
Prenatal Ultrasound Suspicion of Cystic Fibrosis in a Multiethnic Population: Is Extensive CFTR Genotyping Needed?

Abstract
:1. Introduction
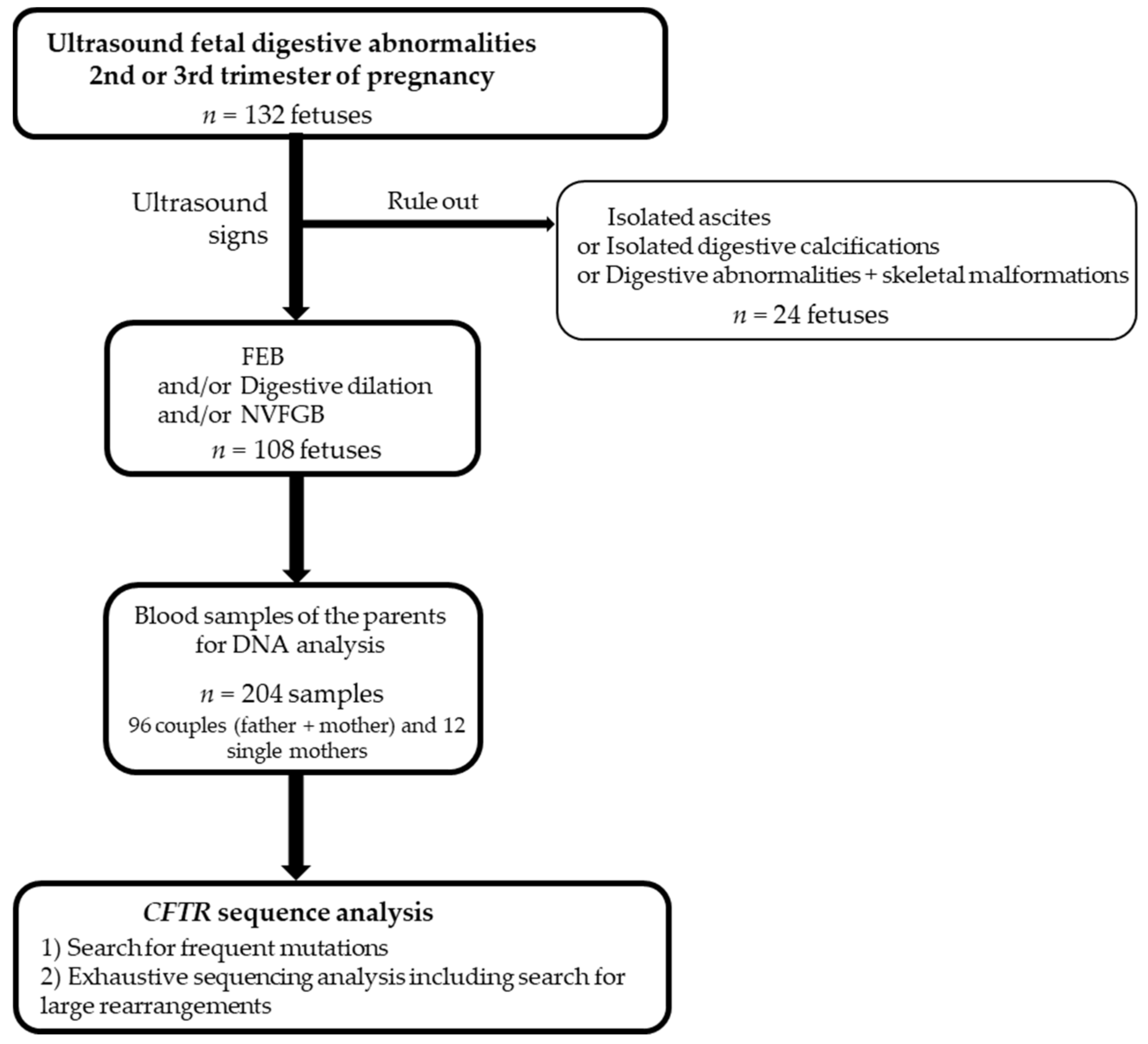
2. Materials and Methods
2.1. Subjects
2.2. Molecular Analysis
3. Results
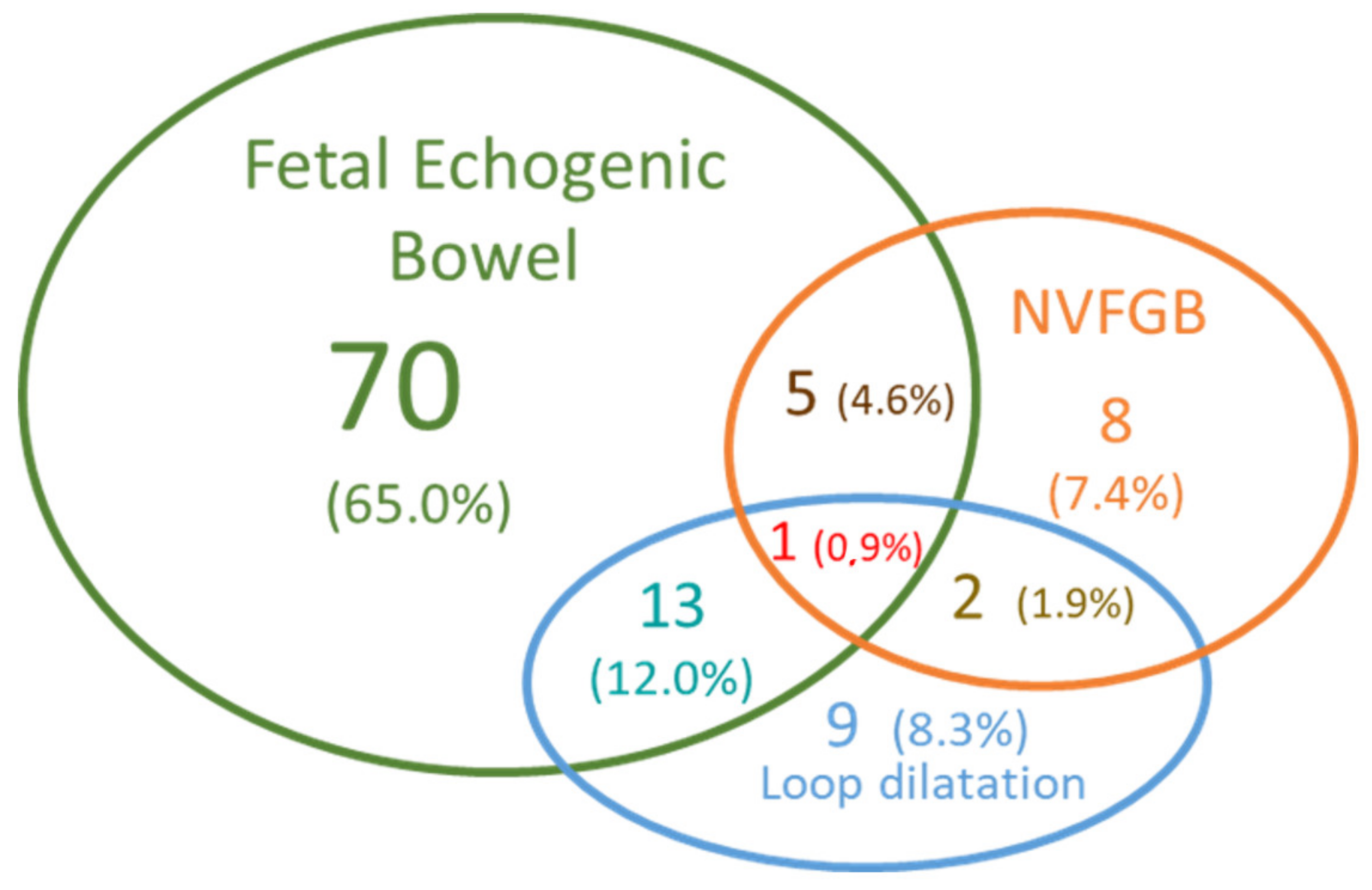
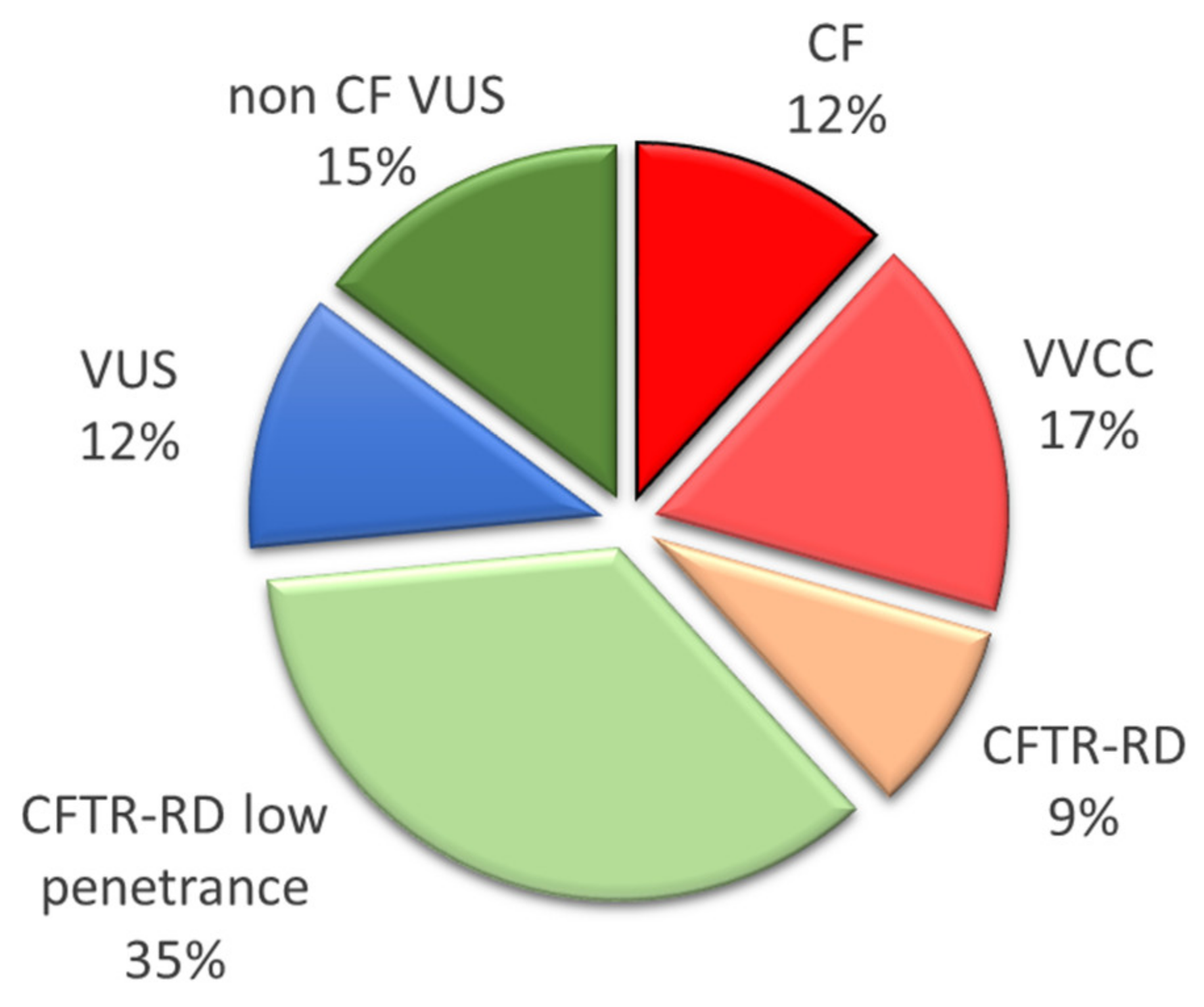
3.1. Fetuses’ Phenotypes and Parental CFTR Genotypes
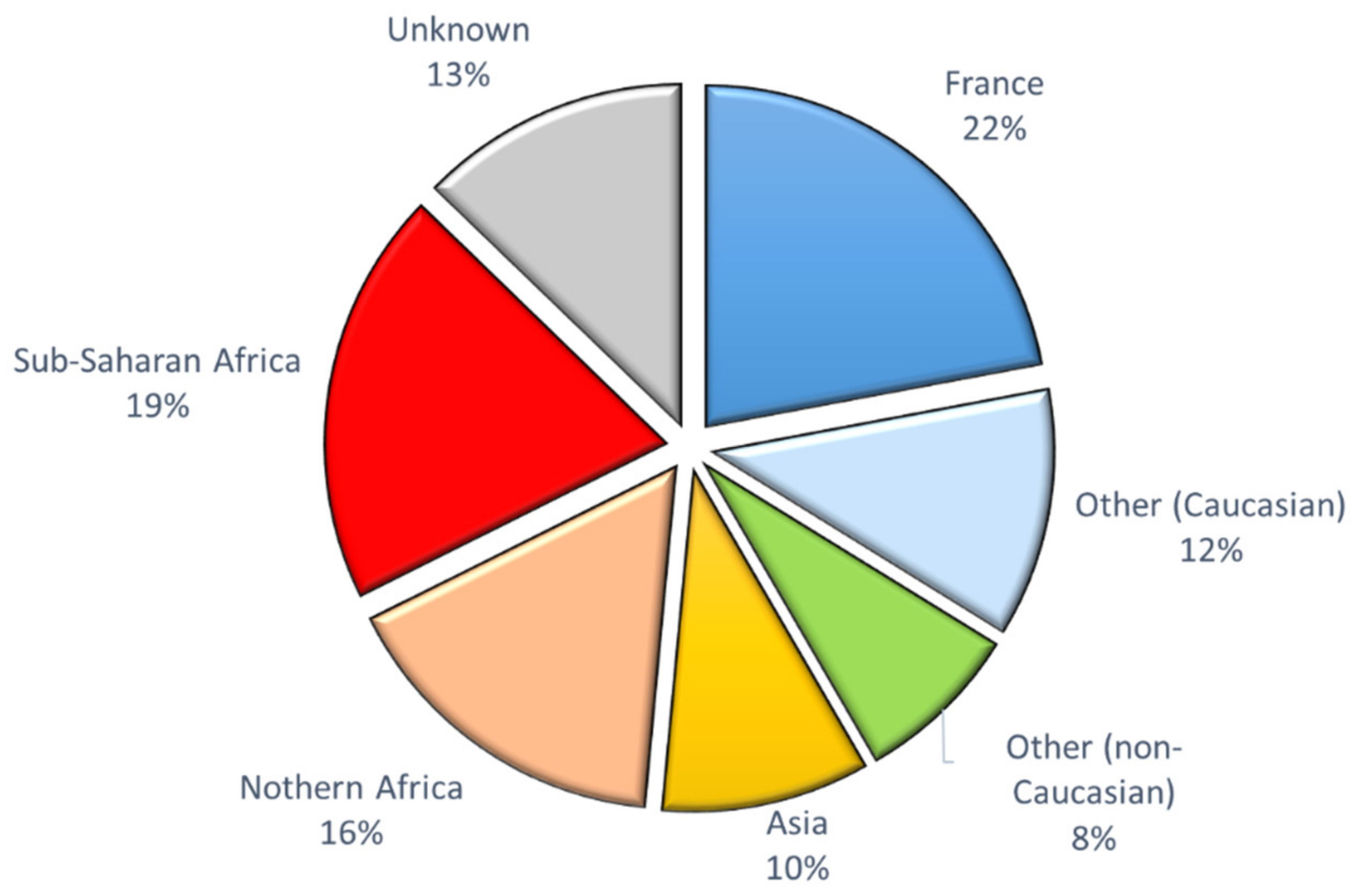
3.2. CFTR Genotypes in Parents from Different Ethnic Groups
4. Discussion
4.1. Ultrasound Digestive Abnormalities
4.2. Low Risk of CF in Our Cohort of Diverse Geographical Origins
4.3. The CF Frequent Mutation Kits Are Not Appropriate in Non-Caucasian Populations
4.4. Difficulties in Interpreting Rare Variants
4.5. The Role of the Laboratory
4.6. The Role of the Geneticist
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsui, L.-C.; Dorfman, R. The Cystic Fibrosis Gene: A Molecular Genetic Perspective. Cold Spring Harb. Perspect. Med. 2013, 3, a009472. [Google Scholar] [CrossRef]
- Castellani, C.; Cuppens, H.; Macek, M.; Cassiman, J.J.; Kerem, E.; Durie, P.; Tullis, E.; Assael, B.M.; Bombieri, C.; Brown, A.; et al. Consensus on the Use and Interpretation of Cystic Fibrosis Mutation Analysis in Clinical Practice. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2008, 7, 179–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Raynal, C.; Corvol, H. Variant Classifications, Databases and Genotype-Phenotype Correlations. Arch. Pédiatrie 2020, 27, eS13–eS18. [Google Scholar] [CrossRef]
- Bell, S.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The Future of Cystic Fibrosis Care: A Global Perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef] [Green Version]
- Corteville, J.E.; Gray, D.L.; Langer, J.C. Bowel Abnormalities in the Fetus—Correlation of Prenatal Ultrasonographic Findings with Outcome. Am. J. Obstet. Gynecol. 1996, 175, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Ghose, I.; Mason, G.C.; Martinez, D.; Harrison, K.L.; Evans, J.A.; Ferriman, E.L.; Stringer, M.D. Hyperechogenic Fetal Bowel: A Prospective Analysis of Sixty Consecutive Cases. BJOG Int. J. Obstet. Gynaecol. 2000, 107, 426–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.; Dommergues, M.; Simon-Bouy, B.; Ferec, C.; Oury, J.F.; Aubry, M.C.; Bessis, R.; Vuillard, E.; Denamur, E.; Bienvenu, T.; et al. Cystic Fibrosis Screening: A Fetus with Hyperechogenic Bowel May Be the Index Case. J. Med. Genet. 1998, 35, 657–660. [Google Scholar] [CrossRef]
- Muller, F.; Simon-Bouy, B.; Girodon, E.; Monnier, N.; Malinge, M.C.; Serre, J.L. French Collaborative Group Predicting the Risk of Cystic Fibrosis with Abnormal Ultrasound Signs of Fetal Bowel: Results of a French Molecular Collaborative Study Based on 641 Prospective Cases. Am. J. Med. Genet. 2002, 110, 109–115. [Google Scholar] [CrossRef]
- Scotet, V.; Braekeleer, M.D.; Audrézet, M.-P.; Quéré, I.; Mercier, B.; Duguépéroux, I.; Andrieux, J.; Blayau, M.; Férec, C. Prenatal Detection of Cystic Fibrosis by Ultrasonography: A Retrospective Study of More than 346,000 Pregnancies. J. Med. Genet. 2002, 39, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Slotnick, R.N.; Abuhamad, A.Z. Prognostic Implications of Fetal Echogenic Bowel. Lancet 1996, 347, 85–87. [Google Scholar] [CrossRef]
- Ameratunga, D.M.; Said, J.M.; Reidy, K.; Palma-Dias, R. Perinatal Outcomes Following the Ultrasound Diagnosis of Echogenic Bowel: An Australian Perspective. Fetal Diagn. Ther. 2012, 31, 179–184. [Google Scholar] [CrossRef]
- D’Amico, A.; Buca, D.; Rizzo, G.; Khalil, A.; Silvi, C.; Makatsariya, A.; Nappi, L.; Liberati, M.; D’Antonio, F. Outcome of Fetal Echogenic Bowel: A Systematic Review and Meta-Analysis. Prenat. Diagn. 2020. [Google Scholar] [CrossRef]
- Masini, G.; Maggio, L.; Marchi, L.; Cavalli, I.; Ledda, C.; Trotta, M.; Pasquini, L. Isolated Fetal Echogenic Bowel in a Retrospective Cohort: The Role of Infection Screening. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 231, 136–141. [Google Scholar] [CrossRef]
- De Becdelièvre, A.; Costa, C.; Jouannic, J.-M.; LeFloch, A.; Giurgea, I.; Martin, J.; Médina, R.; Boissier, B.; Gameiro, C.; Muller, F.; et al. Comprehensive Description of CFTR Genotypes and Ultrasound Patterns in 694 Cases of Fetal Bowel Anomalies: A Revised Strategy. Hum. Genet. 2011, 129, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Dequeker, E.; Stuhrmann, M.; Morris, M.A.; Casals, T.; Castellani, C.; Claustres, M.; Cuppens, H.; des Georges, M.; Ferec, C.; Macek, M.; et al. Best Practice Guidelines for Molecular Genetic Diagnosis of Cystic Fibrosis and CFTR-Related Disorders—Updated European Recommendations. Eur. J. Hum. Genet. EJHG 2009, 17, 51–65. [Google Scholar] [CrossRef]
- Bobadilla, J.L.; Macek, M.; Fine, J.P.; Farrell, P.M. Cystic Fibrosis: A Worldwide Analysis of CFTR Mutations—Correlation with Incidence Data and Application to Screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef]
- Estivill, X.; Bancells, C.; Ramos, C. Geographic Distribution and Regional Origin of 272 Cystic Fibrosis Mutations in European Populations. Hum. Mutat. 1997, 10, 135–154. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Henrie, A.; Hemphill, S.E.; Ruiz-Schultz, N.; Cushman, B.; DiStefano, M.T.; Azzariti, D.; Harrison, S.M.; Rehm, H.L.; Eilbeck, K. ClinVar Miner: Demonstrating Utility of a Web-Based Tool for Viewing and Filtering ClinVar Data. Hum. Mutat. 2018, 39, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- The Clinical and Functional TRanslation of CFTR (CFTR2). Available online: https://cftr2.org/ (accessed on 17 February 2021).
- Claustres, M.; Thèze, C.; des Georges, M.; Baux, D.; Girodon, E.; Bienvenu, T.; Audrezet, M.-P.; Dugueperoux, I.; Férec, C.; Lalau, G.; et al. CFTR-France, a National Relational Patient Database for Sharing Genetic and Phenotypic Data Associated with Rare CFTR Variants. Hum. Mutat. 2017, 38, 1297–1315. [Google Scholar] [CrossRef] [PubMed]
- Espírito Santo, R.; Martins, R.; Valente, S.; Saldanha, J. An Unusual Cause of Neonatal Ascites. BMJ Case Rep. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Ronin, C.; Mace, P.; Stenard, F.; Loundou, A.; Capelle, M.; Mortier, I.; Pellissier, M.C.; Sigaudy, S.; Levy, A.; D’ercole, C.; et al. Antenatal Prognostic Factor of Fetal Echogenic Bowel. Eur. J. Obstet. Gynecol. Reprod. Biol. 2017, 212, 166–170. [Google Scholar] [CrossRef]
- Duguépéroux, I.; Scotet, V.; Audrézet, M.-P.; Saliou, A.-H.; Collet, M.; Blayau, M.; Schmitt, S.; Kitzis, A.; Fresquet, F.; Müller, F.; et al. Nonvisualization of Fetal Gallbladder Increases the Risk of Cystic Fibrosis. Prenat. Diagn. 2012, 32, 21–28. [Google Scholar] [CrossRef]
- Lap, C.C.; Voskuilen, C.S.; Pistorius, L.R.; Mulder, E.J.H.; Visser, G.H.A.; Manten, G.T.R. Reference Curves for the Normal Fetal Small Bowel and Colon Diameters; Their Usefulness in Fetuses with Suspected Dilated Bowel. J. Matern. Fetal Neonatal Med. 2020, 33, 633–638. [Google Scholar] [CrossRef] [Green Version]
- Blazer, S.; Zimmer, E.Z.; Bronshtein, M. Nonvisualization of the Fetal Gallbladder in Early Pregnancy: Comparison with Clinical Outcome. Radiology 2002, 224, 379–382. [Google Scholar] [CrossRef]
- Pasquo, E.D.; Kuleva, M.; Rousseau, A.; Vitucci, A.; Sonigo, P.; Chardot, C.; Salomon, L.J.; Ville, Y. Outcome of Non-Visualization of Fetal Gallbladder on Second-Trimester Ultrasound: Cohort Study and Systematic Review of Literature. Ultrasound Obstet. Gynecol. 2019, 54, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Bergougnoux, A.; Jouannic, J.-M.; Verneau, F.; Bienvenu, T.; Gaitch, N.; Raynal, C.; Girodon, E. Isolated Nonvisualization of the Fetal Gallbladder Should Be Considered for the Prenatal Diagnosis of Cystic Fibrosis. Fetal Diagn. Ther. 2019, 45, 312–316. [Google Scholar] [CrossRef]
- Audrézet, M.P.; Munck, A. Newborn Screening for CF in France: An Exemplary National Experience. Arch. Pédiatrie 2020, 27, eS35–eS40. [Google Scholar] [CrossRef]
- Palomaki, G.E.; Fitzsimmons, S.C.; Haddow, J.E. Clinical Sensitivity of Prenatal Screening for Cystic Fibrosis via CFTR Carrier Testing in a United States Panethnic Population. Genet. Med. 2004, 6, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Mutesa, L.; Bours, V. Diagnostic Challenges of Cystic Fibrosis in Patients of African Origin. J. Trop. Pediatr. 2009, 55, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Owusu, S.K.; Morrow, B.M.; White, D.; Klugman, S.; Vanker, A.; Gray, D.; Zampoli, M. Cystic Fibrosis in Black African Children in South Africa: A Case Control Study. J. Cyst. Fibros. 2020, 19, 540–545. [Google Scholar] [CrossRef]
- Singh, M.; Rebordosa, C.; Bernholz, J.; Sharma, N. Epidemiology and Genetics of Cystic Fibrosis in Asia: In Preparation for the next-Generation Treatments. Respirology 2015, 20, 1172–1181. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.; Pepper, M.S. Cystic Fibrosis on the African Continent. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 653–662. [Google Scholar] [CrossRef] [Green Version]
- Fredj, S.H.; Messaoud, T.; Templin, C.; des Georges, M.; Fattoum, S.; Claustres, M. Cystic Fibrosis Transmembrane Conductance Regulator Mutation Spectrum in Patients with Cystic Fibrosis in Tunisia. Genet. Test. Mol. Biomark. 2009, 13, 577–581. [Google Scholar] [CrossRef]
- Bergougnoux, A.; Délétang, K.; Pommier, A.; Varilh, J.; Houriez, F.; Altieri, J.P.; Koenig, M.; Férec, C.; Claustres, M.; Lalau, G.; et al. Functional Characterization and Phenotypic Spectrum of Three Recurrent Disease-Causing Deep Intronic Variants of the CFTR Gene. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2019, 18, 468–475. [Google Scholar] [CrossRef]
- Strandvik, B.; Björck, E.; Fallström, M.; Gronowitz, E.; Thountzouris, J.; Lindblad, A.; Markiewicz, D.; Wahlström, J.; Tsui, L.C.; Zielenski, J. Spectrum of Mutations in the CFTR Gene of Patients with Classical and Atypical Forms of Cystic Fibrosis from Southwestern Sweden: Identification of 12 Novel Mutations. Genet. Test. 2001, 5, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Kolesár, P.; Minárik, G.; Baldovic, M.; Ficek, A.; Kovács, L.; Kádasi, L. Mutation Analysis of the CFTR Gene in Slovak Cystic Fibrosis Patients by DHPLC and Subsequent Sequencing: Identification of Four Novel Mutations. Gen. Physiol. Biophys. 2008, 27, 299–305. [Google Scholar] [PubMed]
- Lejeune, F.; Maquat, L.E. Mechanistic Links between Nonsense-Mediated MRNA Decay and Pre-MRNA Splicing in Mammalian Cells. Curr. Opin. Cell Biol. 2005, 17, 309–315. [Google Scholar] [CrossRef]
- Gentzsch, M.; Riordan, J.R. Localization of Sequences within the C-terminal Domain of the Cystic Fibrosis Transmembrane Conductance Regulator Which Impact Maturation and Stability*. J. Biol. Chem. 2001, 276, 1291–1298. [Google Scholar] [CrossRef] [Green Version]
- Hughes, E.E.; Stevens, C.F.; Saavedra-Matiz, C.A.; Tavakoli, N.P.; Krein, L.M.; Parker, A.; Zhang, Z.; Maloney, B.; Vogel, B.; DeCelie-Germana, J.; et al. Clinical Sensitivity of Cystic Fibrosis Mutation Panels in a Diverse Population. Hum. Mutat. 2016, 37, 201–208. [Google Scholar] [CrossRef]
- Nappo, S.; Mannucci, L.; Novelli, G.; Sangiuolo, F.; D’Apice, M.R.; Botta, A. Carrier Frequency of CFTR Variants in the Non-Caucasian Populations by Genome Aggregation Database (GnomAD)-Based Analysis. Ann. Hum. Genet. 2020, 84, 463–468. [Google Scholar] [CrossRef]
- Oca, F.; Dreux, S.; Gérard, B.; Simon-Bouy, B.; de Becdelièvre, A.; Ferec, C.; Girodon, E.; Muller, F. Amniotic Fluid Digestive Enzyme Analysis Is Useful for Identifying CFTR Gene Mutations of Unclear Significance. Clin. Chem. 2009, 55, 2214–2217. [Google Scholar] [CrossRef] [Green Version]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ultrasound Signs | Likelihood Ratio (LR) | Number of Fetuses Observed with the Ultrasound Abnormalities in [15] | |
|---|---|---|---|
| CF Fetuses | Non-CF Fetuses | ||
| Isolated signs | |||
| Abdominal calcifications | 0 | 0 | 12 |
| Isolated NVFGB | 0 | 0 | 6 |
| Ascites | 1.19 | 0 | 14 |
| FEB | 0.42 | 8 | 376 |
| Loop dilatation | 3.10 | 3 | 26 |
| Associations of signs | |||
| Meconium peritonitis * | 1.94 | 2 | 26 |
| FEB + loop dilatation | 2.65 | 5 | 42 |
| FEB + NVFGB | 3.87 | 2 | 14 |
| Triad: FEB + loop dilatation + NVFGB | 31.40 | 4 | 4 |
| Origin | cDNA Name | Protein Name | Legacy Name | dbSNP | Nb of Carriers | Classification in CFTR2 | Classification in CFTR-France | ClinVar Miner | Our Interpretation |
|---|---|---|---|---|---|---|---|---|---|
| France | |||||||||
| c.[3718-2477C>T]; [709C>G] | p.[?];[Gln237Glu] | 3849+10kbC>T/Q237E | rs75039782 rs397508784 | 1 | CF (PS); VVCC | VVCC; VVCC | P (1), VUS (1), Lik. B (1), B (1)/ VUS (1) | CF/VVCC, genotype compatible with mild CF | |
| c.1210-34TG[11]T[5] | p.? | TG11T5 | no rs | 2 | VVCC | VUS | VUS (1) | CFTR-RD with low penetrance | |
| c.2900T>C | p.Leu967Ser | L967S | rs1800110 | 1 | VVCC | VUS | P (1), Lik. P (1), VUS (8) | VUS non CF | |
| c.3590A>G | p.His1197Arg | H1197R | no rs | 1 | not reported | not reported | not reported | VUS | |
| c.224G>A | p.Arg75Gln | R75Q | rs1800076 | 2 | non CF | non disease-causing | P (1), Lik. B (5), B (6), VUS (4), | CFTR-RD with low penetrance | |
| Northern Africa | |||||||||
| c.3454G>C | p.Asp1152His | D1152H | rs75541969 | 2 | VVCC | VVCC | P (15), Lik. P (1) | VVCC | |
| c.2991G>C | p.Leu997Phe | L997F | rs1800111 | 2 | non CF | CFTR-RD | not reported | CFTR-RD | |
| (*)c.[1210-34TG[12]T[5]](;) [1210-34TG[11]T[5];1684G>A] | p.[?](;)[?;Val562Ile] | TG12T5/ TG11T5/V562I | rs1800097 | 1 | VVCC; VVCC; non CF | CFTR-RD;VUS;VUS | P (1), Lik. B (1), VUS (6) | CFTR-RD | |
| c.2002C>T | p.Arg668Cys | R668C | rs1800100 | 1 | non CF | CFTR-RD | Lik. B (1), B (3), VUS (11) | CFTR-RD, low penetrance | |
| (*)c.2991G>C/c.1210-34TG[12]T[5]/c.2930C>T | p.Leu997Phe/p.?/p.Ser977Phe | L997F/TG12T5/S977F | rs1800111 rs141033578 | 1 | non CF; VVCC | CFTR-RD; VVCC | not reported/VUS (1), P (2), Lik. P (1) | VVCC/CFTR-RD low penetrance | |
| Sub-Saharan Africa | |||||||||
| c.4230C>A | p.Cys1410* | C1410X | no rs | 2 | not reported | not reported | not reported | CF | |
| c.2290C>T | p.Arg764* | R764X | rs121908810 | 1 | CF (PI) | CF | P (7) | CF | |
| c.846A>T | p.Glu282Asp | E282D | rs142864834 | 1 | not reported | not reported | VUS (4) | VUS | |
| c.4333G>A | p.Asp1445Asn | D1445N | rs148783445 | 1 | not reported | VUS | Lik. P (1), Lik. B (1) VUS (9) | VUS, non CF | |
| c.3517G>A | p.Gly1173Ser | G1173S | rs368393738 | 1 | not reported | not reported | VUS (2) | VUS | |
| c.4243-20A>G | p.? | NA | rs138025486 | 1 | not reported | VUS | Lik. B (3), VUS (3) | VUS, non CF | |
| Other geographical origins (Asia, Europe, mixed origins, unknown) | |||||||||
| Sri Lanka | c.1210-34TG[13]T[5] | p.? | TG13T5 | no rs | 1 | VVCC | VVCC | not reported | VVCC |
| Germany | c.[220C>T;890G>A] | p.Arg74Trp/p.Arg297Gln | R74W/R297Q | rs115545701 rs143486492 | 1 | not reported | VUS | Lik. P (4), Lik. B (2), B (1), VUS (5)/ Lik. B (6), B (1), VUS (2) | VUS non CF |
| Italy | c.1043T>A | p.Met348Lys | M348K | rs142920240 | 1 | not reported | VUS | Lik. B (5), B (1), VUS (3), | VUS non CF |
| Guadeloupe | c.2249C>T | p.Pro750Leu | P750L | rs140455771 | 1 | VVCC | CFTR-RD | P (2), Lik. P (5), VUS (5) | VVCC |
| c.853A>T | p.Ile285Phe | I285F | rs151073129 | 1 | not reported | not reported | Lik. B (2), B (1) | VUS non CF | |
| Reunion Island | c.224G>A | p.Arg75Gln | R75Q | rs1800076 | 1 | non CF | non disease-causing | P (1), Lik. B (5), B (6), VUS (4), | CFTR-RD, low penetrance |
| Mauritius | c.870-1095A>C | p.? | rs540046342 | 1 | not reported | not reported | not reported | VUS | |
| Caucase/Northern Africa | c.2991G>C | p.Leu997Phe | L997F | rs1800111 | 1 | non CF | CFTR-RD | not reported | CFTR-RD low penetrance |
| Unknown | c.1523T>G | p.Phe508Cys | F508C | rs74571530 | 1 | non CF | CFTR-RD | P (2), Lik. B (1), B (5), VUS (3), | CFTR-RD low penetrance |
| c.3705T>G | p.Ser1235Arg | S1235R | rs34911792 | 1 | non CF | VUS | Lik. B (5), B (7), VUS (3) | CFTR-RD low penetrance | |
| c.1210-34TG[12]T[5] | p.? | TG12T5 | no rs | 1 | VVCC | CFTR-RD | not reported | CFTR-RD | |
| Variant | gnomAD Frequency (European Non-Finnish) | Structural Effects Predicted by CYSMA | SIFT/PolyPhen-2 | Mutation Taster | CADD Score | In Silico Splicing Analysis Tools |
|---|---|---|---|---|---|---|
| p.Leu967Ser | 0.10% | Solvent accessibility: buried No steric clashes | T/D | Disease causing | 24.3 | NR |
| p.His1197Arg | 0.00090% | Solvent accessibility: exposed No steric clashes | T/B | Polymorphism | 12.3 | NR |
| p.Glu282Asp | 0.000% | Solvent accessibility: buried No steric clashes | T/D | Disease causing | 24.5 | NR |
| p.Asp1445Asn | 0.029% | Solvent accessibility: buried No steric clashes | T/D | Disease causing | 26.3 | NR |
| p.Gly1173Ser | 0.000% | Solvent accessibility: wild-type G1173 exposed, mutant G1173S buried Steric clashes | T/B | Polymorphism | 2.1 | NR |
| c.4243-20A>G | 0.0016% | Not predicted | Not predicted | Not predicted | 14.9 | HSF: Putative branch point disappearance, but strong branch points in the vicinity of the affected one → putative low impact on splicing |
| p.Arg74Trp | 0.033% | R74 is involved in the CFTR pore construction Solvent accessibility: wild-type R74 exposed, mutant R74W buried Steric clashes | T/D | Disease causing | 25.3 | NR |
| p.Arg297Gln | 0.12% | Solvent accessibility: wild-type R297 buried, mutant R297Q exposed No steric clashes | T/B | Disease causing | 24.8 | NR |
| p.Met348Lys | 0.021% | Solvent accessibility: wild-type M348 buried, mutant M348K exposed Steric clashes | D/D | Disease causing | 27.3 | HSF: Putative acceptor site creation (weaker than the natural site) |
| p.Ile285Phe | 0.000% * | Solvent accessibility: buried Steric clashes | D/D | Disease causing | 27.8 | NR |
| c.870-1095A>C | 0.013% | Not predicted | Not predicted | Not predicted | 5.0 | ESEfinder3.0: Deep Intronic SC35 site creation → putative low impact on splicing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mekki, C.; Aissat, A.; Mirlesse, V.; Mayer Lacrosniere, S.; Eche, E.; Le Floch, A.; Whalen, S.; Prud’Homme, C.; Remus, C.; Funalot, B.; et al. Prenatal Ultrasound Suspicion of Cystic Fibrosis in a Multiethnic Population: Is Extensive CFTR Genotyping Needed? Genes 2021, 12, 670. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12050670
Mekki C, Aissat A, Mirlesse V, Mayer Lacrosniere S, Eche E, Le Floch A, Whalen S, Prud’Homme C, Remus C, Funalot B, et al. Prenatal Ultrasound Suspicion of Cystic Fibrosis in a Multiethnic Population: Is Extensive CFTR Genotyping Needed? Genes. 2021; 12(5):670. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12050670
Chicago/Turabian StyleMekki, Chadia, Abdel Aissat, Véronique Mirlesse, Sophie Mayer Lacrosniere, Elsa Eche, Annick Le Floch, Sandra Whalen, Cecile Prud’Homme, Christelle Remus, Benoit Funalot, and et al. 2021. "Prenatal Ultrasound Suspicion of Cystic Fibrosis in a Multiethnic Population: Is Extensive CFTR Genotyping Needed?" Genes 12, no. 5: 670. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12050670