
Molecular Characterization of the miR156/MsSPL Model in Regulating the Compound Leaf Development and Abiotic Stress Response in Alfalfa



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Abiotic Stress Treatment
2.2. RNA Isolation, Illumina Library Construction, and Sequencing
2.3. Bioinformatics Analysis of RNA-Seq Data
2.4. Identification of Alfalfa SPL Genes
2.5. SPL Gene Bioinformatics Analysis in Alfalfa
2.6. Prediction of the miR156 Target Site and Promoter Cis-Element
2.7. Expression Analysis of MsSPLs in Different Tissues
2.8. RNA Isolation and qRT-PCR Verification
2.9. Statistical Analysis
3. Results
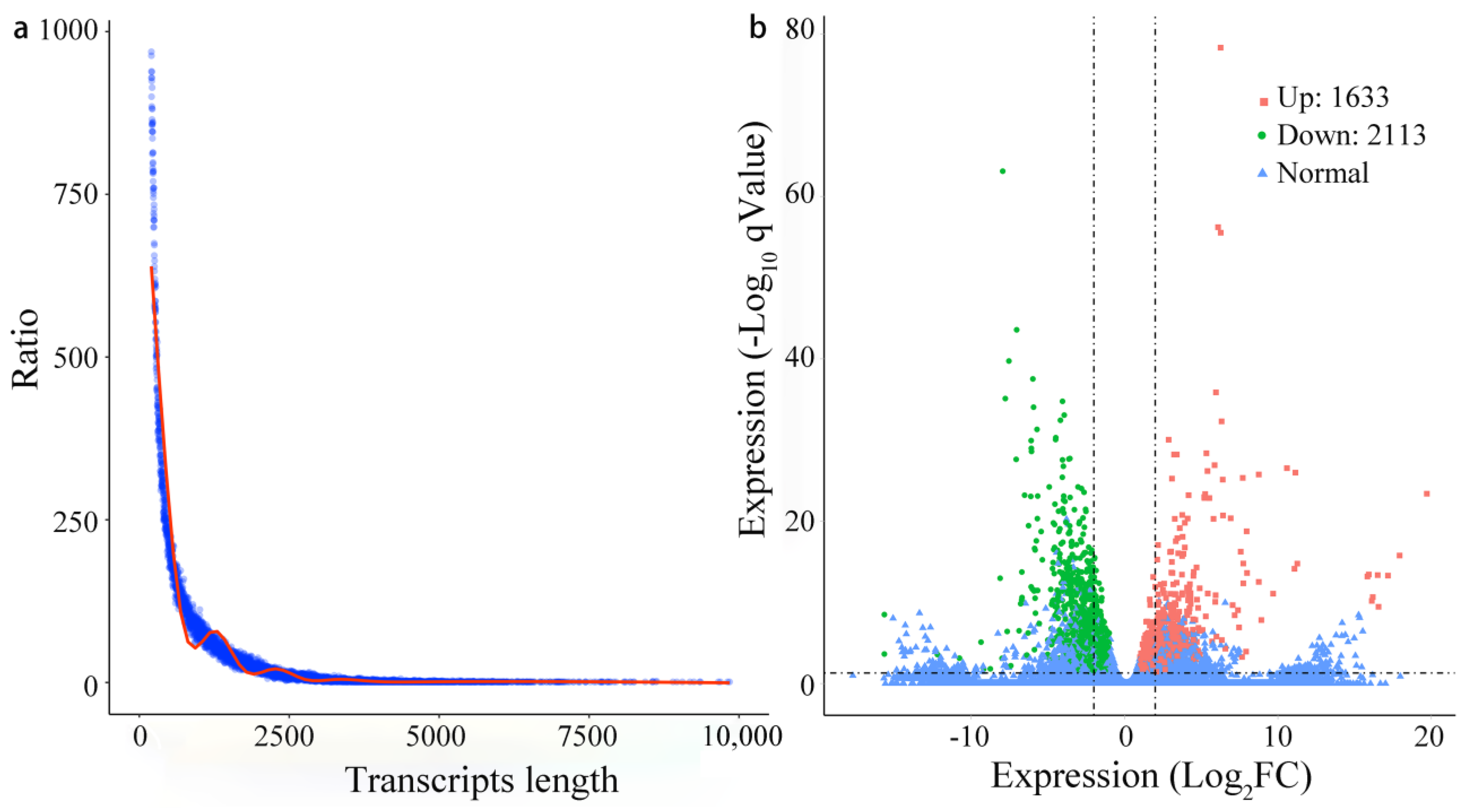
3.1. Illumina Sequencing, Mapping, and Annotation
3.2. Functional Annotation and Transcription Factor Identification
3.3. Identification of MsSPL Genes in Alfalfa
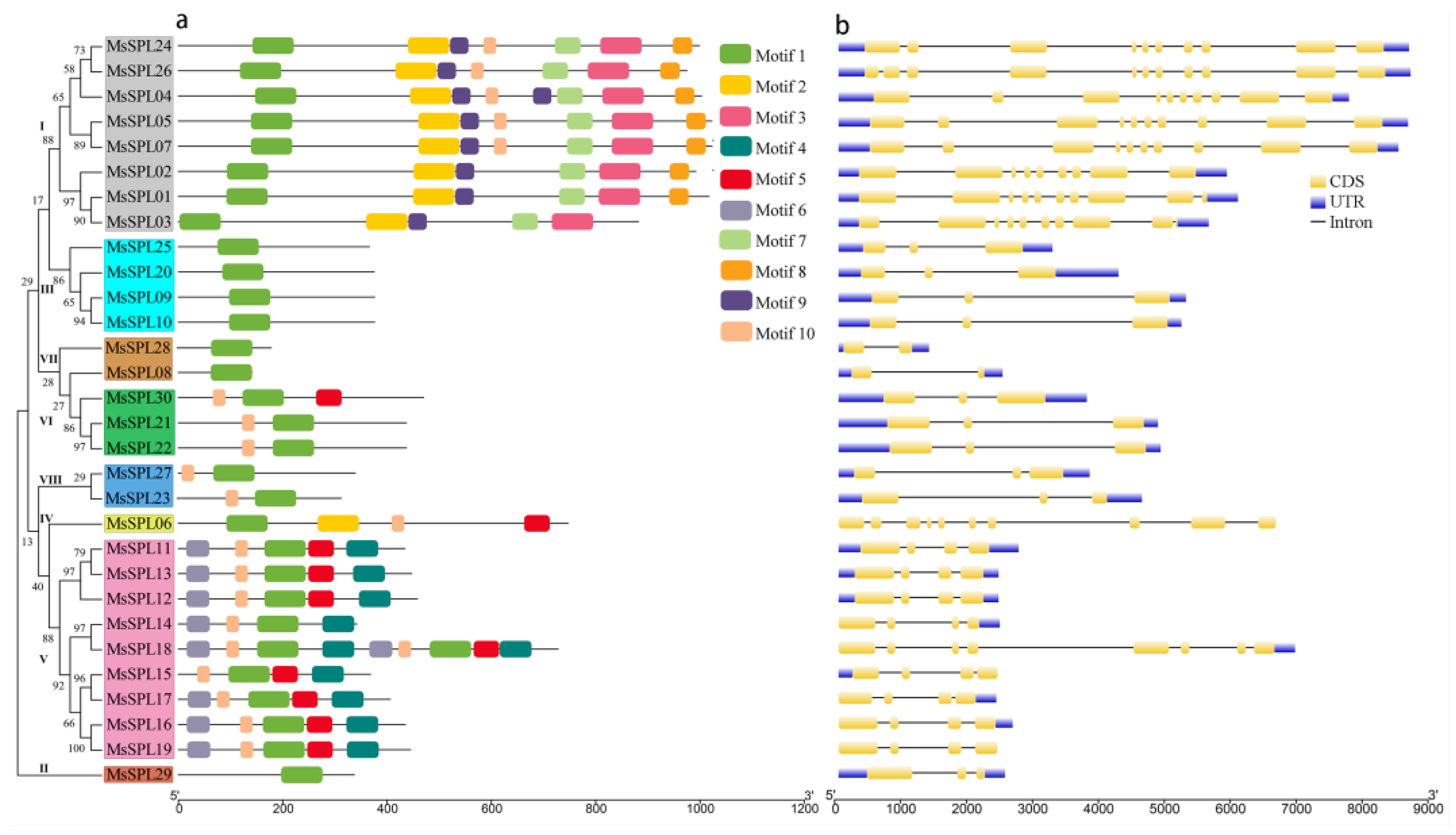
3.4. Phylogenetic, Gene Structure, Conserved Motif, and Domain Analyses
3.5. Multiple Sequence Alignment and Prediction of the miR156 Target Site of MsSPLs
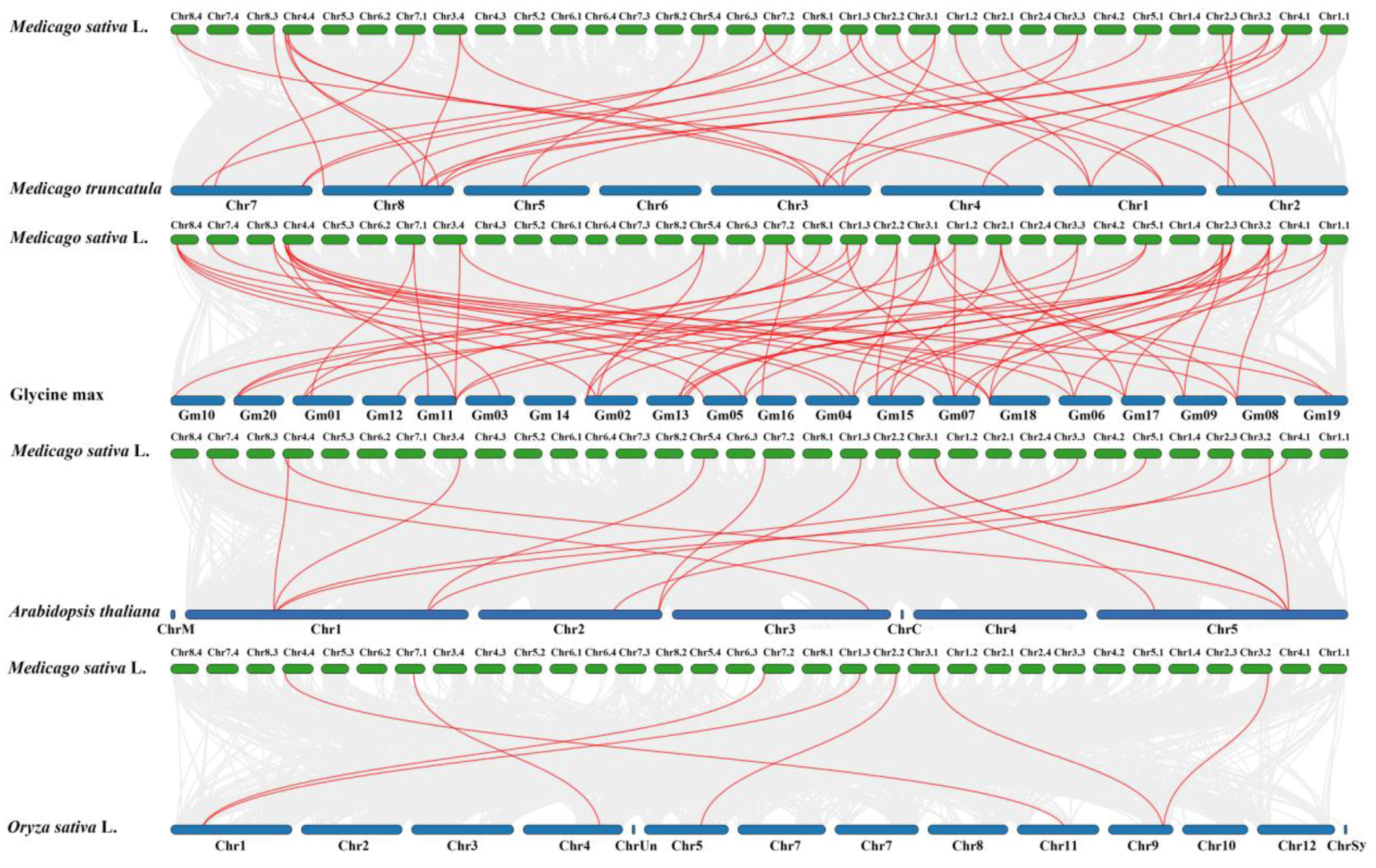
3.6. Chromosomal Distributions, Synteny, and Evolutionary Analyses of the MsSPL
3.7. Cis-element Analysis of the Promoter
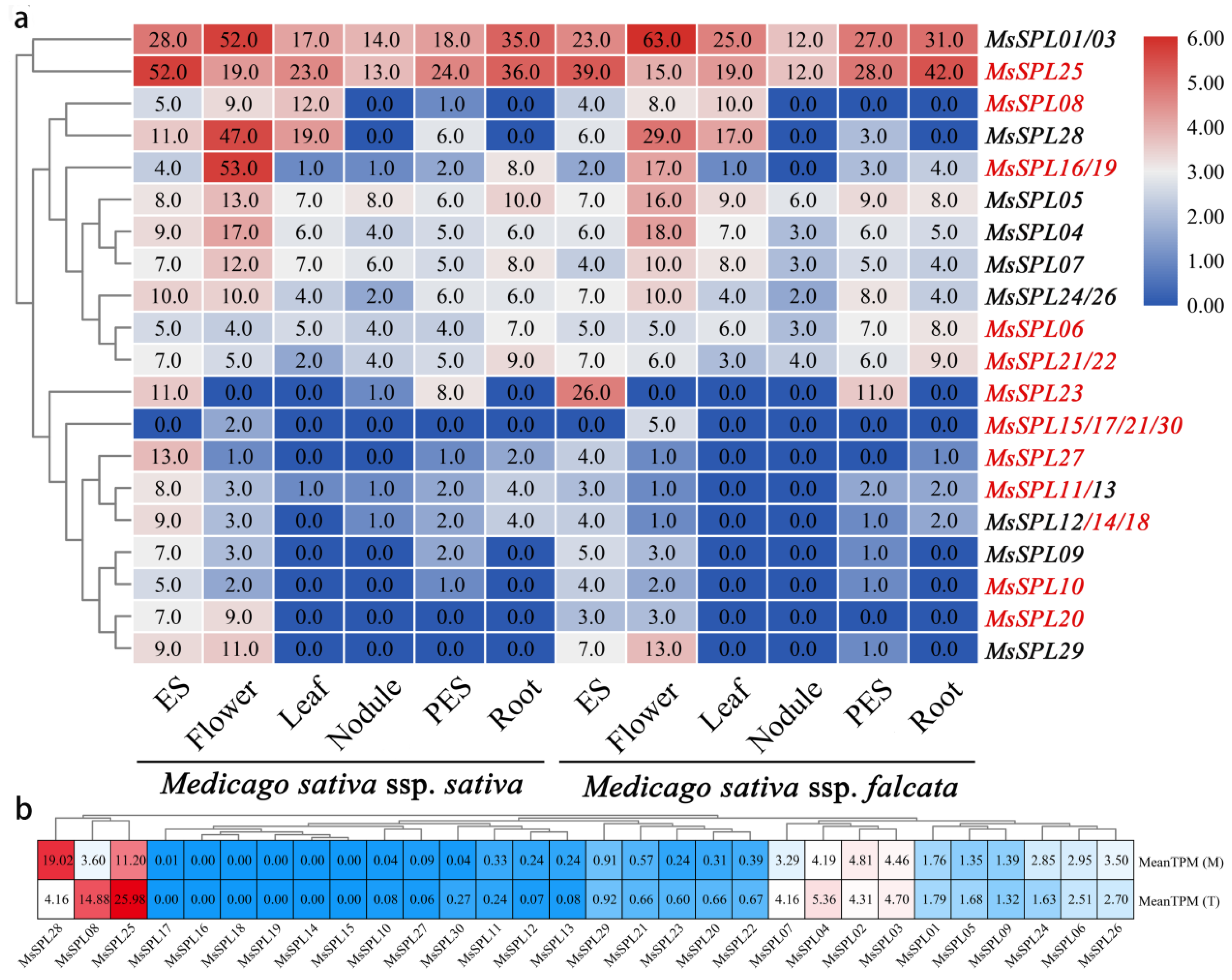
3.8. Transcriptome and qRT-PCR Analysis Revealed Diverse Expression Patterns of MsSPL
3.9. Paralogous of MsSPL08 Isolation and Functional Analysis in M. truncatula
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bourgaud, F.; Gravot, A.; Milesi, S.; Gontier, E. Production of plant secondary metabolites: A historical perspective. Plant Sci. 2001, 161, 839–851. [Google Scholar] [CrossRef]
- Wang, J.-W. The Multifaceted Roles of miR156-targeted SPL Transcription Factors in Plant Developmental Transitions. In Plant Transcription Factors; Elsevier: Amsterdam, The Netherlands, 2016; pp. 281–293. [Google Scholar]
- Warman, L.; Moles, A.; Edwards, W. Not so simple after all: Searching for ecological advantages of compound leaves. Oikos 2010, 120, 813–821. [Google Scholar] [CrossRef]
- Mielmann, A. The utilisation of lucerne (Medicago sativa): A review. Br. Food J. 2013, 115, 590–600. [Google Scholar] [CrossRef]
- Hojilla-Evangelista, M.P.; Selling, G.W.; Hatfield, R.; Digman, M. Extraction, composition, and functional properties of dried alfalfa (Medicago sativa L.) leaf protein. J. Sci. Food Agric. 2017, 97, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zeng, Y.; Yang, Y.; Huang, L.; Tang, B.; Zhang, H.; Hao, F.; Liu, W.; Li, Y.; Liu, Y.; et al. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat. Commun. 2020, 11, 2494. [Google Scholar] [CrossRef]
- He, L.; Liu, Y.; He, H.; Liu, Y.; Qi, J.; Zhang, X.; Li, Y.; Mao, Y.; Zhou, S.; Zheng, X.; et al. A molecular framework underlying the compound leaf pattern of Medicago truncatula. Nat. Plants 2020, 6, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Birkenbihl, R.P.; Jach, G.; Saedler, H.; Huijser, P. Functional Dissection of the Plant-specific SBP-Domain: Overlap of the DNA-binding and Nuclear Localization Domains. J. Mol. Biol. 2005, 352, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Hobert, O. Gene Regulation by Transcription Factors and MicroRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef]
- Yamasaki, K.; Kigawa, T.; Inoue, M.; Tateno, M.; Yamasaki, T.; Yabuki, T.; Aoki, M.; Seki, E.; Matsuda, T.; Nunokawa, E.; et al. A Novel Zinc-binding Motif Revealed by Solution Structures of DNA-binding Domains of Arabidopsis SBP-family Transcription Factors. J. Mol. Biol. 2004, 337, 49–63. [Google Scholar] [CrossRef]
- Kropat, J.; Tottey, S.; Birkenbihl, R.P.; Depège, N.; Huijser, P.; Merchant, S. A regulator of nutritional copper signaling in Chlamydomonas is an SBP domain protein that recognizes the GTAC core of copper response element. Proc. Natl. Acad. Sci. USA 2005, 102, 18730–18735. [Google Scholar] [CrossRef] [Green Version]
- Riese, M.; Höhmann, S.; Saedler, H.; Münster, T.; Huijser, P. Comparative analysis of the SBP-box gene families in P. patens and seed plants. Gene 2007, 401, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Guo, W.; Zhang, B. Genome-wide identification and characterization of SPL transcription factor family and their evolution and expression profiling analysis in cotton. Sci. Rep. 2018, 8, 762. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.; Feyissa, B.A.; Amyot, L.; Aung, B.; Hannoufa, A. MicroRNA156 improves drought stress tolerance in alfalfa (Medicago sativa) by silencing SPL13. Plant Sci. 2017, 258, 122–136. [Google Scholar] [CrossRef]
- Aung, B.; Gruber, M.Y.; Amyot, L.; Omari, K.W.; Bertrand, A.; Hannoufa, A. Ectopic expression of LjmiR156 delays flowering, enhances shoot branching, and improves forage quality in alfalfa. Plant Biotechnol. Rep. 2015, 9, 379–393. [Google Scholar] [CrossRef]
- Aung, B.; Gruber, M.Y.; Amyot, L.; Omari, K.; Bertrand, A.; Hannoufa, A. MicroRNA156 as a promising tool for alfalfa improvement. Plant Biotechnol. J. 2015, 13, 779–790. [Google Scholar] [CrossRef]
- E Silva, G.F.F.; Silva, E.M.; da Silva Azevedo, M.; Guivin, M.A.C.; Ramiro, D.A.; Figueiredo, C.R.; Carrer, H.; Peres, L.E.P.; Nogueira, F.T.S. microRNA156-targeted SPL/SBP box transcription factors regulate tomato ovary and fruit development. Plant J. 2014, 78, 604–618. [Google Scholar] [CrossRef]
- Liu, N.; Tu, L.; Wang, L.; Hu, H.; Xu, J.; Zhang, X. MicroRNA 157-targeted SPL genes regulate floral organ size and ovule production in cotton. BMC Plant Biol. 2017, 17, 7. [Google Scholar] [CrossRef] [Green Version]
- Yu, N.; Cai, W.-J.; Wang, S.; Shan, C.-M.; Wang, L.-J.; Chen, X.-Y. Temporal Control of Trichome Distribution by MicroRNA156-Targeted SPL Genes in Arabidopsis thaliana. Plant Cell 2010, 22, 2322–2335. [Google Scholar] [CrossRef] [Green Version]
- Cardon, G.H.; Hohmann, S.; Nettesheim, K.; Saedler, H.; Huijser, P. Functional analysis of the Arabidopsis thaliana SBP-box gene SPL3: A novel gene involved in the floral transition. Plant J. 1997, 12, 367–377. [Google Scholar] [CrossRef]
- Gandikota, M.; Birkenbihl, R.P.; Höhmann, S.; Cardon, G.H.; Saedler, H.; Huijser, P. The miRNA156/157 recognition element in the 3′ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 2007, 49, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Hu, T.; Zhao, J.; Park, M.-Y.; Earley, K.W.; Wu, G.; Yang, L.; Poethig, R.S. Developmental Functions of miR156-Regulated SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) Genes in Arabidopsis thaliana. PLoS Genet. 2016, 12, e1006263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, L.; Chen, J.; Huang, X.; Gong, H.; Luo, J.; Hou, Q.; Zhou, T.; Lu, T.; Zhu, J.; Shangguan, Y.; et al. OsSPL13 controls grain size in cultivated rice. Nat. Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wang, Y.; Xue, D.; Wang, J.; Yan, M.; Liu, G.; Dong, G.; Zeng, D.; Lu, Z.; Zhu, X.; et al. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 2010, 42, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Austin, R.S.; Amyot, L.; Hannoufa, A. Comparative transcriptome investigation of global gene expression changes caused by miR156 overexpression in Medicago sativa. BMC Genom. 2016, 17, 658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadege, M.; Wen, J.; He, J.; Tu, H.; Kwak, Y.; Eschstruth, A.; Cayrel, A.; Endre, G.; Zhao, P.X.; Chabaud, M.; et al. Large-scale insertional mutagenesis using the Tnt1 retrotransposon in the model legume Medicago truncatula. Plant J. 2008, 54, 335–347. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Blanc, G.; Wolfe, K. Widespread Paleopolyploidy in Model Plant Species Inferred from Age Distributions of Duplicate Genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [Green Version]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- O’Rourke, J.A.; Fu, F.; Bucciarelli, B.; Yang, S.S.; Samac, D.A.; Lamb, J.F.S.; Monteros, M.J.; Graham, M.A.; Gronwald, J.W.; Krom, N.; et al. The Medicago sativa gene index 1.2: A web-accessible gene expression atlas for investigating expression differences between Medicago sativa subspecies. BMC Genom. 2015, 16, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Min, X.; Jin, X.; Zhang, Z.; Wei, X.; Ndayambaza, B.; Wang, Y.; Liu, W. Genome-Wide Identification of NAC Transcription Factor Family and Functional Analysis of the Abiotic Stress-Responsive Genes in Medicago sativa L. J. Plant Growth Regul. 2019, 39, 324–337. [Google Scholar] [CrossRef]
- Zhu, H.; Choi, H.-K.; Cook, D.R.; Shoemaker, R.C. Bridging Model and Crop Legumes through Comparative Genomics. Plant Physiol. 2005, 137, 1189–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornacchione, M.V.; Suarez, D.L. Evaluation of Alfalfa (Medicago sativa L.) Populations’ Response to Salinity Stress. Crop Sci. 2016, 57, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Bar, M.; Ori, N. Leaf development and morphogenesis. Development 2014, 141, 4219–4230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaño-Pachón, D.M.; Ruzicic, S.; Dreyer, I.; Mueller-Roeber, B. PlnTFDB: An integrative plant transcription factor database. BMC Bioinform. 2007, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Jeyakumar, J.M.J.; Ali, A.; Wang, W.-M.; Thiruvengadam, M. Characterizing the Role of the miR156-SPL Network in Plant Development and Stress Response. Plants 2020, 9, 1206. [Google Scholar] [CrossRef]
- Wang, H.; Lu, Z.; Xu, Y.; Kong, L.; Shi, J.; Liu, Y.; Fu, C.; Wang, X.; Wang, Z.-Y.; Zhou, C.; et al. Genome-wide characterization of SPL family in Medicago truncatula reveals the novel roles of miR156/SPL module in spiky pod development. BMC Genom. 2019, 20, 552. [Google Scholar] [CrossRef]
- Preston, J.C.; Hileman, L.C. Functional Evolution in the Plant SQUAMOSA-PROMOTER BINDING PROTEIN-LIKE (SPL) Gene Family. Front. Plant Sci. 2013, 4, 80. [Google Scholar] [CrossRef] [Green Version]
- Kavas, M.; Kizildoğan, A.K.; Abanoz, B. Comparative genome-wide phylogenetic and expression analysis of SBP genes from potato (Solanum tuberosum). Comput. Biol. Chem. 2017, 67, 131–140. [Google Scholar] [CrossRef]
- Tripathi, R.K.; Goel, R.; Kumari, S.; Dahuja, A. Genomic organization, phylogenetic comparison, and expression profiles of the SPL family genes and their regulation in soybean. Dev. Genes Evol. 2017, 227, 101–119. [Google Scholar] [CrossRef]
- Cardon, G.; Höhmann, S.; Klein, J.; Nettesheim, K.; Saedler, H.; Huijser, P. Molecular characterisation of the Arabidopsis SBP-box genes. Gene 1999, 237, 91–104. [Google Scholar] [CrossRef]
- Xie, K.; Wu, C.; Xiong, L. Genomic organization, differential expression, and interaction of SQUAMOSA promoter-binding-like transcription factors and microRNA156 in rice. Plant Physiol. 2006, 142, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, F.; Guan, C.; Jiao, Y. Molecular Mechanisms of Leaf Morphogenesis. Mol. Plant 2018, 11, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, M.; Ori, N. Compound leaf development in model plant species. Curr. Opin. Plant Biol. 2015, 23, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xue, H.; Zhang, F.; Jiang, Q.; Yang, S.; Yue, P.; Wang, F.; Zhang, Y.; Li, L.; He, P.; et al. The miR156/SPL module regulates apple salt stress tolerance by activating MdWRKY100 expression. Plant Biotechnol. J. 2020, 19, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Ye, M.; Sang, M.; Wu, R. A Regulatory Network for miR156-SPL Module in Arabidopsis thaliana. Int. J. Mol. Sci. 2019, 20, 6166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feyissa, B.A.; Arshad, M.; Gruber, M.Y.; Kohalmi, S.E.; Hannoufa, A. The interplay between miR156/SPL13 and DFR/WD40–1 regulate drought tolerance in alfalfa. BMC Plant Biol. 2019, 19, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Feyissa, B.A.; Renaud, J.; Nasrollahi, V.; Kohalmi, S.E.; Hannoufa, A. Transcriptome-IPMS analysis reveals a tissue-dependent miR156/SPL13 regulatory mechanism in alfalfa drought tolerance. BMC Genom. 2020, 21, 1–18. [Google Scholar] [CrossRef]
- Matthews, C.; Arshad, M.; Hannoufa, A. Alfalfa response to heat stress is modulated by microRNA156. Physiol. Plant. 2019, 165, 830–842. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Libraries | Raw Data | Clean Data | Mapped Reads (M) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Raw Reads | Raw Bases (Mbp) | Clean Reads | Clean Bases (Mbp) | Average Read Length (bp) | Q30 Bases Ratio (%) | GC Count (%) | Total Mapped (%) | Mutiple Mapped (%) | Uniquely Mapped (%) | |
| T1 | 40,751,582 | 6112.74 | 3,9151,710 | 5738.75 | 146.58 | 93.78% | 44.99% | 90.10% | 54.27% | 35.83% |
| T2 | 41,100,934 | 6165.14 | 3,9622,384 | 5650.63 | 142.61 | 97.19% | 44.17% | 91.42% | 57.70% | 33.72% |
| T3 | 39,494,852 | 5924.23 | 3,7603,146 | 5377.32 | 143 | 92.58% | 43.84% | 93.24% | 58.61% | 34.63% |
| M1 | 44,693,794 | 6704.07 | 4,2885,164 | 6120.67 | 142.72 | 92.96% | 44.52% | 91.86% | 59.18% | 32.68% |
| M2 | 43,172,140 | 6475.82 | 4,1189,956 | 5880.61 | 142.77 | 92.70% | 44.15% | 94.86% | 61.61% | 33.25% |
| M3 | 46,502,916 | 6975.44 | 4,4872,238 | 6447.54 | 143.69 | 94.16% | 45.09% | 89.87% | 57.35% | 32.52% |
| Gene Name | Accession ID | Protein Length (aa) | Protein MW (kDa) | Protein GRAVY a | pIb | Arabidopsis Ortholog Locus | M. truncatula Ortholog Locus |
|---|---|---|---|---|---|---|---|
| MsSPL01 | MS.gene026366.t1 | 1017 | 113.32 | −0.558 | 7.71 | ATSPL14 | Medtr1g035010.1 |
| MsSPL02 | MS.gene040800.t1 | 992 | 110.44 | −0.571 | 7.61 | ATSPL14 | Medtr1g035010.1 |
| MsSPL03 | MS.gene061710.t1 | 882 | 97.83 | −0.564 | 7.52 | ATSPL14 | Medtr1g035010.1 |
| MsSPL04 | MS.gene20896.t1 | 1003 | 111.77 | −0.479 | 6.28 | ATSPL1 | Medtr1g086250.1 |
| MsSPL05 | MS.gene98509.t1 | 1025 | 113.06 | −0.356 | 7.12 | ATSPL12 | Medtr2g046550.1 |
| MsSPL06 | MS.gene96180.t1 | 747 | 83.81 | −0.267 | 6.75 | ATSPL7 | Medtr2g020620.1 |
| MsSPL07 | MS.gene062030.t1 | 1025 | 113.03 | −0.359 | 7.28 | ATSPL12 | Medtr2g046550.1 |
| MsSPL08 | MS.gene047335.t1 | 141 | 16.58 | −1.294 | 7.2 | ATSPL3 | Medtr2g014200.1 |
| MsSPL09 | MS.gene06231.t1 | 376 | 41.68 | −0.679 | 7.03 | ATSPL13B | Medtr3g099080.1 |
| MsSPL10 | MS.gene055507.t1 | 376 | 41.65 | −0.655 | 7.09 | ATSPL13B | Medtr3g099080.1 |
| MsSPL11 | MS.gene75620.t1 | 434 | 48.02 | −0.832 | 8.61 | ATSPL2 | Medtr3g085180.1 |
| MsSPL12 | MS.gene77585.t1 | 458 | 50.72 | −0.772 | 8.54 | ATSPL2 | Medtr3g085180.1 |
| MsSPL13 | MS.gene045290.t1 | 447 | 49.63 | −0.828 | 8.62 | AT SPL2 | Medtr3g085180.1 |
| MsSPL14 | MS.gene09220.t1 | 342 | 38.34 | −0.796 | 8.44 | ATSPL11 | Medtr8g080690.1 |
| MsSPL15 | MS.gene09219.t1 | 368 | 41.73 | −0.767 | 7.88 | AT SPL10 | Medtr8g080680.1 |
| MsSPL16 | MS.gene09218.t1 | 435 | 48.61 | −0.627 | 5.31 | ATSPL11 | Medtr8g080670.1 |
| MsSPL17 | MS.gene023425.t1 | 406 | 46.03 | −0.768 | 7.35 | ATSPL10 | Medtr8g080680.1 |
| MsSPL18 | MS.gene09027.t1 | 728 | 82.29 | −0.775 | 8.33 | AT SPL2 | Medtr8g080680.1 |
| MsSPL19 | MS.gene09028.t1 | 445 | 49.57 | −0.637 | 5.73 | ATSPL2 | Medtr8g080670.1 |
| MsSPL20 | MS.gene030453.t1 | 375 | 41.52 | −0.788 | 8.57 | ATSPL13B | Medtr8g096780.1 |
| MsSPL21 | MS.gene071085.t1 | 437 | 49.48 | −0.707 | 6.6 | AtSPL6 | Medtr5g046670.1 |
| MsSPL22 | MS.gene029630.t1 | 437 | 49.45 | −0.706 | 6.55 | AtSPL6 | Medtr5g046670.1 |
| MsSPL23 | MS.gene36181.t1 | 314 | 34.69 | −0.725 | 8.55 | ATSPL5 | Medtr7g444860.1 |
| MsSPL24 | MS.gene28650.t1 | 999 | 110.7 | −0.393 | 6.08 | ATSPL1 | Medtr7g110320.1 |
| MsSPL25 | MS.gene99828.t1 | 366 | 40.84 | −0.846 | 8.62 | ATSPL13B | Medtr7g028740.1 |
| MsSPL26 | MS.gene072658.t1 | 975 | 107.92 | −0.407 | 6.38 | ATSPL1 | Medtr7g110320.1 |
| MsSPL27 | MS.gene04608.t1 | 339 | 36.68 | −0.707 | 8.92 | AtSPL9 | Medtr7g092930.1 |
| MsSPL28 | MS.gene36036.t1 | 180 | 20.58 | −1.187 | 9.2 | AT SPL4 | Medtr8g463140.1 |
| MsSPL29 | MS.gene000259.t1 | 337 | 37.43 | −0.971 | 8.61 | AT SPL8 | Medtr8g005960.1 |
| MsSPL30 | MS.gene063508.t1 | 470 | 53.66 | −0.564 | 6.54 | AtSPL6 | Medtr4g109770.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, X.; Luo, K.; Liu, W.; Zhou, K.; Li, J.; Wei, Z. Molecular Characterization of the miR156/MsSPL Model in Regulating the Compound Leaf Development and Abiotic Stress Response in Alfalfa. Genes 2022, 13, 331. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020331
Min X, Luo K, Liu W, Zhou K, Li J, Wei Z. Molecular Characterization of the miR156/MsSPL Model in Regulating the Compound Leaf Development and Abiotic Stress Response in Alfalfa. Genes. 2022; 13(2):331. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020331
Chicago/Turabian StyleMin, Xueyang, Kai Luo, Wenxian Liu, Keyou Zhou, Junyi Li, and Zhenwu Wei. 2022. "Molecular Characterization of the miR156/MsSPL Model in Regulating the Compound Leaf Development and Abiotic Stress Response in Alfalfa" Genes 13, no. 2: 331. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020331