proteins SYNGAP1, NLGN3, DLGAP1, DLGAP2 and SHANK2. Genes Brain Behav. 2020, e12723. [Google Scholar] [CrossRef]
- STRING Database. Available online: https://string-db.org/ (accessed on 6 February 2021).
- Verpelli, C.; Schmeisser, M.J.; Sala, C.; Boeckers, T.M. Scaffold proteins at the postsynaptic density. Adv. Exp. Med. Biol. 2012, 970, 29–61. [Google Scholar] [CrossRef]
- Sala, C.; Vicidomini, C.; Bigi, I.; Mossa, A.; Verpelli, C. Shank synaptic scaffold proteins: Keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 2015, 135, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.; Fananas, L.; Parellada, M.; Krebs, M.O.; Rouleau, G.A.; Fatjo-Vilas, M. Genetic variability in scaffolding proteins and risk for schizophrenia and autism-spectrum disorders: A systematic review. J. Psychiatry Neurosci. 2018, 43, 223–244. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.C.; Ferri, S.L.; Grewal, M.; Smernoff, Z.; Bucan, M.; Weiner, J.A.; Abel, T.; Brodkin, E.S. The Role of Synaptic Cell Adhesion Molecules and Associated Scaffolding Proteins in Social Affiliative Behaviors. Biol. Psychiatry 2020, 88, 442–451. [Google Scholar] [CrossRef]
- Kaizuka, T.; Takumi, T. Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J. Biochem. 2018, 163, 447–455. [Google Scholar] [CrossRef]
- Park, E.; Na, M.; Choi, J.; Kim, S.; Lee, J.R.; Yoon, J.; Park, D.; Sheng, M.; Kim, E. The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the beta PIX guanine nucleotide exchange factor for Rac1 and Cdc42. J. Biol. Chem. 2003, 278, 19220–19229. [Google Scholar] [CrossRef] [Green Version]
- Sarowar, T.; Grabrucker, A.M. Actin-Dependent Alterations of Dendritic Spine Morphology in Shankopathies. Neural Plast. 2016, 2016, 8051861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeidan-Chulia, F.; Salmina, A.B.; Malinovskaya, N.A.; Noda, M.; Verkhratsky, A.; Moreira, J.C. The glial perspective of autism spectrum disorders. Neurosci. Biobehav. Rev. 2014, 38, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Bakos, J.; Bacova, Z.; Grant, S.G.; Castejon, A.M.; Ostatnikova, D. Are Molecules Involved in Neuritogenesis and Axon Guidance Related to Autism Pathogenesis? Neuromol. Med. 2015, 17, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Reichova, A.; Zatkova, M.; Bacova, Z.; Bakos, J. Abnormalities in interactions of Rho GTPases with scaffolding proteins contribute to neurodevelopmental disorders. J. Neurosci. Res. 2018, 96, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Cukier, H.N.; Dueker, N.D.; Slifer, S.H.; Lee, J.M.; Whitehead, P.L.; Lalanne, E.; Leyva, N.; Konidari, I.; Gentry, R.C.; Hulme, W.F.; et al. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol. Autism 2014, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Fu, X.; Tang, Z.; Li, C.; Xu, Y.; Zhang, F.; Zhou, D.; Zhu, C. Altered expression of the CSMD1 gene in the peripheral blood of schizophrenia patients. BMC Psychiatry 2019, 19, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crippa, M.; Malatesta, P.; Bonati, M.T.; Trapasso, F.; Fortunato, F.; Annesi, G.; Larizza, L.; Labate, A.; Finelli, P.; Perrotti, N.; et al. A familial t(4;8) translocation segregates with epilepsy and migraine with aura. Ann. Clin. Transl. Neurol. 2020, 7, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Harich, B.; Klein, M.; Ockeloen, C.W.; van der Voet, M.; Schimmel-Naber, M.; de Leeuw, N.; Schenck, A.; Franke, B. From man to fly—Convergent evidence links FBXO25 to ADHD and comorbid psychiatric phenotypes. J. Child Psychol. Psychiatry 2020, 61, 545–555. [Google Scholar] [CrossRef] [PubMed]
- YUE Lab Computational and Functional Genomics/Epigenomics. Available online: http://3dgenome.fsm.northwestern.edu/view.php (accessed on 27 February 2021).
- Luedi, P.P.; Dietrich, F.S.; Weidman, J.R.; Bosko, J.M.; Jirtle, R.L.; Hartemink, A.J. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007, 17, 1723–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Present Work Patients | Literature Patients | ||||||||||||
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Shi et al., 2017 [7] | Burnside et al., 2013 pt1 [9] | Burnside et al., 2013 pt2 [9] | Chien et al., 2010 [10] | Wu et al., 2010 [11] | Total Number of Patients | |
| Age at diagnosis | 13 | 4 | 16 | 2 | 13 | 38 | 12 | 5 | 2 | 4 | 12 | 1 | |
| Sex | M | M | M | M | M | M | F | M | F | M | M | F | |
| Coordinates (GRCh37/hg19) | chr8:176475-1892812 | chr8:191530-2308985 | chr8:221611-4767606 | chr8:1531691-1603483 | chr8:1731454-3846288 | chr8:1731454-1853939 | chr8:1774139-1857463 | chr8:158048-6004205 | chr8:1- 3623904 | chr8:1-4832134 | chr8:1-2400000 | chr8:21000-2067000 | |
| Size | 1.72 Mb | 2.12 Mb | 4.55 Mb | 71.79 kb | 2.1 Mb | 123 kb | 83.33 kb | 6.0 Mb | 3.6 Mb | 4.8 Mb | 2.4 Mb | 2.1 Mb | |
| Inheritance | Maternal | de novo | de novo | paternal (father with learning disability and stuttering) | inherited (present in the unaffected brother) | de novo | de novo | de novo | |||||
| Coding gene(s) content | ORF4F21, ZNF596, FBXO25, TDRP, ERICH1, DLGAP2, CLN8, ARHGEF10 | ORF4F21, ZNF596, FBXO25, TDRP, ERICH1, DLGAP2, CLN8, ARHGEF10, KBTBD11, MYOM2 | FBXO25, TDRP, ERICH1, DLGAP2, CLN8, ARHGEF10, KBTBD11, MYOM2, CSMD1 | DLGAP2 | CLN8, ARHGEF10,KBTBD11, MYOM2, CSMD1 | CLN8, ARHGEF10 | ARHGEF10 | ORF4F21, ZNF596, FBXO25, TDRP, ERICH1, DLGAP2, CLN8, ARHGEF10, KBTBD11, MYOM2, CSMD1 | ORF4F21, ZNF596, FBXO25, TDRP, ERICH1, DLGAP2, CLN8, ARHGEF10, KBTBD11, MYOM2, CSMD1 | ORF4F21, ZNF596, FBXO25, TDRP, ERICH1, DLGAP2, CLN8, ARHGEF10, KBTBD11, MYOM2, CSMD1 | ORF4F21, ZNF596, FBXO25, TDRP, ERICH1, DLGAP2, CLN8, ARHGEF10, KBTBD11, MYOM2 | ORF4F21, ZNF596, FBXO25, TDRP, ERICH1, DLGAP2, CLN8, ARHGEF10, KBTBD11, MYOM2 | |
| ID | mild ID | mild ID | mild DD at 3y. IQ = 91 at 13y | + | + | + | 6/12 | ||||||
| DD | + | - | - | + | + | + | + | + | + | + | + | 9/12 | |
| Language and speech delay | + | + | + | + | + | - | + | 6/12 | |||||
| Motor impairment | Motor instability, dyspraxia | Motor instability, balance/coordination problems, limb hypotonia | − | Fine and gross coordination problems | Balanced/coordination problems | Balanced/coordination problems | 5/12 | ||||||
| ASD | + | + | + | − | + | − | − | + | 5/12 | ||||
| Behavioral abnormalities/ ADHD | Hyperkinetic behavior, irritability | Hyperkinetic behavior, ADHD | Hyperactivity, aggressiveness, impulsiveness, stereotype behavior | Hyperkinetic behavior, hyperactivity, attention deficit, aggressiveness | ADHD | − | − | ADHD | 6/12 | ||||
| Microcephaly | + | − | + | Microcephaly, brachycephaly | + | − | + | 5/12 | |||||
| Fingers/ toes anomalies | Shortened 4th toe of left foot, bilateral broad 1st toe, finger hyperlaxity | Bilateral clinodactyly of the 5th finger | Nail hypoplasia | 3/12 | |||||||||
| Dysmophisms | + (not specified) | − | Bitemporal narrowing, hypotelorism, prograthism, premature graying of hair | − | Low-set ears, narrow palpebral fissures, thin vermillion of the upper lip | − | low-set ears with bilateral prominence of the antitragus, epicanthal folds and a long philtrum | Mild maxillary, flattening medial epicanthal folds with upslanting palpebral fissures, preauricular pit and bilateral prominence of the antitragus | Palpable metopic ridge, upslanting palpebral, fissures, nystagmus, aniridia, low-set and posteriorly rotated ears with overfolded helices, small upturned nose, and downturned corners of the mouth | − | Hypertelorism, long philtrum, malformed ears | 7/12 | |
| Epilepsy | − | − | + | + | + | 3/12 | |||||||
| Other | Myopia, strabismus, skeletal anomalies | Xerosis cutis, skin anomalies, mild hepatomegaly, cerebral parenchymal anomalies | Short stature, eutrophic skin, | Hippocampal anomalies, heart anomalies | Perinatal distress, jaundice, flat feet, frequent respiratory infections | Scoliosis | Emmetropia, scoliosis, horshoe kidney, hypereosinophily, teeth cavities, hypotyroidism | Coartaction of the aorta | |||||
| Other CNVs (GRCh37/ hg19)/ additional genetic data | 5p15.2 microdeletion coordinates 12260192-12849583 (maternal) | 45,XY,-8[8]/ 46,XY,r(8)(p23.2q24.3)[42] | Absence of mutations in CLN8 coding sequence on the other allele | 6q26 deletion coordinates 163274414-163399757 | |||||||||
| Human Gene Symbol/OMIM Entry | Gene Full Name | Main Biological Activity | Reference | OMIM Disease Association | pLI Score |
|---|---|---|---|---|---|
| OR4F21 | Olfactory receptor family 4 subfamily F member 21 | Olfactory receptor | np | np | np |
| ZNF596 | Zinc finger protein 596 | Transcriptional regulation | np | np | 0 |
| FBXO25 /*609098 | F-box protein 25 | Substrate-recognition component of the SCF (SKP1-CUL1-F- box protein)-type E3 ubiquitin ligase complex. | [24] | np | 0 |
| TDRP | Testis development related protein | Contributes to normal sperm motility | [26] | np | 0 |
| ERICH1 | Glutamate-rich 1 | Unknown | np | np | 0 |
| DLGAP2 /*605438 | Discs, large (Drosophila) homolog-associated protein 2 | Role in the molecular organization of synapses and neuronal cell signaling. Adapter protein linking ion channel to the sub-synaptic cytoskeleton | [16] | np | 1 |
| CLN8 /*607837 | Ceroid-lipofuscinosis, neuronal 8 (epilepsy, progressive with mental retardation) | Lipid synthesis, transport, or sensing | [23] | Ceroid lipofuscinosis 8, AR (# 600143) Ceroid lipofuscinosis, neuronal, 8, Northern epilepsy variant, AR (# 610003) | 0 |
| ARHGEF10 /*608136 | Rho guanine nucleotide exchange factor (GEF) 10 | Membrane trafficking and neural morphogenesis | [14,15] | ?Slowed nerve conduction velocity, AD (# 608236) | 0 |
| KBTBD11 /*618794 | Kelch repeat and BTB (POZ) domain containing 11 | Protein degradation, thereby affecting differentiation, homeostasis, metabolism, cell signaling, and oxidative stress response | [25] | np | 0.06 |
| MYOM2 /*603509 | Myomesin 2 | Major component of the vertebrate myofibrillar M band | [27] | np | 0 |
| CSMD1 /*608397 | CUB and Sushi multiple domains 1 | Role in central nervous system development | [12] | np | 1 |
| DECIPHER Patients | TOTAL NUMBER OF PATIENTS | |||||||||
| 253667 | 277848 | 288405 | 294526 | 295087 | 338097 | 337882 | 351690 | 391615 | ||
| Age at diagnosis | 8 | 5 | nd | nd | 10 | nd | <1 | 13 | nd | |
| Sex | F | M | nd | M | F | M | M | M | M | |
| Coordinates (GRCh37/hg19) | chr8:1588355-1656051 | chr8:176814-1734772 | chr8:738801-1369613 | chr8:1111157-2908829 | chr8:191560-5353580 | chr8:974820-2067679 | chr8:1731454-1853939 | chr8:194617-2170951 | chr8:907664-1368668 | |
| Size | 68 kb | 1.56 Mb | 633 kb | 1.80 Mb | 5.16 Mb | 1.09 Mb | 122 kb | 1.98 Mb | 463 kb | |
| Inheritance | inherited (parent with the same phenotype) | maternal | de novo | maternal | ||||||
| Coding gene(s) content | DLGAP2 | ZNF596, FBXO25, TDRP, ERICH1, DLGAP2, CLN8 | DLGAP2 | DLGAP2, CLN8, ARHGEF10, KBTBD11, MYOM2, CSMD1 | ZNF596, FBXO25, TDRP, ERICH1, DLGAP2, CLN8, ARHGEF10, KBTBD11, MYOM2, CSMD1 | DLGAP2, CLN8, ARHGEF10, KBTBD11, MYOM2 | CLN8, ARHGEF10 | ZNF596, FBXO25, TDRP, ERICH1, DLGAP2, CLN8, ARHGEF10, KBTBD11, MYOM2 | DLGAP2 | |
| ID | + | Learning disability | + | + | + | + | + | 13/21 | ||
| DD | 9/21 | |||||||||
| Language and speech delay | + | 7/21 | ||||||||
| Motor impairment | 5/21 | |||||||||
| ASD | + | 6/21 | ||||||||
| Behavioral problems/ADHD | + | ADHD | 8/21 | |||||||
| Microcephaly | + | 6/21 | ||||||||
| Fingers/toes anomalies | Clinodactyly of the 5th finger | Short phalanx of fingers, sandal gap | Short middle phalanx finger | 6/21 | ||||||
| Dysmophisms | Epicanthus | 8/21 | ||||||||
| Epilepsy | + | 4/21 | ||||||||
| Other | Spotty hyperpigmentation | Coarctation of the aorta | ||||||||
| Other CNVs (GRCh37/hg19)/ additional genetic data | 4q26 duplication coordinates 119606253-120003254 | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catusi, I.; Garzo, M.; Capra, A.P.; Briuglia, S.; Baldo, C.; Canevini, M.P.; Cantone, R.; Elia, F.; Forzano, F.; Galesi, O.; et al. 8p23.2-pter Microdeletions: Seven New Cases Narrowing the Candidate Region and Review of the Literature. Genes 2021, 12, 652. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12050652
Catusi I, Garzo M, Capra AP, Briuglia S, Baldo C, Canevini MP, Cantone R, Elia F, Forzano F, Galesi O, et al. 8p23.2-pter Microdeletions: Seven New Cases Narrowing the Candidate Region and Review of the Literature. Genes. 2021; 12(5):652. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12050652
Chicago/Turabian StyleCatusi, Ilaria, Maria Garzo, Anna Paola Capra, Silvana Briuglia, Chiara Baldo, Maria Paola Canevini, Rachele Cantone, Flaviana Elia, Francesca Forzano, Ornella Galesi, and et al. 2021. "8p23.2-pter Microdeletions: Seven New Cases Narrowing the Candidate Region and Review of the Literature" Genes 12, no. 5: 652. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12050652