
Investigating the Genetic Profile of the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia (ALS-FTD) Continuum in Patients of Diverse Race, Ethnicity and Ancestry

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
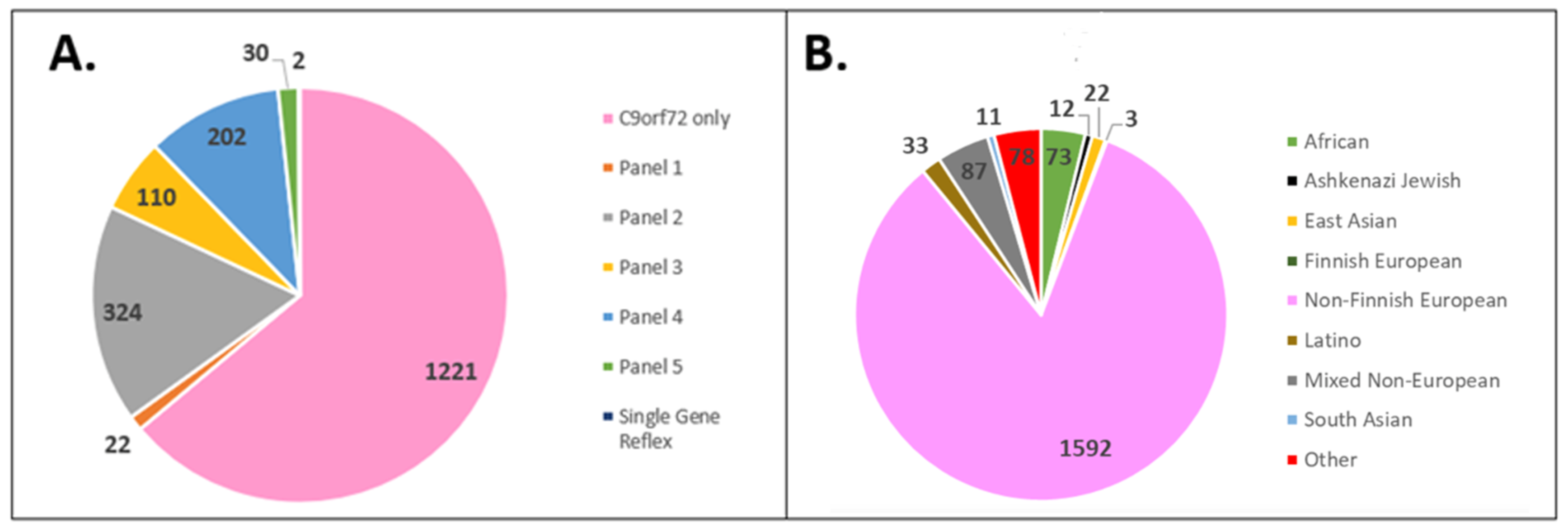
3.1. Cohort Demographics
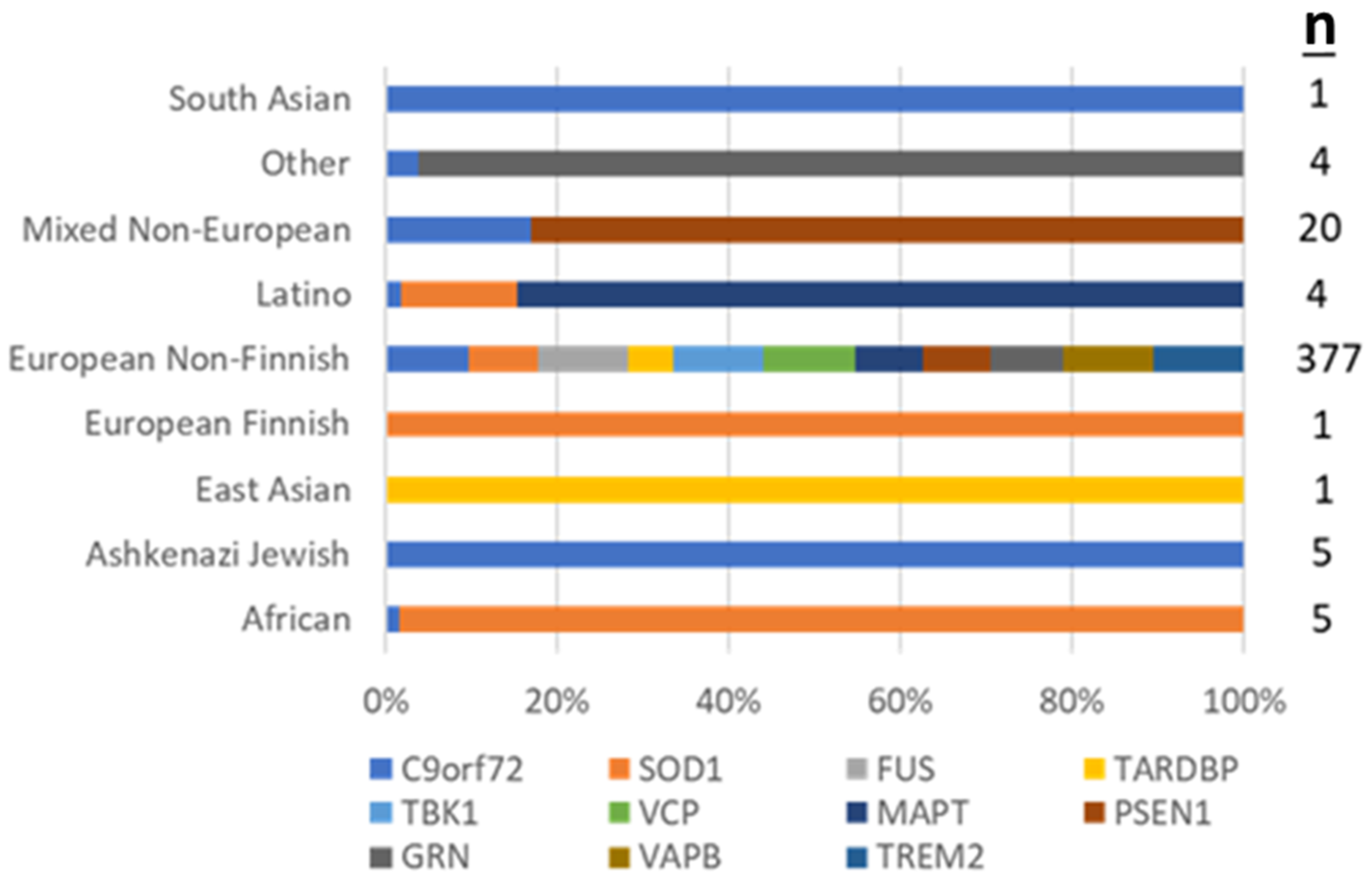
3.2. Positive Test Outcomes across REA Groups
3.3. Uncertain Result Outcomes across REA Groups
3.4. Age at Testing across REA Groups
3.5. Testing Ordered across REA Groups
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Siddique, N.; Siddique, T. Amyotrophic Lateral Sclerosis Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Broce, I.J.; Castruita, P.A.; Yokoyama, J.S. Moving Toward Patient-Tailored Treatment in ALS and FTD: The Potential of Genomic Assessment as a Tool for Biological Discovery and Trial Recruitment. Front. Neurosci. 2021, 15, 150. [Google Scholar] [CrossRef]
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Rechtman, L.; Jordan, H.; Wagner, L.; Horton, D.K.; Kaye, W. Racial and ethnic differences among amyotrophic lateral sclerosis cases in the United States. Amyotroph. Lateral Scler. Front. Degener. 2014, 16, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, D.; Jetté, N.; Fiest, K.M.; Roberts, J.I.; Pearson, D.; Smith, E.E.; Roach, P.; Kirk, A.; Pringsheim, T.; Maxwell, C.J. The Prevalence and Incidence of Frontotemporal Dementia: A Systematic Review. Can. J. Neurol. Sci. J. Can. Des Sci. Neurol. 2016, 43, S96–S109. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.L.; Johnson, N.J.; Chen, J.T.; Cudkowicz, M.E.; Weisskopf, M.G. Race/ethnicity, socioeconomic status, and ALS mortality in the United States. Neurology 2016, 87, 2300–2308. [Google Scholar] [CrossRef] [Green Version]
- Marin, B.; Logroscino, G.; Boumédiene, F.; Labrunie, A.; Couratier, P.; Babron, M.-C.; Leutenegger, A.-L.; Preux, P.-M.; Beghi, E. Clinical and demographic factors and outcome of amyotrophic lateral sclerosis in relation to population ancestral origin. Eur. J. Epidemiol. 2015, 31, 229–245. [Google Scholar] [CrossRef]
- Qadri, S.; Langefeld, C.D.; Milligan, C.; Caress, J.B.; Cartwright, M.S. Racial differences in intervention rates in individuals with ALS. Neurology 2019, 92, e1969–e1974. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Roggenbuck, J.; Fong, J.C. Genetic Testing for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Clin. Lab. Med. 2020, 40, 271–287. [Google Scholar] [CrossRef]
- Roggenbuck, J.; Quick, A.; Kolb, S.J. Genetic testing and genetic counseling for amyotrophic lateral sclerosis: An update for clinicians. Genet. Med. 2017, 19, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Test. Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD) Panel—PreventionGenetics. Available online: https://www.preventiongenetics.com/testInfo?val=Amyotrophic+Lateral+Sclerosis+%28ALS%29+and+Frontotemporal+Dementia+%28FTD%29+Panel (accessed on 24 November 2021).
- Mok, K.; Traynor, B.J.; Schymick, J.; Tienari, P.J.; Laaksovirta, H.; Peuralinna, T.; Myllykangas, L.; Chiò, A.; Shatunov, A.; Boeve, B.F.; et al. The chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiol. Aging 2012, 33, 209.e3–209.e8. [Google Scholar] [CrossRef]
- Pliner, H.A.; Mann, D.M.; Traynor, B.J. Searching for Grendel: Origin and global spread of the C9ORF72 repeat expansion. Acta Neuropathol. 2014, 127, 391–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chio, A.; Restagno, G.; Nicolaou, N.; Sánchez, J.S.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Liu, X.; He, J.; Gao, F.-B.; Gitler, A.D.; Fan, D. The epidemiology and genetics of Amyotrophic lateral sclerosis in China. Brain Res. 2018, 1693, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Nel, M.; Agenbag, G.M.; Henning, F.; Cross, H.M.; Esterhuizen, A.; Heckmann, J.M. C9orf72 repeat expansions in South Africans with amyotrophic lateral sclerosis. J. Neurol. Sci. 2019, 401, 51–54. [Google Scholar] [CrossRef]
- Chio, A.; Battistini, S.; Calvo, A.; Caponnetto, C.; Conforti, F.L.; Corbo, M.; Giannini, F.; Mandrioli, J.; Mora, G.; Sabatelli, M.; et al. Genetic counselling in ALS: Facts, uncertainties and clinical suggestions. J. Neurol. Neurosurg. Psychiatry 2013, 85, 478–485. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.-J.; Baek, W.; Ki, C.-S.; Kim, H.Y.; Koh, S.-H.; Kim, J.-W.; Kim, S.H. Screening of the SOD1, FUS, TARDBP, ANG, and OPTN mutations in Korean patients with familial and sporadic ALS. Neurobiol. Aging 2012, 33, 1017.e17–1017.e23. [Google Scholar] [CrossRef]
- Alavi, A.; Nafissi, S.; Rohani, M.; Zamani, B.; Sedighi, B.; Shamshiri, H.; Fan, J.-B.; Ronaghi, M.; Elahi, E. Genetic analysis and SOD1 mutation screening in Iranian amyotrophic lateral sclerosis patients. Neurobiol. Aging 2013, 34, 1516.e1–1516.e8. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Desaro, P.; Johnston, A.; Ross, O.A.; Wszolek, Z.K.; Ertekin-Taner, N.; Graff-Radford, N.R.; Rademakers, R.; Boylan, K. Novel p.Ile151Val mutation in VCP in a patient of African American descent with sporadic ALS. Neurology 2011, 77, 1102–1103. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Liu, M.-S.; Li, X.-G.; Cui, L.-Y. Screening of VCP mutations in Chinese amyotrophic lateral sclerosis patients. Neurobiol. Aging 2013, 34, 1519.e3–1519.e4. [Google Scholar] [CrossRef]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Cui, L.-Y.; Sun, Q.; Li, X.-G.; Liu, M.-S.; Xu, Y.; Zhou, Y.; Yang, X.-Z. De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurobiol. Aging 2013, 34, 1312.e1–1312.e8. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.; Pitout, I.L.; Fletcher, S.; Wilton, S.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.G.; Jackson, C.E.; Kasarskis, E.J.; England, J.D.; Forshew, D.; Johnston, W.; Kalra, S.; Katz, J.S.; Mitsumoto, H.; Rosenfeld, J.; et al. Practice Parameter update: The care of the patient with amyotrophic lateral sclerosis: Multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009, 73, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Klepek, H.; Nagaraja, H.; Goutman, S.A.; Quick, A.; Kolb, S.J.; Roggenbuck, J. Lack of consensus in ALS genetic testing practices and divergent views between ALS clinicians and patients. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 216–221. [Google Scholar] [CrossRef]
- Vajda, A.; McLaughlin, R.L.; Heverin, M.; Thorpe, O.; Abrahams, S.; Al-Chalabi, A.; Hardiman, O. Genetic testing in ALS. Neurology 2017, 88, 991–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, K.C.; Doyle, C.; Chiò, A.; Traynor, B.J. Use of Genetic Testing in Amyotrophic Lateral Sclerosis by Neurologists. JAMA Neurol. 2017, 74, 125–126. [Google Scholar] [CrossRef]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nat. Cell Biol. 2016, 538, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Popejoy, A.B.; Ritter, D.I.; Crooks, K.; Currey, E.; Fullerton, S.M.; Hindorff, L.A.; Koenig, B.; Ramos, E.M.; Sorokin, E.P.; Wand, H.; et al. The clinical imperative for inclusivity: Race, ethnicity, and ancestry (REA) in genomics. Hum. Mutat. 2018, 39, 1713–1720. [Google Scholar] [CrossRef]
- Ndugga-Kabuye, M.K.; Issaka, R.B. Inequities in multi-gene hereditary cancer testing: Lower diagnostic yield and higher VUS rate in individuals who identify as Hispanic, African or Asian and Pacific Islander as compared to European. Fam. Cancer 2019, 18, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Landry, L.G.; Rehm, H.L. Association of Racial/Ethnic Categories with the Ability of Genetic Tests to Detect a Cause of Cardiomyopathy. JAMA Cardiol. 2018, 3, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Morales, J.; Welter, D.; Bowler, E.H.; Cerezo, M.; Harris, L.W.; McMahon, A.C.; Hall, P.; Junkins, H.A.; Milano, A.; Hastings, E.; et al. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 2018, 19, 21. [Google Scholar] [CrossRef]
- Hoffman-Andrews, L. The known unknown: The challenges of genetic variants of uncertain significance in clinical practice. J. Law Biosci. 2017, 4, 648–657. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Ashida, S.; Goodman, M.S.; Stafford, J.; Lachance, C.; Kaphingst, K.A. Perceived familiarity with and importance of family health history among a medically underserved population. J. Community Genet. 2012, 3, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Marcum, C.S.; Myers, M.; Koehly, L.M. Racial differences in family health history knowledge of type 2 diabetes: Exploring the role of interpersonal mechanisms. Transl. Behav. Med. 2018, 8, 540–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, T.; Seo, J.; Griffith, J.; Baxter, M.; James, A.; Kaphingst, K.A. “You don’t have to keep everything on paper”: African American women’s use of family health history tools. J. Community Genet. 2013, 4, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Brand, D.; Polak, M.; Glass, J.D.; Fournier, C.N. Comparison of Phenotypic Characteristics and Prognosis between Black and White Patients in a Tertiary ALS Clinic. Neurology 2020, 96, e840–e844. [Google Scholar] [CrossRef]
- Goldstein, O.; Gana-Weisz, M.; Nefussy, B.; Vainer, B.; Nayshool, O.; Bar-Shira, A.; Traynor, B.J.; Drory, V.E.; Orr-Urtreger, A. High frequency of C9orf72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis patients from two founder populations sharing the same risk haplotype. Neurobiol. Aging 2018, 64, 160.e1–160.e7. [Google Scholar] [CrossRef]
- Roggenbuck, J.; Rich, K.A.; Vicini, L.; Palettas, M.; Schroeder, J.; Zaleski, C.; Lincoln, T.; Drury, L.; Glass, J.D. Amyotrophic Lateral Sclerosis Genetic Access Program. Neurol. Genet. 2021, 7, e615. [Google Scholar] [CrossRef]
- Caswell-Jin, J.L.; Gupta, T.; Hall, E.; Petrovchich, I.M.; Mills, M.A.; Kingham, K.E.; Koff, R.; Chun, N.M.; Levonian, P.; Lebensohn, A.P.; et al. Racial/ethnic differences in multiple-gene sequencing results for hereditary cancer risk. Genet. Med. 2018, 20, 234–239. [Google Scholar] [CrossRef]
- Peterson, R.E.; Kuchenbaecker, K.; Walters, R.K.; Chen, C.-Y.; Popejoy, A.B.; Periyasamy, S.; Lam, M.; Iyegbe, C.; Strawbridge, R.J.; Brick, L.; et al. Genome-wide Association Studies in Ancestrally Diverse Populations: Opportunities, Methods, Pitfalls, and Recommendations. Cell 2019, 179, 589–603. [Google Scholar] [CrossRef]
- Couratier, P.; Lautrette, G.; Luna, J.; Corcia, P. Phenotypic variability in amyotrophic lateral sclerosis. Rev. Neurol. 2021, 177, 536–543. [Google Scholar] [CrossRef]
- EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis; Andersen, P.M.; Abrahams, S.; Borasio, G.D.; de Carvalho, M.; Chio, A.; Van Damme, P.; Hardiman, O.; Kollewe, K.; Morrison, K.E.; et al. EFNS guidelines on the Clinical Management of Amyotrophic Lateral Sclerosis (MALS)—Revised report of an EFNS task force. Eur. J. Neurol. 2011, 19, 360–375. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Awareness of Family Health History as a Risk Factor for Disease—United States, 2004. MMWR Morb. Mortal. Wkly. Rep. 2004, 53, 1044–1047. [Google Scholar]
- Kaphingst, K.; Lachance, C.; Gepp, A.; D’Anna, L.H.; Rios-Ellis, B. Educating Underserved Latino Communities about Family Health History Using Lay Health Advisors. Public Health Genom. 2011, 14, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.R.; Jones, A.R.; Opie-Martin, S.; Shatunov, A.; Iacoangeli, A.; Al Khleifat, A.; Smith, B.N.; Topp, S.; Morrison, K.E.; Shaw, P.J.; et al. Younger age of onset in familial amyotrophic lateral sclerosis is a result of pathogenic gene variants, rather than ascertainment bias. J. Neurol. Neurosurg. Psychiatry 2019, 90, 268–271. [Google Scholar] [CrossRef] [Green Version]
- Popejoy, A.B.; Crooks, K.R.; Fullerton, S.M.; Hindorff, L.A.; Hooker, G.W.; Koenig, B.A.; Pino, N.; Ramos, E.M.; Ritter, D.I.; Wand, H.; et al. Clinical Genetics Lacks Standard Definitions and Protocols for the Collection and Use of Diversity Measures. Am. J. Hum. Genet. 2020, 107, 72–82. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Panel 1 (28 gene ALS/dementia panel): ALS2, ANG, ATXN1, CHCHD10, CHMP2B, DCTN1, ERBB4, FIG4, FUS, HNRNPA1, KIF5A, MATR3, NEFH, NEK1, OPTN, PFN1, PRPH, SETX, SIGMAR1, SOD1, SPG11, SQSTM1, TARDBP, TBK1, TUBA4A, UBQLN2, VAPB, VCP |
| Panel 2 (24 gene ALS/dementia panel): ANG, ANXA11, ARHGEF28, CDH13, CHMP2B, FUS, GRN, HNRNPA1, HNRNPA2B1, KIF5A, MAPT, OPTN, PFN1, PSEN1, PSEN2, SETX, SOD1, SQSTM1, TARDBP, TBK1, TREM2, UBQLN2, VAPB, VCP |
| Panel 3 (11 gene dementia panel): APP, CHMP2B, FUS, GRN, MAPT, PSEN1, PSEN2, SQSTM1, TARDBP, TREM2, UBQLN2 |
| Panel 4 (5 gene ALS panel): FUS, SOD1, TARDBP, TBK1, VCP |
| Panel 5 (3 gene ALS panel): FUS, SOD1, TARDBP |
| Single Gene Reflexes: SOD1 and FIG4 |
| REA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| African/ African American (n = 73) | Ashkenazi Jewish (n = 12) | East Asian (n = 22) | European Finnish (n = 3) | European Non-Finnish (n = 1592) | Latino (n = 33) | Mixed Non-European (n = 87) | South Asian (n = 11) | Other (n = 78) | p-Value | |
| Positive | 5 (6.8%) | 5 (41.7%) | 0 (0.0%) | 0 (0.0%) | 339 (21.3%) | 2 (6.1%) | 17 (19.5%) | 1 (9.1%) | 3 (3.8%) | <0.001 |
| Negative | 68 (93.2%) | 7 (58.3%) | 22 (100.0%) | 3 (100.0%) | 1246 (78.3%) | 31 (93.9%) | 68 (78.2%) | 10 (90.9%) | 75 (96.2%) | |
| Intermediate | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 7 (0.4%) | 0 (0.0%) | 2 (2.3%) | 0 (0.0%) | 0 (0.0%) | |
| REA Group | p-Value | ||
|---|---|---|---|
| Result | European (n = 1595) | Underrepresented (n = 316) | <0.001 |
| Positive | 339 (21.3%) | 33 (10.4%) | |
| Negative | 1249 (78.3%) | 281 (88.9%) | |
| Intermediate | 7 (0.4%) | 2 (0.6%) | |
| REA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Results (n, %) | African/ African American (n = 24) | Ashkenazi Jewish (n = 5) | East Asian (n = 10) | European Finnish (n = 1) | European Non-Finnish (n = 575) | Latino (n = 20) | Mixed Non-European (n = 26) | South Asian (n = 8) | Other (n = 19) | p-Value |
| Positive | 4 (16.7) | 0 (0.0) | 0 (0.0) | 1 (100.0) | 38 (6.6) | 2 (10.0) | 1 (3.8) | 0 (0.0) | 1 (5.3) | 0.012 |
| Negative | 19 (79.2) | 5 (100.0) | 8 (80.0) | 0 (0.0) | 486 (84.5) | 17 (85.0) | 23 (88.5) | 6 (75.0) | 13 (68.4) | |
| Variant of Uncertain Significance | 1 (4.2) | 0 (0.0) | 2 (20.0) | 0 (0.0) | 51 (8.9) | 1 (5.0) | 2 (7.7) | 2 (25.0) | 5 (26.3) | |
| Test (n, %) | European (n = 377) | Underrepresented (n = 44) | p-Value |
|---|---|---|---|
| C9 Positive | 339 (90) | 33 (75) | 0.007 |
| Multigene Panel Positive | 38 (10) | 11 (25) |
| Positive | Intermediate | Negative | p-Value | |
|---|---|---|---|---|
| n | 372 | 9 | 1530 | 0.006 |
| Age at testing (Mean, SD) | 58.76 (11.54) | 59.56 (14.29) | 59.56 (14.29) |
| Positive | Non-Positive | p-Value | |
|---|---|---|---|
| n | 47 | 641 | 0.063 |
| Age at testing (Mean, SD) | 57.26 (12.70) | 60.78 (12.35) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesaros, M.; Lenz, S.; Lim, W.; Brown, J.; Drury, L.; Roggenbuck, J. Investigating the Genetic Profile of the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia (ALS-FTD) Continuum in Patients of Diverse Race, Ethnicity and Ancestry. Genes 2022, 13, 76. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13010076
Mesaros M, Lenz S, Lim W, Brown J, Drury L, Roggenbuck J. Investigating the Genetic Profile of the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia (ALS-FTD) Continuum in Patients of Diverse Race, Ethnicity and Ancestry. Genes. 2022; 13(1):76. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13010076
Chicago/Turabian StyleMesaros, Maysen, Steven Lenz, Woobeen Lim, Jordan Brown, Luke Drury, and Jennifer Roggenbuck. 2022. "Investigating the Genetic Profile of the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia (ALS-FTD) Continuum in Patients of Diverse Race, Ethnicity and Ancestry" Genes 13, no. 1: 76. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13010076