
Identification of Compound Heterozygous Variants in LRP4 Demonstrates That a Pathogenic Variant outside the Third β-Propeller Domain Can Cause Sclerosteosis

 , , and
, , and 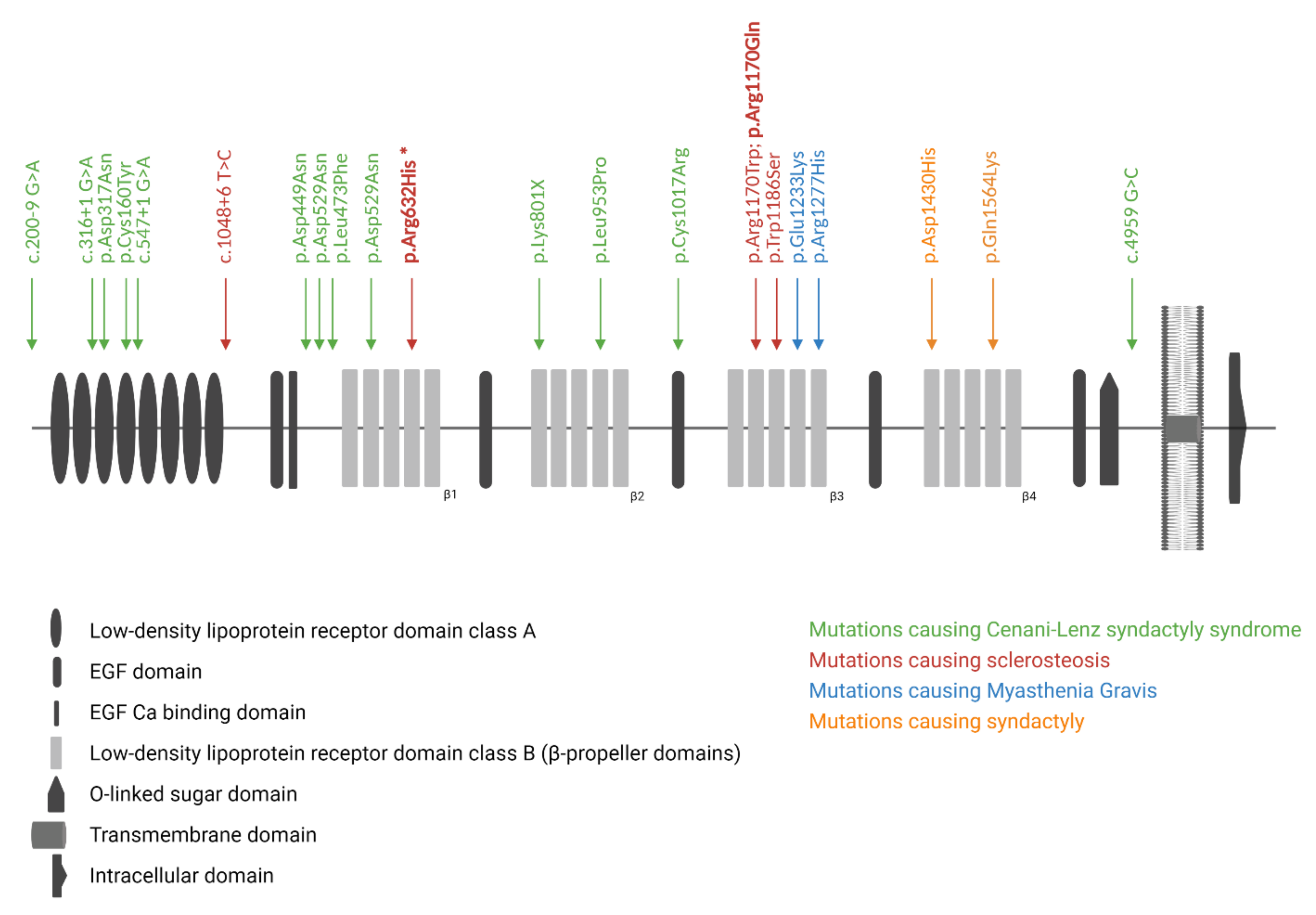{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Material
2.2. Mutation Analysis
2.3. Expression Constructs and In Vitro Mutagenesis
2.4. Luciferase Reporter Assay
2.5. Statistical Analysis
3. Results
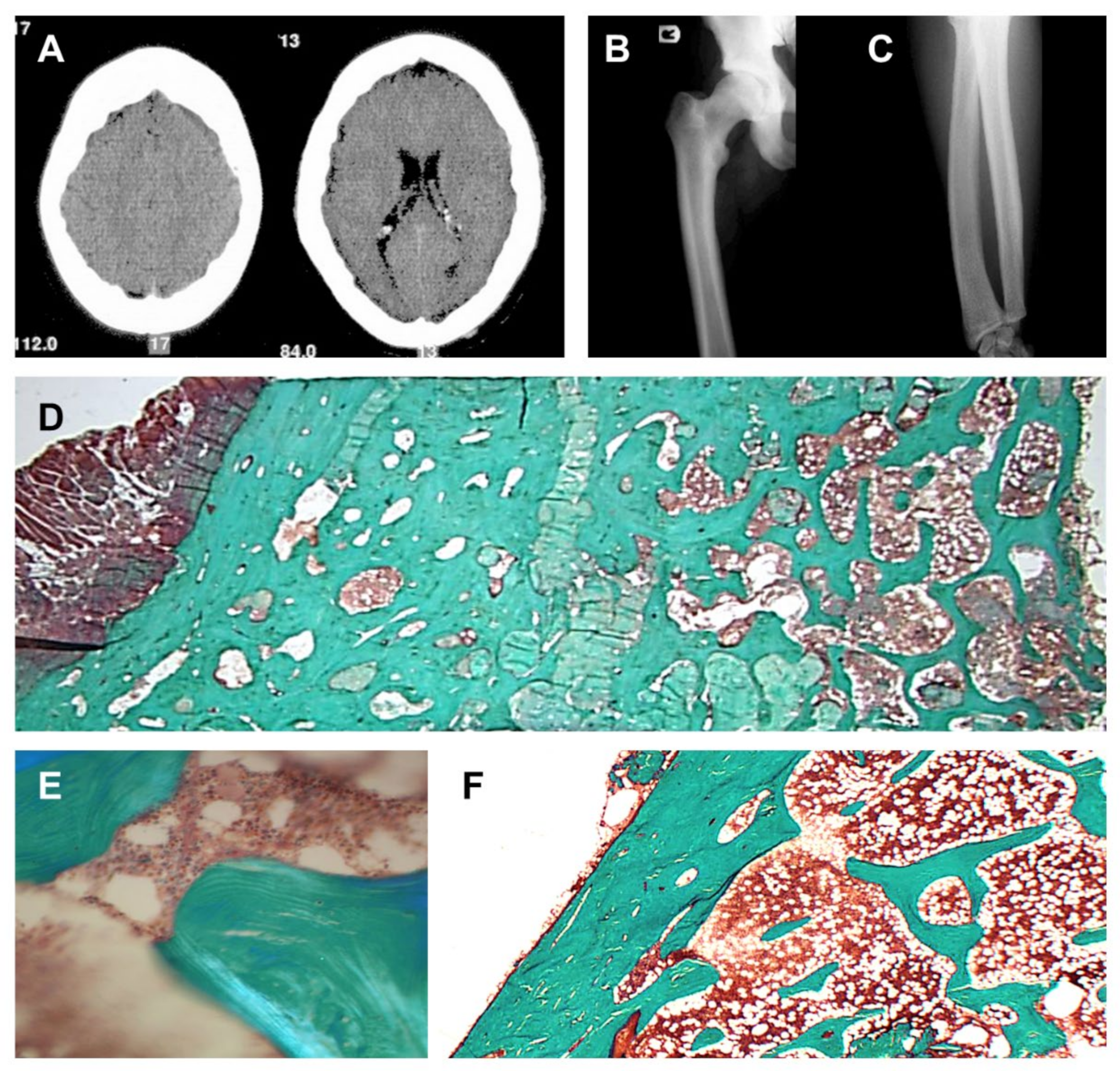
3.1. Clinical Description
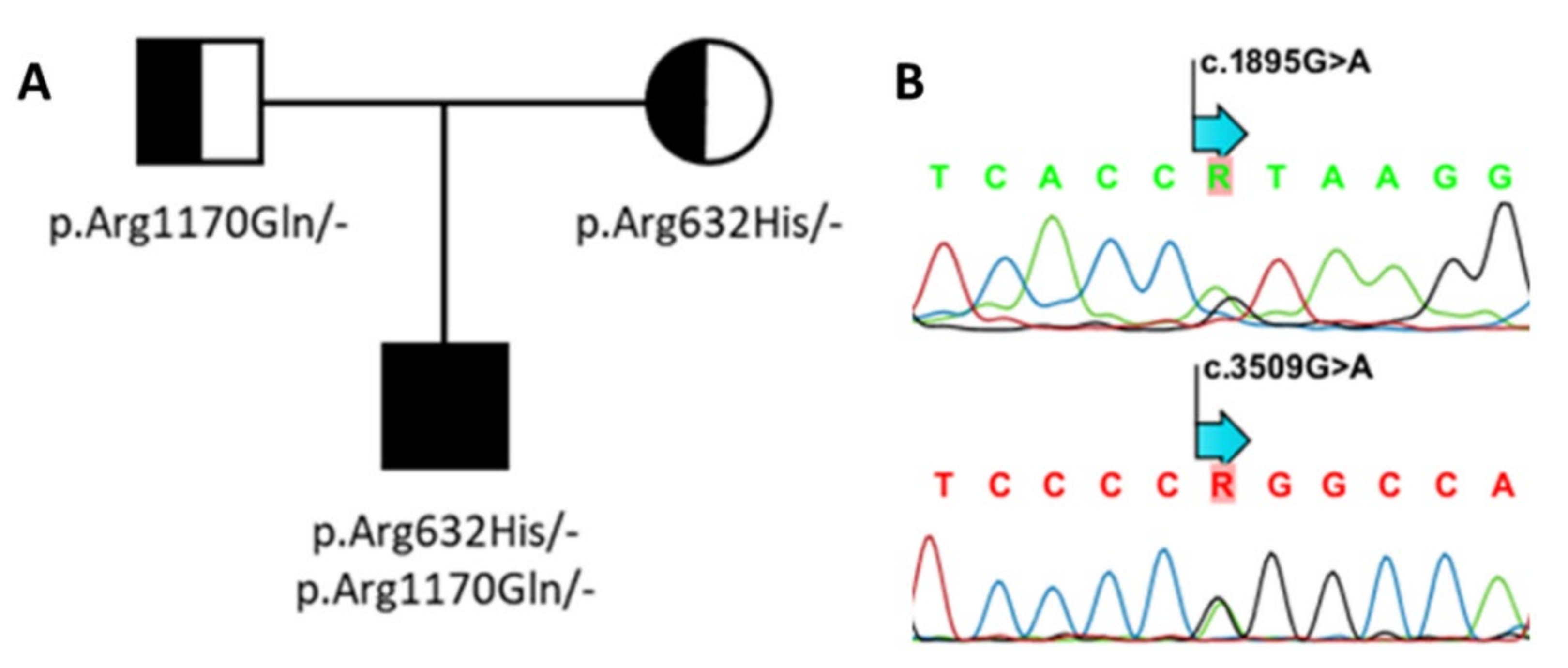
3.2. Genetic Screening of SOST and LRP4
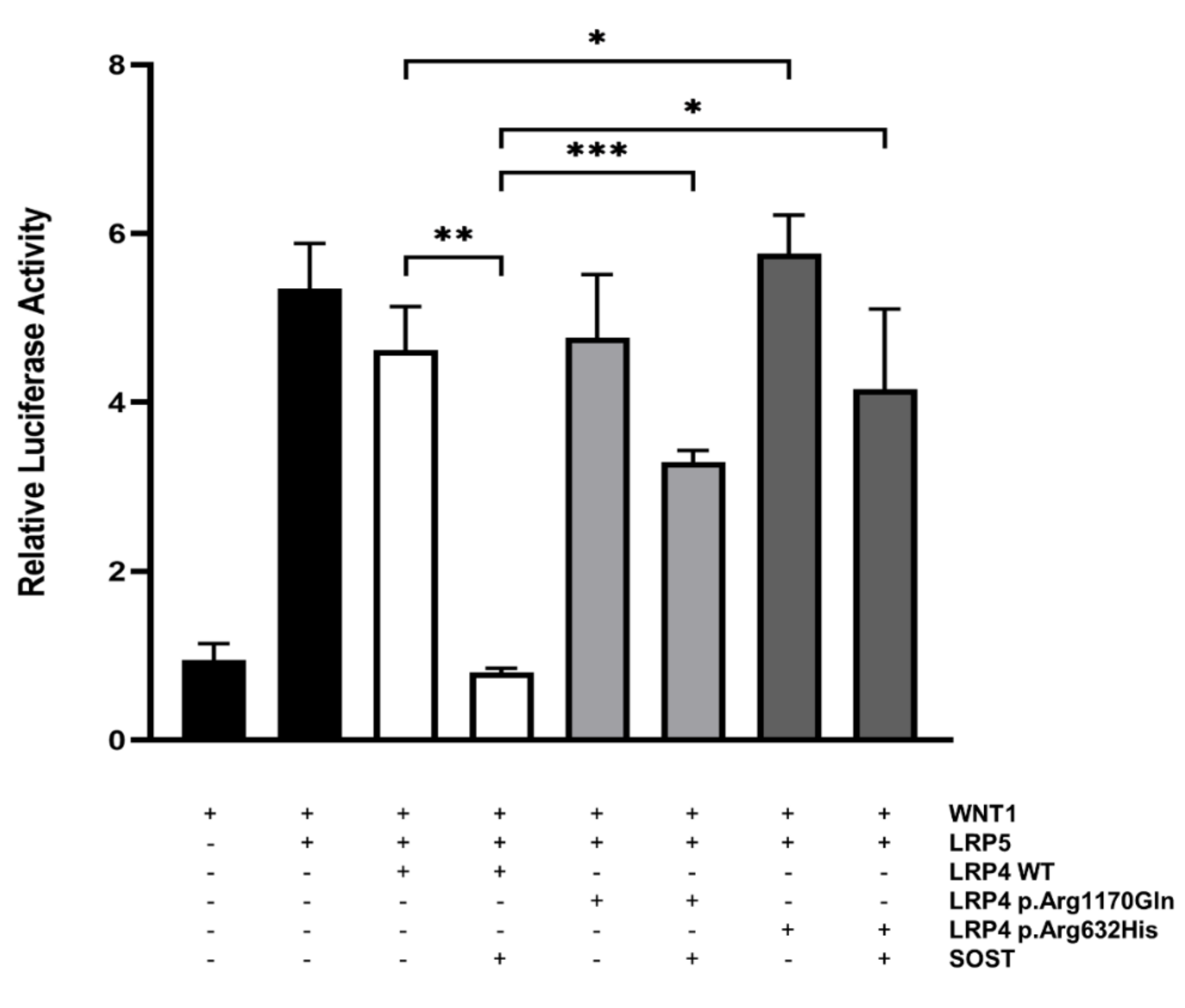
3.3. Functional Evaluation of p.Arg632His LRP4
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beighton, P.; Davidson, J.; Durr, L.; Hamersma, H. Sclerosteosis—An autosomal recessive disorder. Clin. Genet. 1977, 11, 1–7. [Google Scholar] [CrossRef]
- Hamersma, H.; Gardner, J.; Beighton, P. The natural history of sclerosteosis. Clin. Genet. 2003, 63, 192–197. [Google Scholar] [CrossRef]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.; et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Piters, E.; Culha, C.; Moester, M.; Van Bezooijen, R.; Adriaensen, D.; Mueller, T.; Weidauer, S.; Jennes, K.; de Freitas, F.; Löwik, C.; et al. First missense mutation in the SOST gene causing sclerosteosis by loss of sclerostin function. Hum. Mutat. 2010, 31, E1526–E1543. [Google Scholar] [CrossRef] [Green Version]
- Semënov, M.; Tamai, K.; He, X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 2005, 280, 26770–26775. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin Binds to LRP5/6 and Antagonizes Canonical Wnt Signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [Green Version]
- Leupin, O.; Piters, E.; Halleux, C.; Hu, S.; Kramer, I.; Morvan, F.; Bouwmeester, T.; Schirle, M.; Bueno-Lozano, M.; Fuentes, F.J.; et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 2011, 286, 19489–19500. [Google Scholar] [CrossRef] [Green Version]
- Huybrechts, Y.; Mortier, G.; Boudin, E.; Van Hul, W. WNT signaling and bone: Lessons from skeletal dysplasias and disorders. Front. Endocrinol. 2020, 11, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.Y.; Dieckmann, M.; Herz, J.; Niemeier, A. Lrp4, a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo. PLoS ONE 2009, 4, e7930. [Google Scholar] [CrossRef]
- Xiong, L.; Jung, J.-U.; Wu, H.; Xia, W.-F.; Pan, J.-X.; Shen, C.; Mei, L.; Xiong, W.-C. Lrp4 in osteoblasts suppresses bone formation and promotes osteoclastogenesis and bone resorption. Proc. Natl. Acad. Sci. USA 2015, 112, 3487–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holdsworth, G.; Slocombe, P.; Doyle, C.; Sweeney, B.; Veverka, V.; Le Riche, K.; Franklin, R.J.; Compson, J.; Brookings, D.; Turner, J.; et al. Characterization of the interaction of sclerostin with the low density lipoprotein receptor-related protein (LRP) family of Wnt co-receptors. J. Biol. Chem. 2012, 287, 26464–26477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fijalkowski, I.; Geets, E.; Steenackers, E.; Van Hoof, V.; Ramos, F.J.; Mortier, G.; Fortuna, A.M.; Van Hul, W.; Boudin, E. A Novel Domain-Specific Mutation in a Sclerosteosis Patient Suggests a Role of LRP4 as an Anchor for Sclerostin in Human Bone. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2016, 31, 874–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, M.P.; Deepak Amalnath, S.; McAlister, W.H.; Pedapati, R.; Muthupillai, V.; Duan, S.; Huskey, M.; Bijanki, V.N.; Mumm, S. Sclerosteosis: Report of type 1 or 2 in three Indian Tamil families and literature review. Bone 2018, 116, 321–332. [Google Scholar] [CrossRef]
- Ohkawara, B.; Cabrera-Serrano, M.; Nakata, T.; Milone, M.; Asai, N.; Ito, K.; Ito, M.; Masuda, A.; Ito, Y.; Engel, A.G.; et al. LRP4 third beta-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum. Mol. Genet. 2014, 23, 1856–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudin, E.; Yorgan, T.; Fijalkowski, I.; Sonntag, S.; Steenackers, E.; Hendrickx, G.; Peeters, S.; De Maré, A.; Vervaet, B.; Verhulst, A.; et al. The Lrp4R1170Q Homozygous Knock-In Mouse Recapitulates the Bone Phenotype of Sclerosteosis in Humans. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Bullock, W.A.; Hoggatt, A.M.; Horan, D.J.; Elmendorf, A.J.; Sato, A.Y.; Bellido, T.; Loots, G.G.; Pavalko, F.M.; Robling, A.G. Lrp4 Mediates Bone Homeostasis and Mechanotransduction through Interaction with Sclerostin In Vivo. iScience 2019, 20, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukowska-Olech, E.; Sowińska-Seidler, A.; Szczałuba, K.; Jamsheer, A. A novel biallelic splice-site variant in the LRP4 gene causes sclerosteosis 2. Birth Defects Res. 2020, 112, 652–659. [Google Scholar] [CrossRef]
- Li, Y.; Pawlik, B.; Elcioglu, N.; Aglan, M.; Kayserili, H.; Yigit, G.; Percin, F.; Goodman, F.; Nürnberg, G.; Cenani, A.; et al. LRP4 Mutations Alter Wnt/β-Catenin Signaling and Cause Limb and Kidney Malformations in Cenani-Lenz Syndrome. Am. J. Hum. Genet. 2010, 86, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Sukenik Halevy, R.; Chien, H.-C.; Heinz, B.; Bamshad, M.J.; Nickerson, D.A.; Kircher, M.; Ahituv, N.; University of Washington Center for Mendelian Genomics. Mutations in the fourth β-propeller domain of LRP4 are associated with isolated syndactyly with fusion of the third and fourth fingers. Hum. Mutat. 2018, 39, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Chavassieux, P.; Portero-Muzy, N.; Roux, J.-P.; Garnero, P.; Chapurlat, R. Are Biochemical Markers of Bone Turnover Representative of Bone Histomorphometry in 370 Postmenopausal Women? J. Clin. Endocrinol. Metab. 2015, 100, 4662–4668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pangrazio, A.; Boudin, E.; Piters, E.; Damante, G.; Iacono, N.L.; D’Elia, A.V.; Vezzoni, P.; Van Hul, W.; Villa, A.; Sobacchi, C. Identification of the first deletion in the LRP5 gene in a patient with Autosomal Dominant Osteopetrosis type I. Bone 2011, 49, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Coldefy, A.-S.; Hubbard, S.R.; Burden, S.J. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J. Biol. Chem. 2011, 286, 40624–40630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weatherbee, S.D.; Anderson, K.V.; Niswander, L.A. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development 2006, 133, 4993–5000. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huybrechts, Y.; Boudin, E.; Hendrickx, G.; Steenackers, E.; Hamdy, N.; Mortier, G.; Martínez Díaz-Guerra, G.; Bracamonte, M.S.; Appelman-Dijkstra, N.M.; Van Hul, W. Identification of Compound Heterozygous Variants in LRP4 Demonstrates That a Pathogenic Variant outside the Third β-Propeller Domain Can Cause Sclerosteosis. Genes 2022, 13, 80. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13010080
Huybrechts Y, Boudin E, Hendrickx G, Steenackers E, Hamdy N, Mortier G, Martínez Díaz-Guerra G, Bracamonte MS, Appelman-Dijkstra NM, Van Hul W. Identification of Compound Heterozygous Variants in LRP4 Demonstrates That a Pathogenic Variant outside the Third β-Propeller Domain Can Cause Sclerosteosis. Genes. 2022; 13(1):80. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13010080
Chicago/Turabian StyleHuybrechts, Yentl, Eveline Boudin, Gretl Hendrickx, Ellen Steenackers, Neveen Hamdy, Geert Mortier, Guillermo Martínez Díaz-Guerra, Milagros Sierra Bracamonte, Natasha M. Appelman-Dijkstra, and Wim Van Hul. 2022. "Identification of Compound Heterozygous Variants in LRP4 Demonstrates That a Pathogenic Variant outside the Third β-Propeller Domain Can Cause Sclerosteosis" Genes 13, no. 1: 80. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13010080