
Investigation of Genetic Causes in Patients with Congenital Heart Disease in Qatar: Findings from the Sidra Cardiac Registry

 , and
, and
Abstract
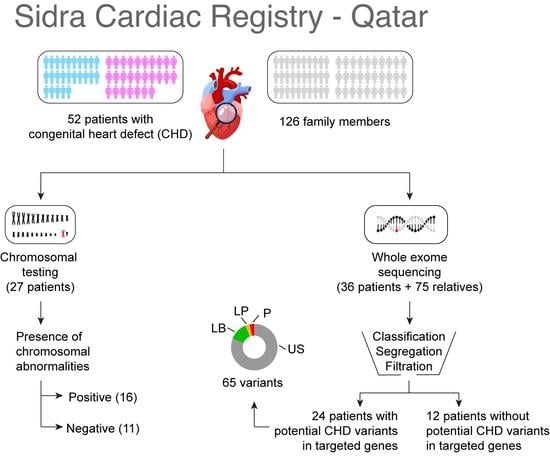
:
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Study Participants and Data Collection
2.3. Whole Exome Sequencing (WES)
2.4. Sequence Quality, Alignment, and Variant Calling
- (a)
- Allele frequencies, obtained from the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/) (accessed on 1 January 2022), Qatar 1000 Genomes [20,21], and Greater Middle East (GME) Variome Project (http://igm.ucsd.edu/gme/) (accessed on 1 January 2022) [22].
- (b)
- Variant pathogenicity prediction scores, including Sorting Intolerant From Tolerant (SIFT), Polymorphism Phenotyping (PolyPhen), and Combined Annotation Dependent Depletion (CADD).
- (c)
- Variant phenotypic and abnormality-related information using the available databases, mainly the Human Gene Mutation Database (HGMD®), ClinVar (https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/) (accessed on 1 January 2022) and Genome-Wide Association Study (GWAS) Catalog.
- (d)
- Information on the phenotypic abnormalities of the captured genes, obtained from Human Phenotype Ontology (HPO|) (https://hpo.jax.org/app/) (accessed on 1 January 2022) [23].
2.5. Single-Nucleotide Variant (SNV) Segregation Analysis and Filtration
2.6. Statistical Analysis
3. Results
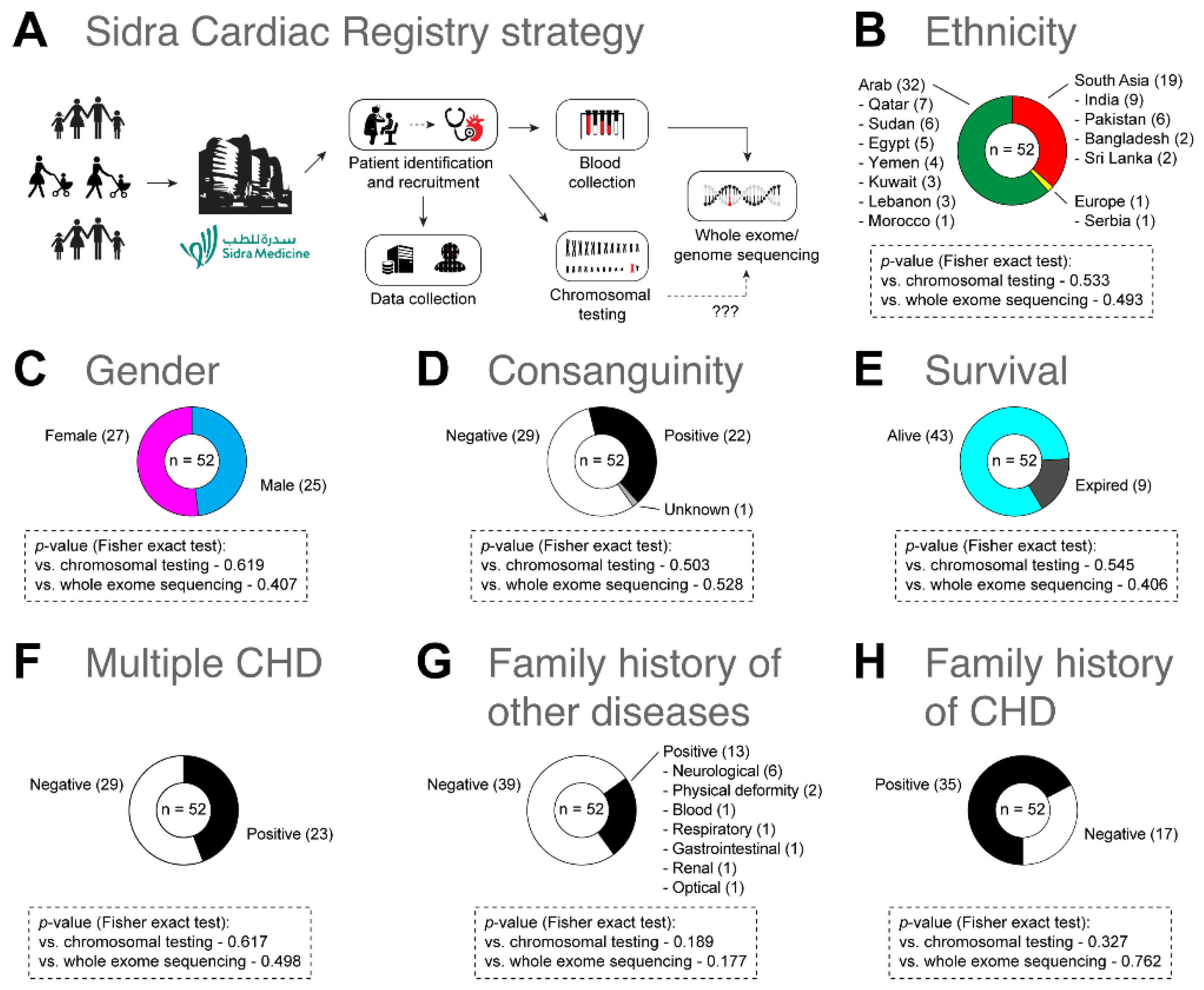
3.1. Demographic and Clinical Characteristics
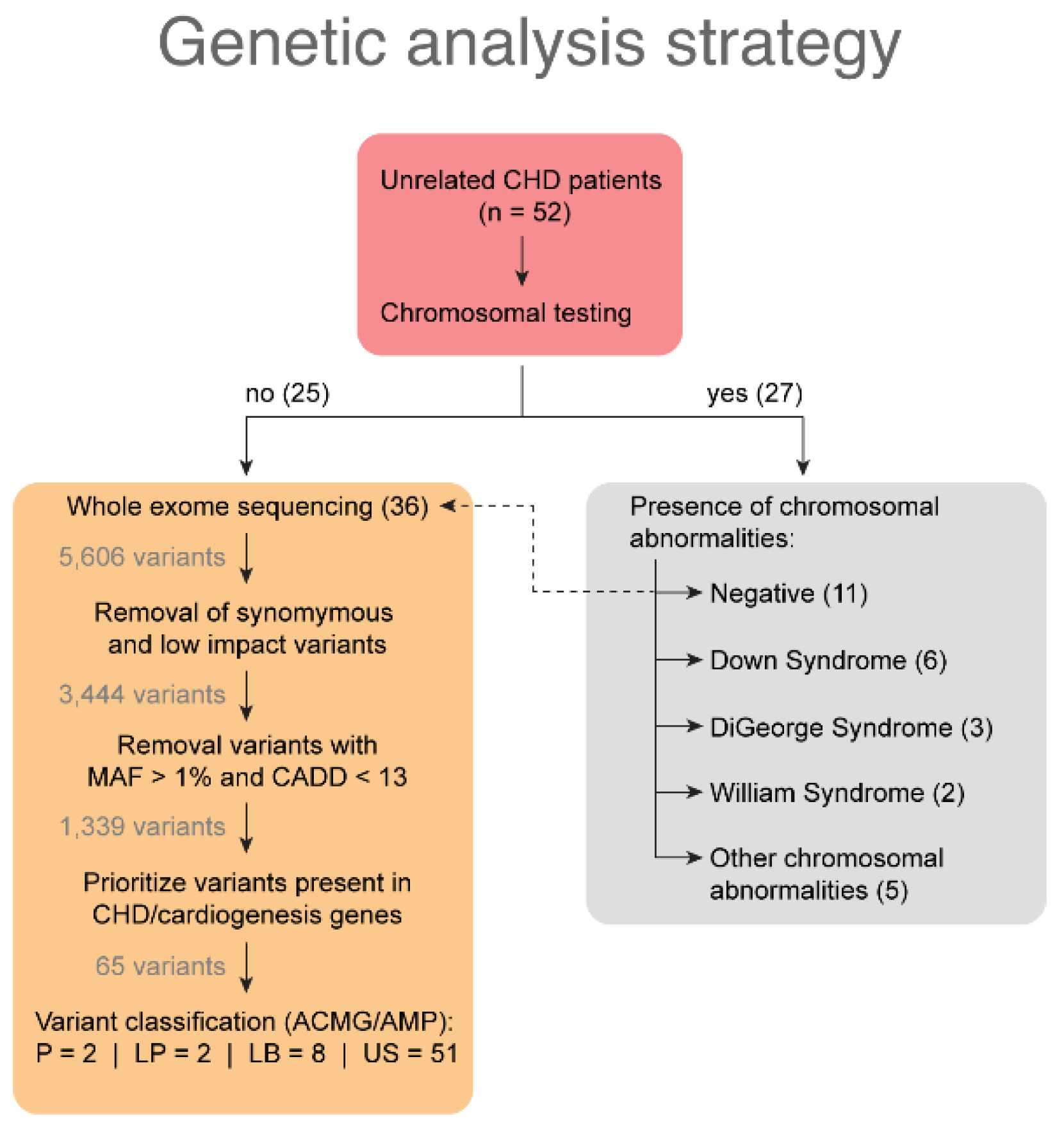
3.2. Clinical Cytogenetic Testing
3.3. Variants Identified by Whole Exome Sequencing Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A. Genetic basis for congenital heart disease: Revisited: A scientific statement from the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.; Seidman, C.E. Genetics of congenital heart disease: The glass half empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadmouri, G.O.; Nair, P.; Obeid, T.; Al Ali, M.T.; Al Khaja, N.; Hamamy, H.A. Consanguinity and reproductive health among Arabs. Reprod. Health 2009, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdan, M.A.; Chedid, F.; Bekdache, G.N.; Begam, M.; Alsafi, W.; Sabri, Z.; Mirghani, H.M. Perinatal outcome of congenital heart disease in a population with high consanguinity. J. Perinat. Med. 2015, 43, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Aburawi, E.H.; Aburawi, H.E.; Bagnall, K.M.; Bhuiyan, Z.A. Molecular insight into heart development and congenital heart disease: An update review from the Arab countries. Trends Cardiovasc. Med. 2015, 25, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Robida, A.; Folger, G.M.; Hajar, H.A. Incidence of congenital heart disease in Qatari children. Int. J. Cardiol. 1997, 60, 19–22. [Google Scholar] [CrossRef]
- Iyad, A.; Fares, A.; Laila, T. Incidence of congenital heart disease in jordanian children born at jordan university hospital: A seven-year retrospective study. Jordan Med. J. 2017, 51, 109–117. [Google Scholar]
- Bitar, F.F.; Baltaji, N.; Dbaibo, G.; Yunis, K.; Obeid, M. Congenital heart disease at a tertiary care center in Lebanon. Middle East J. Anaesthesiol. 1999, 15, 159–164. [Google Scholar]
- Subramanyan, R.; Joy, J.; Venugopalan, P.; Sapru, A.; Khusaiby, S.A. Incidence and spectrum of congenital heart disease in Oman. Ann. Trop. Paediatr. 2000, 20, 337–341. [Google Scholar] [CrossRef]
- Zaqout, M.; Aslem, E.S.; Oweida, F.S.; De Wolf, D. Prevalence of congenital heart disease among Palestinian children born in the Gaza Strip. Cardiol. Young 2014, 24, 905–909. [Google Scholar] [CrossRef]
- Al-Fahham, M.M.; Ali, Y.A. Pattern of congenital heart disease among Egyptian children: A 3-year retrospective study. Egypt Heart J. 2021, 73, 11. [Google Scholar] [CrossRef]
- Hammami, O.; Salem, B.; Boujemaa, Z.; Chebbi, Y.; Aoun, S.; Meddeb, I.; Abid, F.; Gandoura, N. Epidemiologic and clinical features of congenital heart diseases in children at the Bizerta Hospital. Tunis. Med. 2007, 85, 829–833. [Google Scholar]
- World Health Organization, Geneva, Switzerland. ICD-10 Coding Manual-List of All Reportable Congenital Malformations; New York State Department of Health: Albany, NY, USA, 2020.
- Sundaresan, V.; Mambetisaeva, E.; Andrews, W.; Annan, A.; Knöll, B.; Tear, G.; Bannister, L. Dynamic expression patterns of Robo (Robo1 and Robo2) in the developing murine central nervous system. J. Comp. Neurol. 2004, 468, 467–481. [Google Scholar] [CrossRef]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.; Abecasis, G.R.; Kang, H.M. Unified representation of genetic variants. Bioinformatics 2015, 31, 2202–2204. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Razali, R.M.; Rodriguez-Flores, J.; Ghorbani, M.; Naeem, H.; Aamer, W.; Aliyev, E.; Jubran, A.; Clark, A.G.; Fakhro, K.A.; Mokrab, Y. Thousands of Qatari genomes inform human migration history and improve imputation of Arab haplotypes. Nat. Commun. 2021, 12, 5929. [Google Scholar] [CrossRef]
- Fakhro, K.A.; Staudt, M.R.; Ramstetter, M.D.; Robay, A.; Malek, J.A.; Badii, R.; Al-Marri, A.A.-N.; Khalil, C.A.; Al-Shakaki, A.; Chidiac, O.; et al. The Qatar Genome: A Population-specific Tool for Precision Medicine in the Middle East. Hum. Genome Var. 2016, 3, 16016. [Google Scholar] [CrossRef]
- Scott, E.M.; Halees, A.; Itan, Y.; Spencer, E.G.; He, Y.; Azab, M.A.; Gabriel, S.B.; Belkadi, A.; Boisson, B.; Abel, L. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 2016, 48, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Doğan, T. HPO2GO: Prediction of human phenotype ontology term associations for proteins using cross ontology annotation co-occurrences. PeerJ 2018, 6, e5298. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Dadvand, P.; Rankin, J.; Shirley, M.D.; Rushton, S.; Pless-Mulloli, T. Descriptive epidemiology of congenital heart disease in Northern England. Paediatr. Perinat. Epidemiol. 2009, 23, 58–65. [Google Scholar] [CrossRef]
- Hartman, R.J.; Rasmussen, S.A.; Botto, L.D.; Riehle-Colarusso, T.; Martin, C.L.; Cragan, J.D.; Shin, M.; Correa, A. The contribution of chromosomal abnormalities to congenital heart defects: A population-based study. Pediatr. Cardiol. 2011, 32, 1147–1157. [Google Scholar] [CrossRef]
- Pradat, P. Epidemiology of major congenital heart defects in Sweden, 1981–1986. J. Epidemiol. Community Health 1992, 46, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Abbag, F.I. Congenital heart diseases and other major anomalies in patients with Down syndrome. Saudi Med. J. 2006, 27, 219. [Google Scholar]
- Stoll, C.; Dott, B.; Alembik, Y.; Roth, M.-P. Associated congenital anomalies among cases with Down syndrome. Eur. J. Med. Genet. 2015, 58, 674–680. [Google Scholar] [CrossRef]
- Cirillo, E.; Giardino, G.; Grasso, F.; Gallo, V.; Pignata, C. DiGeorge Syndrome. In Genetic Syndromes: A Comprehensive Reference Guide; Springer: Berlin/Heidelberg, Germany, 2022; pp. 1–7. [Google Scholar]
- Griffith, E.; Alfonso, N.; Hehmeyer, K.; Pope, K. Genetic syndromes and their associations with congenital heart disease. Prog. Pediatric Cardiol. 2022, 65, 101521. [Google Scholar] [CrossRef]
- Sherer, D.; Dalloul, M.; Pinard, V.; Sheu, J.; Abulafia, O. Fetal trisomy 8 mosaicism associated with truncus arteriosus Type I. Ultrasound Obstet. Gynecol. 2017, 50, 541–542. [Google Scholar] [CrossRef]
- Alkuraya, F.S.; Harris, D.J. Trisomy 8 mosaicism in a patient with heterotaxia. Birth Defects Res. Part A Clin. Mol. Teratol. 2005, 73, 58–60. [Google Scholar] [CrossRef]
- Belengeanu, V.; Boia, M.; Farcas, S.; Popa, C.; Stoian, M.; Belengeanu, A. Trisomy 8 mosaicism with atypical phenotypic features. J. Pediatr. 2010, 13, 36–39. [Google Scholar]
- Fujimoto, A.; Allanson, J.; Crowe, C.A.; Lipson, M.H.; Johnson, V.P. Natural history of mosaic trisomy 14 syndrome. Am. J. Med. Genet. 1992, 44, 189–196. [Google Scholar] [CrossRef]
- Fran Lynch, M.; Fernandes, C.J.; Shaffer, L.G.; Potocki, L. Trisomy 14 mosaicism: A case report and review of the literature. J. Perinatol. 2004, 24, 121–123. [Google Scholar] [CrossRef]
- Kunst, G.; Gillbe, C. General anesthesia for cardiac catheterization in a child with trisomy 14 mosaicism. Anesth. Analg. 2005, 100, 1860. [Google Scholar] [CrossRef]
- Tunca, Y.; Wilroy, R.S.; Kadandale, J.S.; Martens, P.R.; Gunther, W.M.; Tharapel, A.T. Hypomelanosis of Ito and a ‘mirror image’whole chromosome duplication resulting in trisomy 14 mosaicism. In Annales de Genetique; Elsevier: Rio de Janriro, Brazil, 2000; pp. 39–43. [Google Scholar]
- Lin, A.E.; Westgate, M.-N.; van der Velde, M.E.; Lacro, R.V.; Holmes, L.B. Adams-Oliver syndrome associated with cardiovascular malformations. Clin. Dysmorphol. 1998, 7, 235–241. [Google Scholar] [CrossRef]
- Algaze, C.; Esplin, E.D.; Lowenthal, A.; Hudgins, L.; Tacy, T.A.; Selamet Tierney, E.S. Expanding the phenotype of cardiovascular malformations in Adams–Oliver syndrome. Am. J. Med. Genet. A 2013, 161, 1386–1389. [Google Scholar] [CrossRef]
- Irving, M.; Hanson, H.; Turnpenny, P.; Brewer, C.; Ogilvie, C.M.; Davies, A.; Berg, J. Deletion of the distal long arm of chromosome 10; is there a characteristic phenotype? A report of 15 de novo and familial cases. Am. J. Med. Genet. A 2003, 123, 153–163. [Google Scholar] [CrossRef]
- Yatsenko, S.; Kruer, M.; Bader, P.; Corzo, D.; Schuette, J.; Keegan, C.; Nowakowska, B.; Peacock, S.; Cai, W.; Peiffer, D. Identification of critical regions for clinical features of distal 10q deletion syndrome. Clin. Genet. 2009, 76, 54–62. [Google Scholar] [CrossRef]
- Medioni, C.; Bertrand, N.; Mesbah, K.; Hudry, B.; Dupays, L.; Wolstein, O.; Washkowitz, A.J.; Papaioannou, V.E.; Mohun, T.J.; Harvey, R.P. Expression of Slit and Robo genes in the developing mouse heart. Dev. Dyn. 2010, 239, 3303–3311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruszka, P.; Tanpaiboon, P.; Neas, K.; Crosby, K.; Berger, S.I.; Martinez, A.F.; Addissie, Y.A.; Pongprot, Y.; Sittiwangkul, R.; Silvilairat, S. Loss of function in ROBO1 is associated with tetralogy of Fallot and septal defects. J. Med. Genet. 2017, 54, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Faiyaz-Ul-Haque, M.; Zaidi, S.; Wahab, A.; Eltohami, A.; Al-Mureikhi, M.; Al-Thani, G.; Peltekova, V.; Tsui, L.C.; Teebi, A.S. Identification of a p. Ser81Arg encoding mutation in SLC2A10 gene of arterial tortuosity syndrome patients from 10 Qatari families. Clin. Genet. 2008, 74, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Callewaert, B.; Willaert, A.; Kerstjens-Frederikse, W.; De Backer, J.; Devriendt, K.; Albrecht, B.; Ramos-Arroyo, M.; Doco-Fenzy, M.; Hennekam, R.; Pyeritz, R. Arterial tortuosity syndrome: Clinical and molecular findings in 12 newly identified families. Hum. Mutat. 2008, 29, 150–158. [Google Scholar] [CrossRef]
- Tan, H.L.; Glen, E.; Töpf, A.; Hall, D.; O’Sullivan, J.J.; Sneddon, L.; Wren, C.; Avery, P.; Lewis, R.J.; ten Dijke, P. Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum. Mutat. 2012, 33, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Corsten-Janssen, N.; Kerstjens-Frederikse, W.S.; du Marchie Sarvaas, G.J.; Baardman, M.E.; Bakker, M.K.; Bergman, J.E.; Hove, H.D.; Heimdal, K.R.; Rustad, C.F.; Hennekam, R.C. The cardiac phenotype in patients with a CHD7 mutation. Circ. Cardiovasc. Genet. 2013, 6, 248–254. [Google Scholar] [CrossRef] [Green Version]
- Jongmans, M.; Admiraal, R.; Van Der Donk, K.; Vissers, L.; Baas, A.; Kapusta, L.; van Hagen, J.M.; Donnai, D.; De Ravel, T.; Veltman, J. CHARGE syndrome: The phenotypic spectrum of mutations in the CHD7 gene. J. Med. Genet. 2006, 43, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Tillman, S. Congenital Heart Defects and the Expression of Ccdc141. Honors Thesis, University of Mississippi, Oxford, MI, USA, 2021. [Google Scholar]
- Zhang, J.; An, X.; Sun, X.; Yu, K.; Gong, T. Screening of Candidate Key Genes Associated with Congenital Heart Disease Using Bioinformatics Data Analysis. In Proceedings of the 2020 International Conference on Modeling, Big Data Analytics and Simulation (MBDAS2020), Xiamen, China, 20–21 December 2020; Journal of Physics: Conference Series; Volume 1813, p. 012038. [Google Scholar]
- Kim, D.S.; Burt, A.A.; Crosslin, D.R.; Robertson, P.D.; Ranchalis, J.E.; Boyko, E.J.; Nickerson, D.A.; Furlong, C.E.; Jarvik, G.P. Novel common and rare genetic determinants of paraoxonase activity: FTO, SERPINA12, and ITGAL [S]. J. Lipid Res. 2013, 54, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Bittencourt, M.I. Description of a New GLA Gene Variant in a Patient with Hypertrophic Cardiomyopathy. Is it Fabry Disease? Arq. Bras. Cardiol. 2019, 113, 85–86. [Google Scholar] [CrossRef]
- Lee, S.R.; Han, J. Mitochondrial mutations in cardiac disorders. Mitochondrial Dyn. Cardiovasc. Med. 2017, 982, 81–111. [Google Scholar]
- Majamaa-Voltti, K.; Peuhkurinen, K.; Kortelainen, M.-L.; Hassinen, I.E.; Majamaa, K. Cardiac abnormalities in patients with mitochondrial DNA mutation 3243A> G. BMC Cardiovasc. Disord. 2002, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Savarese, M.; Maggi, L.; Vihola, A.; Jonson, P.H.; Tasca, G.; Ruggiero, L.; Bello, L.; Magri, F.; Giugliano, T.; Torella, A. Interpreting Genetic Variants in Titin in Patients with Muscle Disorders. JAMA Neurol. 2018, 75, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Faiyaz-Ul-Haque, M.; Zaidi, S.H.E.; Al-Sanna, N.; Alswaid, A.; Momenah, T.; Kaya, N.; Al-Dayel, F.; Bouhoaigah, I.; Saliem, M.; Tsui, L.-C. A Novel Missense and a Recurrent Mutation in Slc2a10 Gene of Patients Affected with Arterial Tortuosity Syndrome. Atherosclerosis 2009, 203, 466–471. [Google Scholar] [CrossRef]
- [VCV000773847.2]. National Center for Biotechnology Information. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/variation/VCV000773847.2 (accessed on 1 June 2022).
- National Center for Biotechnology Information. [Vcv000167010.9]. ClinVar. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/variation/VCV000167010.9 (accessed on 1 June 2022).
- [VCV000722668.2]. National Center for Biotechnology Information. ClinVar. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/variation/VCV000722668.2 (accessed on 1 June 2022).
- Information, National Center for Biotechnology. Clinvar. [Vcv000722667.2]. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/variation/VCV000722667.2 (accessed on 1 June 2022).
- Zhu, T.; Gong, X.; Bei, F.; Ma, L.; Sun, J.; Wang, J.; Qiu, G.; Sun, J.; Sun, Y.; Zhang, Y. Primary Immunodeficiency-Related Genes in Neonatal Intensive Care Unit Patients with Various Genetic Immune Abnormalities: A Multicentre Study in China. Clin. Transl. Immunol. 2021, 10, e1266. [Google Scholar] [CrossRef]
- Sun, Y.; Sun, J.; Li, N.; Cai, C.; Gong, X.; Ma, L. Phenotypic Spectrum of Typical Charge Syndrome in a Chinese Male Neonate: A Case Report. Transl. Pediatr. 2020, 9, 180. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. [VCV000136948.9]. Clinvar. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/variation/VCV000136948.9 (accessed on 1 June 2022).
- [VCV000707217.7]. National Center for Biotechnology Information. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/variation/VCV000707217.7 (accessed on 1 June 2022).
- [VCV000129948.9]. National Center for Biotechnology Information. ClinVar. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/variation/VCV000129948.9 (accessed on 1 June 2022).
- Kanthi, A.; Hebbar, M.; Bielas, S.L.; Girisha, K.M.; Shukla, A. Bi-Allelic C. 181_183deltgt in Btb Domain of Klhl7 Is Associated with Overlapping Phenotypes of Crisponi/Ciss1-Like and Bohring-Opitz Like Syndrome. Eur. J. Med. Genet. 2019, 62, 103528. [Google Scholar] [CrossRef]
- [VCV000216489.13]. National Center for Biotechnology Information. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/variation/VCV000216489.13 (accessed on 1 June 2022).
- National Center for Biotechnology Information. [VCV000258032.9]. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/variation/VCV000258032.9 (accessed on 1 June 2022).
- [VCV000010738.32]. National Center for Biotechnology Information. ClinVar. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/variation/VCV000010738.32 (accessed on 1 June 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Cardiac Phenotype | Extracardiac Phenotype | Chromosomal Abnormality | Genes Encompassed | Associated Condition | Interpretation of Test Results | Parental Testing |
|---|---|---|---|---|---|---|---|
| Cardio-1.A | ASD, VSD | Hypotonia, dysmorphic features, developmental delay | 47, XY, +21 | Gain of one full copy of chromosome 21 | Down Syndrome | Pathogenic | No record |
| Cardio-3.A | TGV | Vertebral abnormalities, anal atresia, cardiac abnormalities, tracheoesophageal fistula, renal anomalies, limb defects | Chromosome 6 deletion * | Unknown | Unknown | Unknown | No record |
| Cardio-4.A | TOF, PA | Thymus hypoplasia, compromised immune system, absent left kidney, idiopathic left club foot, global developmental delay with central hypotonia | 22q11.2 deletion | 40 genes (TBX1 and COMT) | DiGeorge Syndrome | Pathogenic | Mother is normal; father has no records |
| Cardio-6.A | VSA | Congenital nasolacrimal duct obstruction, esotropia, failure to thrive, hypothyroidism, supraventricular tachycardia, thrombocytopenia | 22q11.2 deletion | 40 genes (TBX1 and COMT) | DiGeorge Syndrome | Pathogenic | No records |
| Cardio-9.A | ASD, DORV, PA, Hypoplastic mitral valve and left ventricle | Hypotension, acidosis, bradycardia, severe developmental delay, seizures | 9q34.3 deletion | SNAPC4, PMPCA, INPP5E, SEC16A, NOTCH1 | Adams Oliver Syndrome | Likely Pathogenic | Mother is normal; father is inconclusive |
| Cardio-28.A | TOF | Acute renal failure, fluid overload, skin pigmentation, undescended testicles | Mosaic 47, XY, +14 | Gain of one full copy of chromosome 14 in some somatic cells | Mosaic trisomy 14 | Pathogenic | Parents are normal |
| Cardio-30.A | TOF | Bilateral hydronephrosis, hypotonia, dysmorphic features | 47, XY, +21 | Gain of one full copy of chromosome 21 | Down Syndrome | Pathogenic | No records |
| Cardio-32.A | VSD | Dysphagia, gastroesophageal reflux disease, failure to thrive, global developmental delay, hypotonia, central sleep apnea, right ankle contracture, asymmetric leg length | 7q11.23 deletion,10q26.3 deletion, | ELN, LIMK1, BAZ1B, CLIP2, GTF2IRD, NSUN5, CLDN4, EIF4H, LAT2, MLXIPL, TBL2, WBSCR18, WBSCR22, WBSCR27—Not defined for 10q26.3 deletion | William Syndrome and 10q26.3 deletion | Pathogenic | Parents are normal |
| Cardio-33.A | ASD | Polydactyly, dysmorphic features, delayed motor development | 47, XY, +21 | Gain of one full copy of chromosome 21 | Down Syndrome | Pathogenic | Parents are normal |
| Cardio-37.A | TOF, PS | Failure to thrive, hypothyroidism | 47, XY +8 22q11.21 deletion | Gain of one full copy of chromosome 8 | DiGeorge syndrome and mosaic trisomy 8 | Pathogenic | No records |
| Cardio-39.A | VSD | Dysmorphic features, hypotonia | 47, XY, +21 | Gain of one full copy of chromosome 21 | Down Syndrome | Pathogenic | No records |
| Cardio-44.A | AS, PS | Anal stenosis, dysmorphic features | 7q11.23 deletion | ELN, LIMK1, BAZ1B, CLIP2, GTF2IRD, NSUN5, CLDN4, EIF4H, LAT2, MLXIPL, TBL2, WBSCR18, WBSCR22, WBSCR27 | William Syndrome | Pathogenic | No records |
| Cardio-45.A | HLHS | Depressed nasal bridge, developmental delay | 8p11.21 duplication | Not reported | Not specified | Unknown | Mother is normal |
| Cardio-56.A | ASD, PA, TAPVD, Heterotaxy | Bowel obstruction, osteomyelitis, asplenia | 16p11.2 deletion | SH2B1 gene | Not specified | Likely benign | Mother is normal; father has the same deletion |
| Cardio-60.A | ASD | Laryngomalacia, swallowing dysfunction | 47, XX, +21 | Gain of one full copy of chromosome 21 | Down Syndrome | Pathogenic | No records |
| Cardio-62.A | ASD | Hypotonia, dysmorphic features | 47, XY, +21 | Gain of one full copy of chromosome 21 | Down Syndrome | Pathogenic | No records |
| Gene | Amino Acid Change | Nucleotide Change | Variant Type | Variant Impact | Familial Segregation | Zygosity | Inheritance | ACMG/AMP Classification | CHD Phenotype | Extra Phenotype | Patient |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ROBO1 | - | c.2883-1G>T | SNP | splice acceptor | de novo | Heterozygous | None | Pathogenic | Multiple CHD | None | Cardio-12.A |
| SMAD6 | - | c.817+1G>C | SNP | splice site donor | de novo | Heterozygous | None | Likely pathogenic | Shone’s complex | None | Cardio-15.A |
| SLC2A10 | p.Ser81Arg | c.243C>G | SNP | Missense | AR | Homozygous | Both parents | Likely pathogenic | ATS | None | Cardio-5.A |
| CHD7 | p.Arg2098 * | c.6292C>T | SNP | stop gained | de novo | Heterozygous | None | Pathogenic | TOF | Prolonged QT interval, hearing loss | Cardio-27.A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okashah, S.; Vasudeva, D.; El Jerbi, A.; Khodjet-El-khil, H.; Al-Shafai, M.; Syed, N.; Kambouris, M.; Udassi, S.; Saraiva, L.R.; Al-Saloos, H.; et al. Investigation of Genetic Causes in Patients with Congenital Heart Disease in Qatar: Findings from the Sidra Cardiac Registry. Genes 2022, 13, 1369. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13081369
Okashah S, Vasudeva D, El Jerbi A, Khodjet-El-khil H, Al-Shafai M, Syed N, Kambouris M, Udassi S, Saraiva LR, Al-Saloos H, et al. Investigation of Genetic Causes in Patients with Congenital Heart Disease in Qatar: Findings from the Sidra Cardiac Registry. Genes. 2022; 13(8):1369. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13081369
Chicago/Turabian StyleOkashah, Sarah, Dhanya Vasudeva, Aya El Jerbi, Houssein Khodjet-El-khil, Mashael Al-Shafai, Najeeb Syed, Marios Kambouris, Sharda Udassi, Luis R. Saraiva, Hesham Al-Saloos, and et al. 2022. "Investigation of Genetic Causes in Patients with Congenital Heart Disease in Qatar: Findings from the Sidra Cardiac Registry" Genes 13, no. 8: 1369. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13081369