
Clinical Risk Factors for Aortic Root Dilation in Patients with 22q11.2 Deletion Syndrome: A Longitudinal Single-Center Study
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Statistical Analysis
3. Results
3.1. Patients and Demographics
3.2. Comparison between ARD and Non-ARD 22q11.2DS Patients
3.3. Genetic Aspects
3.4. Multiple Logistic Regression Analysis
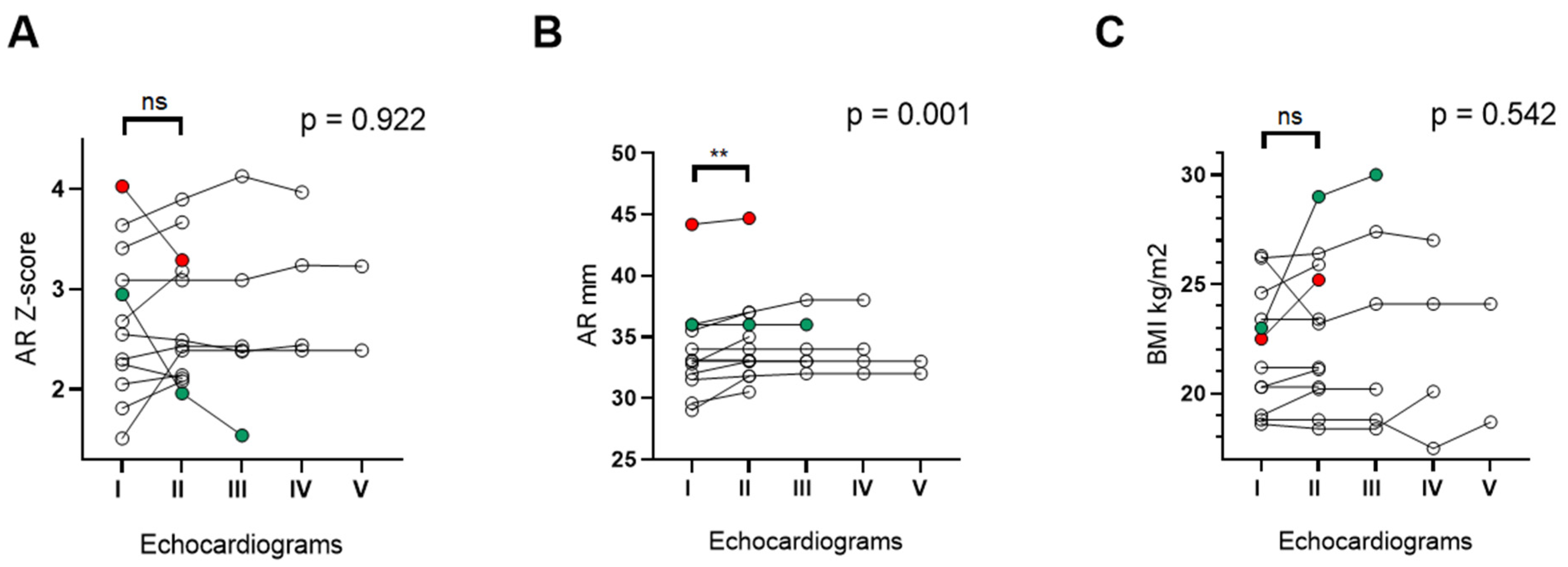
3.5. Changes in Aortic Root Size over Time
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 Deletion Syndrome. Nat. Rev. Dis. Prim. 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald-McGinn, D.M.; LaRossa, D.; Goldmuntz, E.; Sullivan, K.; Eicher, P.; Gerdes, M.; Moss, E.; Wang, P.; Solot, C.; Schultz, P.; et al. The 22q11.2 Deletion: Screening, Diagnostic Workup, and Outcome of Results; Report on 181 Patients. Genet. Test. 1997, 1, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Marino, B.; Digilio, M.C.; Toscano, A.; Anaclerio, S.; Giannotti, A.; Feltri, C.; de Ioris, M.A.; Angioni, A.; Dallapiccola, B. Anatomic patterns of conotruncal defects associated with deletion 22q11. Genet. Med. 2001, 3, 45–48. [Google Scholar] [CrossRef] [Green Version]
- Momma, K. Cardiovascular Anomalies Associated with Chromosome 22q11.2 Deletion Syndrome. Am. J. Cardiol. 2010, 105, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- McElhinney, D.B.; Clark, B.J.; Weinberg, P.M.; Kenton, M.L.; McDonald-McGinn, D.; Driscoll, D.A.; Zackai, E.H.; Goldmuntz, E. Association of Chromosome 22q11 Deletion with Isolated Anomalies of Aortic Arch Laterality and Branching. J. Am. Coll. Cardiol. 2001, 37, 2114–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastromoro, G.; Calcagni, G.; Vignaroli, W.; Anaclerio, S.; Pugnaloni, F.; Rinelli, G.; Secinaro, A.; Bordonaro, V.; Putotto, C.; Unolt, M.; et al. Crossed pulmonary arteries: An underestimated cardiovascular variant with a strong association with genetic syndromes-A report of 74 cases with systematic review of the literature. Am. J. Med. Genet. A 2022, 188, 2351–2359. [Google Scholar] [CrossRef] [PubMed]
- John, A.S.; McDonald-McGinn, D.M.; Zackai, E.H.; Goldmuntz, E. Aortic Root Dilation in Patients With 22q11.2 Deletion Syndrome. Am. J. Med. Genet. A 2009, 149, 939–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, A.S.; Rychik, J.; Khan, M.; Yang, W.; Goldmuntz, E. 22q11.2 Deletion Syndrome as a Risk Factor for Aortic Root Dilation in Tetralogy of Fallot. Cardiol. Young 2014, 24, 303–310. [Google Scholar] [CrossRef]
- de Rinaldis, C.P.; Butensky, A.; Patel, S.; Edman, S.; Wasserman, M.; McGinn, D.E.; Bailey, A.; Zackai, E.H.; Crowley, T.B.; McDonald-McGinn, D.M.; et al. Aortic Root Dilation in Patients with 22q11.2 Deletion Syndrome Without Intracardiac Anomalies. Pediatr. Cardiol. 2021, 42, 1594–1600. [Google Scholar] [CrossRef]
- Lin, A.E.; Basson, C.T.; Goldmuntz, E.; Magoulas, P.L.; McDermott, D.A.; McDonald-McGinn, D.M.; McPherson, E.; Morris, C.A.; Noonan, J.; Nowak, C.; et al. Adults with Genetic Syndromes and Cardiovascular Abnormalities: Clinical History and Management. Genet. Med. 2008, 10, 469–494. [Google Scholar] [CrossRef]
- Yetman, A.T.; Graham, T. The Dilated Aorta in Patients With Congenital Cardiac Defects. J. Am. Coll. Cardiol. 2009, 53, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, K. Aortic Dilatation in Complex Congenital Heart Disease. Cardiovasc. Diagn. 2018, 8, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Lamandé, S.R.; Bateman, J.F. Genetic Disorders of the Extracellular Matrix. Anat. Rec. (Hoboken) 2020, 303, 1527–1542. [Google Scholar] [CrossRef] [Green Version]
- Fung, W.L.A.; Butcher, N.J.; Costain, G.; Andrade, D.M.; Boot, E.; Chow, E.W.C.; Chung, B.; Cytrynbaum, C.; Faghfoury, H.; Fishman, L.; et al. Practical Guidelines for Managing Adults with 22q11.2 Deletion Syndrome. Genet. Med. 2015, 17, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Burnside, R.D. 22q11.21 Deletion Syndromes: A Review of Proximal, Central, and Distal Deletions and Their Associated Features. Cytogenet. Genome Res. 2015, 146, 89–99. [Google Scholar] [CrossRef]
- Mikhail, F.M.; Burnside, R.D.; Rush, B.; Ibrahim, J.; Godshalk, R.; Rutledge, S.L.; Robin, N.H.; Descartes, M.D.; Carroll, A.J. The recurrent distal 22q11.2 microdeletions are often de novo and do not represent a single clinical entity: A proposed categorization system. Genet. Med. 2014, 16, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Lang, R.M.; Badano, L.P.; Victor, M.-A.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for Cardiac Chamber Quantification by Echocardiography in Adults: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 28, e14. [Google Scholar] [CrossRef] [Green Version]
- Devereux, R.B.; de Simone, G.; Arnett, D.K.; Best, L.G.; Boerwinkle, E.; Howard, B.V.; Kitzman, D.; Lee, E.T.; Mosley, T.H.; Weder, A.; et al. Normal Limits in Relation to Age, Body Size and Gender of Two-Dimensional Echocardiographic Aortic Root Dimensions in Persons ≥15 Years of Age. Am. J. Cardiol. 2012, 110, 1189–1194. [Google Scholar] [CrossRef] [Green Version]
- Chiarelli, N.; Ritelli, M.; Zoppi, N.; Colombi, M. Cellular and Molecular Mechanisms in the Pathogenesis of Classical, Vascular, and Hypermobile Ehlers–Danlos Syndromes. Genes 2019, 10, 609. [Google Scholar] [CrossRef] [Green Version]
- Alfano, D.; Altomonte, A.; Cortes, C.; Bilio, M.; Kelly, R.G.; Baldini, A. Tbx1 Regulates Extracellular Matrix-Cell Interactions in the Second Heart Field. Hum. Mol. Genet. 2019, 28, 2295–2308. [Google Scholar] [CrossRef]
- Oppenheimer, A.G.; Fulmer, S.; Shifteh, K.; Chang, J.-K.; Brook, A.; Shanske, A.L.; Shprintzen, R.J. Cervical Vascular and Upper Airway Asymmetry in Velo-Cardio-Facial Syndrome: Correlation of Nasopharyngoscopy with MRA. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stalmans, I.; Lambrechts, D.; De smet, F.; Jansen, S.; Wang, J.; Maity, S.; Kneer, P.; von der Ohe, M.; Swillen, A.; Maes, C.; et al. VEGF: A Modifier of the Del22q11 (DiGeorge) Syndrome? Nat. Med. 2003, 9, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E., Jr.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the Diagnosis and Management of Patients With Thoracic Aortic Disease: Executive Summary. Catheter. Cardiovasc. Interv. 2010, 76, E43–E86. [Google Scholar] [CrossRef] [PubMed]
- Erbel, R.; Aboyans, V.; Boileau, C.; Bossone, E.; Bartolomeo, R.D.; Eggebrecht, H.; Evangelista, A.; Falk, V.; Frank, H.; Gaemperli, O.; et al. 2014 ESC Guidelines on the Diagnosis and Treatment of Aortic Diseases: Document Covering Acute and Chronic Aortic Diseases of the Thoracic and Abdominal Aorta of the AdultThe Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2873–2926. [Google Scholar] [PubMed] [Green Version]
- King, C. Undiagnosed DiGeorge Syndrome Presenting in Middle Age with an Aortic Root Aneurysm and Chronic Dissection. BMJ Case Rep. 2015, 35, bcr2015210697. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, V.; Bella, J.N.; Arnett, D.K.; Roman, M.J.; Oberman, A.; Kitzman, D.W.; Hopkins, P.N.; Paranicas, M.; Rao, D.C.; Devereux, R.B. Aortic Root Dilatation at Sinuses of Valsalva and Aortic Regurgitation in Hypertensive and Normotensive Subjects: The Hypertension Genetic Epidemiology Network Study. Hypertension 2001, 37, 1229–1235. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.-J.; Zeng, Z.; Teoh, K.; Sharma, A.M.; Abouzahr, L.; Cybulsky, I.; Lamy, A.; Semelhago, L.; Lee, R.M.K.W. Perivascular Adipose Tissue Modulates Vascular Function in the Human Internal Thoracic Artery. J. Thorac. Cardiovasc. Surg. 2005, 130, 1130–1136. [Google Scholar] [CrossRef] [Green Version]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef]


{kind=link}
| 22q11.2DS without Major CHD (n = 74) | Non-ARD (n = 50) | ARD (n = 24) | p Value | |
|---|---|---|---|---|
| Male, n (%) | 44 (59.5) | 27 (54) | 17 (70.8) | 0.167 1 |
| Age (years), median (IQR) | 27.5 (23–35) | 26.5 (23–32) | 28.5 (22.5–39) | 0.246 2 |
| BMI (Kg/m²), median (IQR) | 23.7 (20.5–29.1) | 22.2 (21.9–32.3) | 21.45 (20.1–25.3) | 0.005 2 |
| CV risk factors, n (%) | ||||
| Dyslipidemia | 7 (15.6) | 5 (20) | 3 (12.5) | 0.867 1 |
| Hypertension | 11 (14.9) | 8 (19) | 3 (12.5) | 0.715 1 |
| Smoking | 10 (17.8) | 7 (19.4) | 3 (15) | 0.519 1 |
| DMT2 | 3 (4.0) | 2 (4) | 1 (4.1) | 0.601 1 |
| Overweight/obesity | 33 (44.6) | 25 (50) | 8 (33.3) | 0.254 1 |
| Aortic arch/epiaortic vessel anomalies, n (%) | 20 (27) | 9 (18) | 11 (45.8) | 0.012 1 |
| Double aortic arch | 1 (1.4) | 0 | 1 (4.2) | 0.146 1 |
| Right aortic arch (isolated) | 15 (20.3) | 8 (16) | 7 (29.3) | 0.223 1 |
| Right aortic arch with aberrant left subclavian artery | 8 (10.8) | 4 (8) | 4 (16.7) | 0.424 1 |
| Left aortic arch with aberrant right subclavian artery | 5 (6.8) | 1 (2) | 4 (16.7) | 0.019 1 |
| Kommerell diverticulum | 3 (4.1) | 1 (2) | 2 (8.3) | 0.196 1 |
| Vascular ring | 2 (2.7) | 1 (2) | 1 (4.2) | 0.591 1 |
| Crossed pulmonary arteries, n(%) | 19 (25.7) | 10 (20) | 9 (37.5) | 0.107 1 |
| Bicuspid aortic valve, n (%) | 4 (5.4) | 2 (4) | 2 (8.3) | 0.440 1 |
| AAD, n (%) | 19 (25.7) | 6 (12.2) | 13 (54.2) | <0.0001 1 |
| Skeletal/Connective tissue disorders,n (%) | 56 (78.9) | 33 (70.2) | 23 (95.8) | 0.012 1 |
| Scoliosis | 48 (67.6) | 30 (63.8) | 18 (75.0) | 0.341 1 |
| Skeletal anomalies of lower limbs | 28 (40) | 17 (37) | 11 (45.8) | 0.472 1 |
| Skeletal anomalies of upper limbs | 7 (10) | 7 (15.2) | 0 | 0.044 1 |
| Vertebral abnormalities | 7(10) | 3 (6.5) | 4 (16.7) | 0.179 1 |
| Ligamentous laxity | 3 (4.3) | 1 (2.2) | 2 (8.3) | 0.227 1 |
| Variable | ARD | AAD |
|---|---|---|
| Prevalence, n (%) | 24/74 (32.4) | 19/74 (25.7) |
| Degree, n (%) | ||
| Mild | 16/24 (76.7) | 13/19 (68.4) |
| Moderate/severe | 8/24 (33.3) | 6/19 (31.6) |
| Z-score, median (IQR) | 2.43 (2.08–3.01) | 2.7 (2.4–3.2) |
| 22q11.2DS without Major CHD (n = 74) | Non-ARD (n = 50) | ARD (n = 24) | p Value 1 | |||||
|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | |||
| Diagnostic test | FISH | 26 | 35.1% | 19 | 38.0% | 8 | 33.3% | 1.000 |
| SNP-/CGH-array | 47 | 63.5% | 31 | 62.0% | 15 | 62.5% | ||
| MLPA | 1 | 1.4% | 0 | 0.0% | 1 | 4.2% | ||
| Segregation | De novo | 18 | 37.5% | 12 | 37.5% | 6 | 37.5% | 1.000 |
| Parental | 2 | 4.2% | 2 | 6.3% | 0 | 0.0% | ||
| Unknown | 28 | 58.3% | 18 | 56.3% | 10 | 62.5% | ||
| 22q11.2 deleted position | Proximal | 43 | 89.6% | 28 | 87.5% | 15 | 93.8% | 0.593 |
| Central | 2 | 4.2% | 2 | 6.3% | 0 | 0.0% | ||
| Distal | 3 | 6.3% | 2 | 6.3% | 1 | 6.3% | ||
| 22q11.2 deleted size | Typical (2.5–3 Mb) | 35 | 72.9% | 22 | 68.8% | 13 | 81.3% | 0.358 |
| Atypical (<2.5 Mb) | 13 | 27.1% | 10 | 31.3% | 3 | 18.8% | ||
| Additional CNVs | No | 29 | 61.7% | 20 | 62.5% | 9 | 60.0% | 0.777 |
| Yes | 11 | 23.4% | 8 | 25.0% | 3 | 20.0% | ||
| Unknown | 7 | 14.9% | 4 | 12.5% | 3 | 20.0% | ||
| B | p Value | OR | 95% CI per OR | |
|---|---|---|---|---|
| BMI (Kg/m²) | −0.14 | 0.011 | 0.86 | 0.77–0.97 |
| Aortic arch/epiaortic vessel anomalies | 1.11 | 0.076 | 3.02 | 0.89–10.27 |
| Skeletal/Connective tissue disorders | 2.55 | 0.020 | 12.82 | 1.43–115.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putotto, C.; Pulvirenti, F.; Pugnaloni, F.; Isufi, I.; Unolt, M.; Anaclerio, S.; Caputo, V.; Bernardini, L.; Messina, E.; Moretti, C.; et al. Clinical Risk Factors for Aortic Root Dilation in Patients with 22q11.2 Deletion Syndrome: A Longitudinal Single-Center Study. Genes 2022, 13, 2334. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13122334
Putotto C, Pulvirenti F, Pugnaloni F, Isufi I, Unolt M, Anaclerio S, Caputo V, Bernardini L, Messina E, Moretti C, et al. Clinical Risk Factors for Aortic Root Dilation in Patients with 22q11.2 Deletion Syndrome: A Longitudinal Single-Center Study. Genes. 2022; 13(12):2334. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13122334
Chicago/Turabian StylePutotto, Carolina, Federica Pulvirenti, Flaminia Pugnaloni, Ina Isufi, Marta Unolt, Silvia Anaclerio, Viviana Caputo, Laura Bernardini, Elisa Messina, Corrado Moretti, and et al. 2022. "Clinical Risk Factors for Aortic Root Dilation in Patients with 22q11.2 Deletion Syndrome: A Longitudinal Single-Center Study" Genes 13, no. 12: 2334. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13122334