

The Multi-Omic Landscape of Primary Breast Tumors and Their Metastases: Expanding the Efficacy of Actionable Therapeutic Targets

Abstract
:1. Introduction
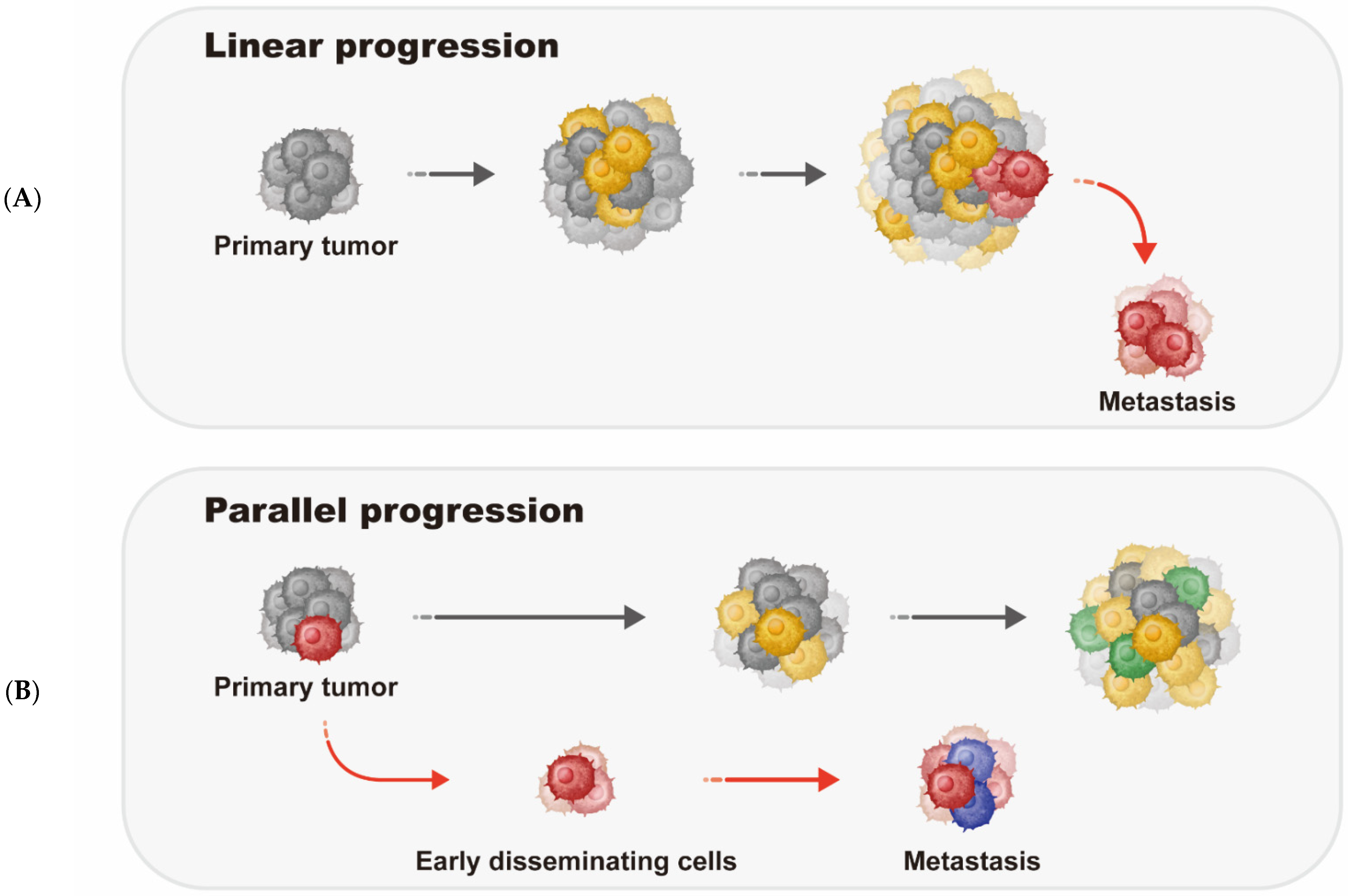
2. Metastatic Breast Cancer Progression Models
3. Breast Tumor Immune Microenvironment
4. Histological and Molecular Subgroups of Breast Cancer
4.1. Histological Breast Cancer Subgroups
4.2. Molecular Subgroups of Breast Cancer
5. Genetic Landscape of Breast Cancer
5.1. Hereditary Germline Mutations
5.2. Somatic Mutation Profiles
5.3. Copy Number Variation in Breast Tumors
5.4. Genetic Profiles of Metastatic Breast Tumors
5.5. Chromosomal Instability (CIN)
6. DNA Methylation Alterations in Breast Cancer
7. Therapeutic Options for Breast Cancer Patients
8. Established Treatment Schemes for Metastatic Breast Cancer Patients
9. Novel Molecular Therapeutic Targets for mBC
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Nikisic, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Esteve, J.; et al. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): Analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Rocca, A.; Melegari, E.; Palleschi, M. Ribociclib and Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 1592. [Google Scholar] [PubMed]
- Swain, S.M.; Miles, D.; Kim, S.B.; Im, Y.H.; Im, S.A.; Semiglazov, V.; Ciruelos, E.; Schneeweiss, A.; Loi, S.; Monturus, E.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): End-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 519–530. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative, G.; Peto, R.; Davies, C.; Godwin, J.; Gray, R.; Pan, H.C.; Clarke, M.; Cutter, D.; Darby, S.; McGale, P.; et al. Comparisons between different polychemotherapy regimens for early breast cancer: Meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet 2012, 379, 432–444. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Lindstrom, L.S.; Karlsson, E.; Wilking, U.M.; Johansson, U.; Hartman, J.; Lidbrink, E.K.; Hatschek, T.; Skoog, L.; Bergh, J. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J. Clin. Oncol. 2012, 30, 2601–2608. [Google Scholar] [CrossRef]
- Karlsson, E.; Sandelin, K.; Appelgren, J.; Zhou, W.; Jirstrom, K.; Bergh, J.; Warnberg, F. Clonal alteration of breast cancer receptors between primary ductal carcinoma in situ (DCIS) and corresponding local events. Eur. J. Cancer 2014, 50, 517–524. [Google Scholar] [CrossRef]
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Gui, P.; Bivona, T.G. Evolution of metastasis: New tools and insights. Trends Cancer 2022, 8, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; McGranahan, N. Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2020, 37, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.E.; Hicks, J. Tracing the tumor lineage. Mol. Oncol. 2010, 4, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Paul, M.R.; Pan, T.C.; Pant, D.K.; Shih, N.N.; Chen, Y.; Harvey, K.L.; Solomon, A.; Lieberman, D.; Morrissette, J.J.; Soucier-Ernst, D.; et al. Genomic landscape of metastatic breast cancer identifies preferentially dysregulated pathways and targets. J. Clin. Investig. 2020, 130, 4252–4265. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: Biology and clinical significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef]
- Kroigard, A.B.; Larsen, M.J.; Laenkholm, A.-V.; Knoop, A.S.; Jensen, J.D.; Bak, M.; Mollenhauer, J.; Kruse, T.A.; Thomassen, M. Clonal expansion and linear genome evolution through breast cancer progression from pre-invasive stages to asynchronous metastasis. Oncotarget 2015, 6, 5634–5649. [Google Scholar] [CrossRef] [PubMed]
- Kroigard, A.B.; Larsen, M.J.; Brasch-Andersen, C.; Laenkholm, A.V.; Knoop, A.S.; Jensen, J.D.; Bak, M.; Mollenhauer, J.; Thomassen, M.; Kruse, T.A. Genomic Analyses of Breast Cancer Progression Reveal Distinct Routes of Metastasis Emergence. Sci. Rep. 2017, 7, 43813. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Karthik, G.M.; Alkodsi, A.; Kjallquist, U.; Stalhammar, G.; Lovrot, J.; Martinez, N.F.; Lagergren, J.; Hautaniemi, S.; Hartman, J.; et al. Evolutionary history of metastatic breast cancer reveals minimal seeding from axillary lymph nodes. J. Clin. Investig. 2018, 128, 1355–1370. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Salemme, V.; Centronze, G.; Cavallo, F.; Defilippi, P.; Conti, L. The crosstalk between tumore cells and the immune microenvironement in breast cancer: Implications for immunotherapy. Front. Oncol. 2021, 11, 610313. [Google Scholar] [CrossRef]
- Zhu, L.; Narloch, J.L.; Onkar, S.; Joy, M.; Broadwater, G.; Luedke, C.; Hall, A.; Kim, R.; Pogue-Geile, K.; Sammons, S.; et al. Metastatic breast cancers have reduced immune cell recruitment but harbor increased macrophages relative to their matched primary tumors. J. Immunother. Cancer 2019, 7, 265. [Google Scholar] [CrossRef]
- Schlam, I.; Church, S.E.; Hether, T.D.; Chaldekas, K.; Hudson, B.M.; White, A.M.; Maisonet, E.; Harris, B.T.; Swain, S.M. The tumor immune microenvironment of primary and metastatic HER2-positive breast cancers utilizing gene expression and spatial proteomic profiling. J. Transk Med. 2021, 19, 480. [Google Scholar] [CrossRef]
- Vos, H.; Lambein, K.; Richard, F.; Marien, B.; Nevelsteen, I.; Punie, K.; Wildiers, K.; Berben, L.; Laenen, A.; Floris, G.; et al. Comparison of the tumor immune microenvironment of primary hormone receptor-negative HER2-positive and triple negative breast cancer. NPJ Breast Cancer 2021, 7, 128. [Google Scholar] [CrossRef]
- Bianchini, G.; Qi, Y.; Alvarez, R.H.; Iwamoto, T.; Coutant, C.; Ibrahim, N.K.; Valero, V.; Cristofanilli, M.; Green, M.C.; Radvanyi, L.; et al. Molecular anatomy of breast cancer stroma and its prognostic value in estrogen receptor-positive and -negative cancers. J. Clin. Oncol. 2010, 28, 4316–4323. [Google Scholar] [CrossRef]
- Karn, T.; Pusztai, L.; Holtrich, U.; Iwamoto, T.; Shiang, C.Y.; Schmidt, M.; Muller, V.; Solbach, C.; Gaetje, R.; Hanker, L.; et al. Homogeneous datasets of triple negative breast cancers enable the identification of novel prognostic and predictive signatures. PLoS ONE 2011, 6, e28403. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; von Minckwitz, G.; Brase, J.C.; Sinn, B.V.; Gade, S.; Kronenwett, R.; Pfitzner, B.M.; Salat, C.; Loi, S.; Schmitt, W.D.; et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J. Clin. Oncol. 2015, 33, 983–991. [Google Scholar] [CrossRef]
- Cimino-Mathews, A.; Thompson, E.; Taube, J.M.; Ye, X.; Lu, Y.; Meeker, A.; Xu, H.; Sharma, R.; Lecksell, K.; Cornish, T.C.; et al. PD-L1 (B7-H1) expression and the immune tumor microenvironment in primary and metastatic breast carcinomas. Hum. Pathol. 2016, 47, 52–63. [Google Scholar] [CrossRef]
- Ogiya, R.; Niikura, N.; Kumaki, N.; Bianchini, G.; Kitano, S.; Iwamoto, T.; Hayashi, N.; Yokoyama, K.; Oshitanai, R.; Terao, M.; et al. Comparison of tumor-infiltrating lymphocytes between primary and metastatic tumors in breast cancer patients. Cancer Sci. 2016, 107, 1730–1735. [Google Scholar] [CrossRef]
- Szekely, B.; Bossuyt, V.; Li, X.; Wali, V.B.; Patwardhan, G.A.; Frederick, C.; Silber, A.; Park, T.; Harigopal, M.; Pelekanou, V.; et al. Immunological differences between primary and metastatic breast cancer. Ann. Oncol. 2018, 29, 2232–2239. [Google Scholar] [CrossRef]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef]
- Janssen, L.M.E.; Ramsay, E.E.; Logsdon, C.D.; Overwijk, W.W. The immune system in cancer metastasis: Friend or foe? J. Immuno. Ther. Cancer 2017, 5, 79. [Google Scholar]
- Weigelt, B.; Reis-Filho, J.S. Histological and molecular types of breast cancer: Is there a unifying taxonomy? Nat. Rev. Clin. Oncol. 2009, 6, 718–730. [Google Scholar] [CrossRef]
- Weigelt, B.; Geyer, F.C.; Reis-Filho, J.S. Histological types of breast cancer: How special are they? Mol. Oncol. 2010, 4, 192–208. [Google Scholar] [CrossRef]
- Brouckaert, O.; Paridaens, R.; Floris, G.; Rakha, E.; Osborne, K.; Neven, P. A critical review why assessment of steroid hormone receptors in breast cancer should be quantitative. Ann. Oncol. 2013, 24, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Campan, M.; Weisenberger, D.J.; Laird, P.W. DNA methylation profiles of female steroid hormone-driven human malignancies. Curr. Top. Microbiol. Immunol. 2006, 310, 141–178. [Google Scholar]
- Li, C.I.; Daling, J.R.; Malone, K.E. Incidence of invasive breast cancer by hormone receptor status from 1992 to 1998. J. Clin. Oncol. 2003, 21, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Mal, R.; Magner, A.; David, J.; Datta, J.; Vallabhaneni, M.; Kassem, M.; Manouchehri, J.; Willingham, N.; Stover, D.; Vandeusen, J.; et al. Estrogen Receptor β (ERbeta): A Ligand Activated Tumor Suppressor. Front Oncol. 2020, 10, 587386. [Google Scholar] [CrossRef] [PubMed]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors α (ERalpha) and β (ERbeta): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef]
- Bocchinfuso, W.P.; Lindzey, J.K.; Hewitt, S.C.; Clark, J.A.; Myers, P.H.; Cooper, R.; Korach, K.S. Induction of mammary gland development in estrogen receptor-α knockout mice. Endocrinology 2000, 141, 2982–2994. [Google Scholar] [CrossRef]
- Forster, C.; Makela, S.; Warri, A.; Kietz, S.; Becker, D.; Hultenby, K.; Warner, M.; Gustafsson, J.A. Involvement of estrogen receptor β in terminal differentiation of mammary gland epithelium. Proc. Natl. Acad. Sci. USA 2002, 99, 15578–15583. [Google Scholar] [CrossRef]
- Rubin, I.; Yarden, Y. The basic biology of HER2. Ann. Oncol. 2001, 12 (Suppl. 1), S3–S8. [Google Scholar] [CrossRef]
- Wang, J.; Xu, B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Almansour, N.M. Triple-Negative Breast Cancer: A Brief Review About Epidemiology, Risk Factors, Signaling Pathways, Treatment and Role of Artificial Intelligence. Front. Mol. Biosci. 2022, 9, 836417. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-like breast cancer: A critical review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef]
- Reynolds, S. Triple-negative Breast Cancer Disproportionately Affects African American and Hispanic Women. Cancer Bull. 2007, 4. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Calza, S.; Hall, P.; Auer, G.; Bjohle, J.; Kronenwett, U.; Liu, E.T.; Miller, L.; Ploner, A.; Smeds, J.; Bergh, J.; et al. Intrinsic molecular signature of breast cancer in a population-cohort of 412 patients. Breast Cancer Res. 2006, 8, R34. [Google Scholar] [CrossRef]
- Johnson, K.S.; Conant, E.F.; Soo, M.S. Molecular Subtypes of Breast Cancer: A Review for Breast Radiologists. J. Breast Imaging 2021, 3, 12–24. [Google Scholar] [CrossRef]
- Pernas, S.; Tolaney, S.M. HER2-positive breast cancer: New therapeutic frontiers and overcoming resistance. Ther. Adv. Med. Oncol. 2019, 11, 1758835919833519. [Google Scholar] [CrossRef]
- Onsum, M.D.; Geretti, E.; Paragas, V.; Kudia, A.J.; Moulis, S.P.; Luus, L.; Wickham, T.J.; McDonagh, C.F.; MacBeath, G.; Hendriks, B.S. Single-cell quantitative HER2 measurement identifies heterogeneity and distinct subgreoups within traditionally defined HER2-positive patients. Am. J. Pathol 2013, 183, 1446–1460. [Google Scholar] [CrossRef]
- Gampenrieder, S.P.; Rinnerthaler, G.; Tinchon, C.; Petzer, C.; Balic, M.; Heibl, S.; Schmitt, C.; Zabernigg, A.F.; Egle, D.; Sandholzer, M.; et al. Landscape of HER2-low metastatic breast cancer (MBC): Results from the Austrian AGMT_MBC-Registry. Breast Cancer Res. 2021, 23, 112. [Google Scholar] [CrossRef] [PubMed]
- van den Ende, N.; Smid, M.; Timmermans, A.; van Brakel, J.B.; Hansum, T.; Foekens, R.; Trapman, A.M.A.C.; Heemskerk-Gerritsen, B.A.M.; Jager, A.; Martens, J.W.M.; et al. HER2-low breast cancer shows a lower immune response compared to HER2-negative vases. Sci. Rep. 2022, 12, 12974. [Google Scholar] [CrossRef] [PubMed]
- Migiletta, F.; Griguolo, G.; Bottosso, M.; Giarratano, T.; Lo Mele, M.; Fassan, M.; Cacciatore, M.; Genovesi, E.; De Bartolo, D.; Vernaci, G.; et al. Evolution of HER2-low expression from primary to recurrent breast cancer. NPJ Breast Cancer 2021, 7, 137. [Google Scholar] [CrossRef] [PubMed]
- Rossi, V.; Sarotto, I.; Maggiorotto, F.; Berchialla, P.; Kubatzki, F.; Tomasi, N.; Redana, S.; Martinello, R.; Valabrega, G.; Aglietta, M.; et al. Moderate Immunohistochemical expression of HER-2 (2+) without HER-2 gene amplification is a negative prognostic factor in early breast cancer. Oncologist 2012, 17, 1418–1425. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, G.M.; Kim, J.H.; Kim, J.Y.; Park, H.S.; Park, S.; Cho, Y.U.; Park, B.W.; Kim, S.I.; Sohn, J. Intermediate HER2 expression is associated with poor prognosis in estrogen receptor-positive breast cancer patients ages 55 years and older. Breast Cancer Res. Treat. 2020, 179, 687–697. [Google Scholar] [CrossRef]
- Agostinetto, E.; Rediti, M.; Fimereli, D.; Debien, V.; Pccart, M.; Aftimos, P.; Sotirou, C.; de Azambuja, E. HER2-low breast cancer: Molecular characteristics and prognosis. Cancers 2021, 13, 2824. [Google Scholar] [CrossRef]
- Denkert, C.; Seither, F.; Schneeweiss, A.; Link, T.; Blohmer, J.-U.; Just, M.; Wimberger, P.; Forberger, A.; Tesch, H.; Jackisch, C.; et al. Clinical and molecular characteristics of HER2-low-positive breast cancer: Pooled analyses of individual patient data from four prospective, neoadjuvant clinical trials. Lancet Oncol. 2021, 22, 1151–1161. [Google Scholar] [CrossRef]
- Cleator, S.; Heller, W.; Coombes, R.C. Triple-negative breast cancer: Therapeutic options. Lancet Oncol. 2007, 8, 235–244. [Google Scholar] [CrossRef]
- Milioli, H.H.; Tishchenko, I.; Riveros, C.; Berretta, R.; Moscato, P. Basal-like breast cancer: Molecular profiles, clinical features and survival outcomes. BMC Med. Genom. 2017, 10, 19. [Google Scholar] [CrossRef]
- Banerjee, S.; Reis-Filho, J.S.; Ashley, S.; Steele, D.; Ashworth, A.; Lakhani, S.R.; Smith, I.E. Basal-like breast carcinomas: Clinical outcome and response to chemotherapy. J. Clin. Pathol. 2006, 59, 729–735. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Gage, M.; Wattendorf, D.; Henry, L.R. Translational advances regarding hereditary breast cancer syndromes. J. Surg. Oncol. 2012, 105, 444–451. [Google Scholar] [CrossRef]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef]
- Narod, S.A.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer 2004, 4, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.M.; Fan, S.; Pestell, R.G.; Goldberg, I.D. BRCA1 gene in breast cancer. J. Cell Physiol. 2003, 196, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsdottir, K.; Ashworth, A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006, 25, 5864–5874. [Google Scholar] [CrossRef]
- Rosen, E.M.; Fan, S.; Isaacs, C. BRCA1 in hormonal carcinogenesis: Basic and clinical research. Endocr.-Relat. Cancer 2005, 12, 533–548. [Google Scholar] [CrossRef]
- Hu, C.; Polley, E.C.; Yadav, S.; Lilyquist, J.; Shimelis, H.; Na, J.; Hart, S.N.; Goldgar, D.E.; Shah, S.; Pesaran, T.; et al. The contribution of germline predisposition gene mutations to clinical subtypes of invasive breast cancer from a clinical genetic testing cohort. J. Natl. Cancer Inst. 2020, 112, 1231–1241. [Google Scholar] [CrossRef]
- Stuttgen, K.; Croessmann, S.; Fetting, J.; Stearns, V.; Nunes, R.; Connolly, R.M.; Park, B.H. Pathogenic germline variants in patients with metastatic breast cancer. JAMA Oncol. 2019, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer Association Consortium. Pathology of tumors associated with pathogenic germline variants in 9 breast cancer susceptibility genes. JAMA Oncol. 2022, 22, e216744. [Google Scholar]
- Couch, F.J.; Shimmelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Guo, X.; Tang, L.; Chen, M.; Luo, X.; Peng, L.; Xu, X.; Wang, S.; Xiao, Z.; Yi, W.; et al. Analysis of BRCA1/2 mutation spectrum and prevalence in selected Chinese breast cancer patients by next-generation sequencing. J. Cancer Res. Clin. Oncol. 2017, 143, 2011–2024. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Chen, S.-Y.; Ren, J.-X.; Pei, Y.-C.; Jiang, C.-W.; Zhao, S.; Xiao, Y.; Xu, X.-E.; Liu, G.-Y.; Hu, X.; et al. Molecular features and functional implications of germline variants in triple-negative breast cancer. J. Natl. Cancer Inst. 2021, 113, 884–892. [Google Scholar] [CrossRef]
- Fostira, F.; Kostantopoulou, I.; Apostolou, P.; Papamentzelopoulou, M.S.; Papadimitriou, C.; Faliakou, E.; Christodoulou, C.; Boukovinas, I.; Razis, E.; Tryfonopoulos, D.; et al. One in three highly selected Greek patients with breast cancer carries a loss-of-function variant in a cancer susceptibility gene. J. Med. Genet. 2019, 57, 53–61. [Google Scholar] [CrossRef]
- Urban, R.Q.; Velasquez, C.E.D.; Gitler, R.; Castillo, M.P.R.; Toporek, M.S.; Morales, A.F.; Garcia, O.M.; Esquivel, L.G.; Mejia, G.T.; Dean, M.; et al. Comprehensive analysis of germline variants in Mexican patients with hereditary breast and ovariant cancer susceptibility. Cancers 2018, 10, 361. [Google Scholar] [CrossRef]
- Gomez-Flores-Ramos, L.; Barraza-Arellano, A.L.; Mojar, A.; Trujillo-Martinez, M.; Grimaldo, L.; Ortiz-Lopez, R.; Trevino, V. Germline variants in cancer genes from young breast cancer Mexican patients. Cancers 2022, 14, 1467. [Google Scholar] [CrossRef]
- Mardis, E.M.; Wilson, R.K. Cancer genome sequencing: A review. Hum. Molec Genet. 2009, 18, R163–R168. [Google Scholar]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analyses of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Paczkowska, M.; Barenboim, J.; Sintupisut, N.; Fox, N.S.; Zhu, H.; Abd-Rabbo, D.; Mee, M.W.; Boutros, P.C.; PCAWG Drivers and Functional Interpretation Working Group; Reimand, J. PCAWG Consortium, Integrative pathway enrichment analysis of multivariate omics data. Nat. Commun. 2020, 11, 735. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Jones, P.A.; Laird, P.W. Cancer epigenetics comes of age. Nat. Genet. 1999, 21, 163–167. [Google Scholar] [CrossRef]
- Shilen, A.; Malkin, D. Copy number variations and cancer. Genome Med. 2009, 1, 62. [Google Scholar] [CrossRef]
- Aradhya, S.; Cherry, A.M. Array-based comparative genomic hybridization: Clinical contexts for targeted and whole-genome designs. Genet. Med. 2007, 9, 553–559. [Google Scholar] [CrossRef]
- Dimitriadou, E.; Vermeesch, J.R.; Array, C.G.H. Fluorescence In Situ Hybridization (FISH). In Springer Protocols Handbooks; Liehr, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar] [CrossRef]
- Hu, L.; Ru, K.; Zhang, L.; Huang, Y.; Zhu, X.; Liu, H.; Zetterberg, A.; Cheng, T.; Miao, W. Fluorescence in situ hybridization (FISH): An increasingly demanded tool for biomarker research and personalized medicine. Biomarker Res. 2014, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Kok, V.C. Genomic mutational profiles of metastatic breast cancer: Obtaining them early and during the continuum of oncological care. Transl. Cancer Res. 2017, 6, S391–S394. [Google Scholar] [CrossRef]
- Roy-Chowdhuri, S.; de Melo Gagliato, D.; Routbort, M.J.; Patel, K.P.; Singh, R.R.; Broaddus, R.; Lazar, A.J.; Sahin, A.; Alvarez, R.H.; Moulder, S.; et al. Multigene clinical mutational profiling of breast carcinoma using next-generation sequencing. Am. J. Clin. Pathol 2015, 144, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184.e7. [Google Scholar] [CrossRef]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.C.; Massard, C.; Levy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, J.; Sokol, E.S.; Hartmaier, R.J.; Trabucco, S.E.; Frampton, G.M.; Goldberg, M.E.; Albacker, L.A.; Daemen, A.; Manning, G. The genomic landscape of metastatic breast cancer: Insights from 11,000 tumors. PLoS ONE 2020, 15, e0231999. [Google Scholar] [CrossRef]
- Ng, C.K.Y.; Bidard, F.C.; Piscuoglio, S.; Geyer, F.C.; Lim, R.S.; de Bruijn, I.; Shen, R.; Pareja, F.; Berman, S.H.; Wang, L.; et al. Genetic Heterogeneity in Therapy-Naive Synchronous Primary Breast Cancers and Their Metastases. Clin. Cancer Res. 2017, 23, 4402–4415. [Google Scholar] [CrossRef]
- Pasha, N.; Turner, N.C. Understanding and overcoming tumor heterogeneity in metastatic breast cancer treatment. Nat. Cancer 2021, 2, 680–692. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef]
- Beelen, K.; Hoefnagel, L.D.; Opdam, M.; Wesseling, J.; Sanders, J.; Vincent, A.D.; van Diest, P.J.; Linn, S.C. PI3K/AKT/mTOR pathway activation in primary and corresponding metastatic breast tumors after adjuvant endocrine therapy. Int. J. Cancer 2014, 135, 1257–1263. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, P.; Spangle, J.M.; Von, T.; Roberts, T.M.; Lin, N.U.; Krop, I.E.; Winer, E.P.; Zhao, J.J. PI3K-p110alpha mediates resistance to HER2-targeted therapy in HER2+, PTEN-deficient breast cancers. Oncogene 2016, 35, 3607–3612. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Lee, E.; Won, H.H. Comprehensive characterization of distinct genetic alterations in metastatic breast cancer across various metastatic sites. NPJ Breast Cancer 2021, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Tijhuis, A.E.; Johnson, S.C.; McClelland, S.E. The emerging links between chromosomal instability (CIN), metastasis, inflammation and tumour immunity. Mol. Cytogenet. 2019, 12, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salgueiro, L.; Buccitelli, C.; Rowald, K.; Somogyi, K.; Kandala, S.; Korbel, J.O.; Sotillo, R. Acquisition of chromosome instability is a mechanism to evade oncogene addiction. EMBO Mol. Med. 2020, 12, e10941. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Cantley, L.C. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef]
- Kou, F.; Wu, L.; Ren, X.; Yang, L. Chromosome Abnormalities: New Insights into Their Clinical Significance in Cancer. Mol. Ther. Oncolytics. 2020, 17, 562–570. [Google Scholar] [CrossRef]
- Mo, H.; Wang, X.; Ma, F.; Qian, Z.; Sun, X.; Yi, Z.; Guan, X.; Li, L.; Liu, B.; Xu, B. Genome-wide chromosomal instability by cell-free DNA sequencing predicts survival in patients with metastatic breast cancer. Breast 2020, 53, 111–118. [Google Scholar] [CrossRef]
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 2022, 185, 563–575.e11. [Google Scholar] [CrossRef]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.K.; Liu, Y.; Lay, F.D.; Liang, G.; Berman, B.P.; Jones, P.A. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012, 22, 2497–2506. [Google Scholar] [CrossRef]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Rice, J.C.; Futscher, B.W. Transcriptional repression of BRCA1 by aberrant cytosine methylation, histone hypoacetylation and chromatin condensation of the BRCA1 promoter. Nucleic Acids Res. 2000, 28, 3233–3239. [Google Scholar] [CrossRef]
- Rice, J.C.; Ozcelik, H.; Maxeiner, P.; Andrulis, I.; Futscher, B.W. Methylation of the BRCA1 promoter is associated with decreased BRCA1 mRNA levels in clinical breast cancer specimens. Carcinogenesis 2000, 21, 1761–1765. [Google Scholar] [CrossRef]
- Matros, E.; Wang, Z.C.; Lodeiro, G.; Miron, A.; Iglehart, J.D.; Richardson, A.L. BRCA1 promoter methylation in sporadic breast tumors: Relationship to gene expression profiles. Breast Cancer Res. Treat. 2005, 91, 179–186. [Google Scholar] [CrossRef]
- Ibragimova, I.; Cairns, P. Assays for hypermethylation of the BRCA1 gene promoter in tumor cells to predict sensitivity to PARP-inhibitor therapy. Methods Mol. Biol. 2011, 780, 277–291. [Google Scholar]
- Kirn, V.; Strake, L.; Thangarajah, F.; Richters, L.; Eischeid, H.; Koitzsch, U.; Odenthal, M.; Fries, J. ESR1-promoter-methylation status in primary breast cancer and its corresponding metastases. Clin. Exp. Metastasis 2018, 35, 707–712. [Google Scholar] [CrossRef]
- Lapidus, R.G.; Nass, S.J.; Butash, K.A.; Parl, F.F.; Weitzman, S.A.; Graff, J.G.; Herman, J.G.; Davidson, N.E. Mapping of ER gene CpG island methylation-specific polymerase chain reaction. Cancer Res. 1998, 58, 2515–2519. [Google Scholar] [PubMed]
- Widschwendter, M.; Siegmund, K.D.; Muller, H.M.; Fiegl, H.; Marth, C.; Muller-Holzner, E.; Jones, P.A.; Laird, P.W. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 2004, 64, 3807–3813. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.A.; Camargo, A.A.; Braun, K.; Slowik, R.; Cavalli, I.J.; Ribeiro, E.M.; Pedrosa Fde, O.; de Souza, E.M.; Costa, F.F.; Klassen, G. Simultaneous CXCL12 and ESR1 CpG island hypermethylation correlates with poor prognosis in sporadic breast cancer. BMC Cancer 2010, 10, 23. [Google Scholar] [CrossRef]
- Zurita, M.; Lara, P.C.; del Moral, R.; Torres, B.; Linares-Fernandez, J.L.; Arrabal, S.R.; Martinez-Galan, J.; Oliver, F.J.; Ruiz de Almodovar, J.M. Hypermethylated 14-3-3-sigma and ESR1 gene promoters in serum as candidate biomarkers for the diagnosis and treatment efficacy of breast cancer metastasis. BMC Cancer 2010, 10, 217. [Google Scholar] [CrossRef] [Green Version]
- Widschwendter, M.; Jones, P.A. DNA methylation and breast carcinogenesis. Oncogene 2002, 21, 5462–5482. [Google Scholar] [CrossRef]
- Jovanovic, J.; Ronneberg, J.A.; Tost, J.; Kristensen, V. The epigenetics of breast cancer. Mol. Oncol. 2010, 4, 242–254. [Google Scholar] [CrossRef]
- Mahmood, N.; Rabbani, S.A. DNA methylation and breast cancer: Mechanistic and therapeutic applications. Trends Cancer Res. 2017, 12, 1–6. [Google Scholar]
- de Ruijter, T.C.; van der Heide, F.; Smits, K.M.; Aarts, M.J.; van Engeland, M.; Heijnen, V.C.G. Prognostic DNA methylation markers for hormone receptor breast cancer: A systematic review. Breast Cancer Res. 2020, 22, 13. [Google Scholar] [CrossRef]
- Pang, J.M.; Dobrovic, A.; Fox, S.B. DNA methylation in ductal carcinoma in situ of the breast. Breast Cancer Res. 2013, 15, 206. [Google Scholar]
- Laird, P.W. The Power and the Promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-throughput DNA methylation using universal bead arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Carvalho, B.; Wu, H.; Brandenburg, S.A.; Jeddeloh, J.A.; Wen, B.; Feinberg, A.P. Comprehensive high-throughput arrays for relative methylation (CHARM). Genome Res. 2008, 18, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, P.E.; van Dijk, C.M.; Tollenaar, R.A.E.M.; et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2011, 44, 40–46. [Google Scholar] [CrossRef]
- Zhou, W.; Dinh, H.Q.; Ramjan, Z.; Weisenberger, D.J.; Nicolet, C.M.; Shen, H.; Laird, P.W.; Berman, B.P. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018, 50, 591–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, F.; Turcan, S.; Rimner, A.; Kaufman, A.; Giri, D.; Morris, L.G.; Shen, R.; Seshan, V.; Mo, Q.; Heguy, A.; et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci. Transl. Med. 2011, 3, 75ra25. [Google Scholar] [CrossRef] [PubMed]
- van der Auwera, I.; Yu, W.; Suo, L.; Van Neste, L.; van Dam, P.; Van Marck, E.A.; Pauwels, P.; Vermeulen, P.B.; Dirix, L.Y.; Van Laere, S.J. Array-based DNA methylation profiling for breast cancer subtype discrimination. PLoS ONE 2010, 5, e12616. [Google Scholar] [CrossRef]
- Hill, V.K.; Ricketts, C.; Bieche, I.; Vacher, S.; Gentle, D.; Lewis, C.; Maher, E.R.; Latif, F. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res. 2011, 71, 2988–2999. [Google Scholar] [CrossRef]
- Dedeurwaerder, S.; Desmedt, C.; Calonne, E.; Singhal, S.K.; Haibe-Kains, B.; Defrance, M.; Michiels, S.; Volkmar, M.; Deplus, R.; Luciani, J.; et al. DNA methylation profiling reveals a predominant immune component in breast cancers. EMBO Mol. Med. 2011, 3, 726–741. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef]
- Weisenberger, D.J. Characterizing DNA methylation alterations from The Cancer Genome Atlas. J. Clin. Investig. 2014, 124, 17–23. [Google Scholar] [CrossRef]
- Nounou, M.I.; ElAmrawy, F.; Ahmed, N.; Abdelraouf, K.; Goda, S.; Syed-Sha-Qhattal, H. Breast Cancer: Conventional Diagnosis and Treatment Modalities and Recent Patents and Technologies. Breast Cancer 2015, 9, 17–34. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the hallmarks of metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Livraghi, L.; Garber, J.E. PARP inhibitors in the management of breast cancer: Current data and future prospects. BMC Med. 2015, 13, 188. [Google Scholar] [CrossRef]
- Liu, X.; Wu, K.; Zheng, D.; Luo, C.; Fan, Y.; Zhong, X.; Zheng, H. Efficacy and Safety of PARP Inhibitors in Advanced or Metastatic Triple-Negative Breast Cancer: A Systematic Review and Meta-Analysis. Front Oncol. 2021, 11, 742139. [Google Scholar] [CrossRef]
- Taylor, A.M.; Chan, D.L.H.; Tio, M.; Patil, S.M.; Traina, T.A.; Robson, M.E.; Khasraw, M. PARP (Poly ADP-Ribose Polymerase) inhibitors for locally advanced or metastatic breast cancer. Cochrane Database Syst. Rev. 2021, 4, CD011395. [Google Scholar]
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De Luca, F.; Galardi, F.; Risi, E.; De Santo, I.; Benelli, M.; et al. Mechanisms of resistance to CDK4/6 inhibitors: Potential implications and biomarkers for clinical practice. Front Oncol. 2019, 9, 666. [Google Scholar] [CrossRef]
- Brandao, M.; Maurer, C.; Ziegelmann, P.K.; Ponde, N.F.; Ferreira, A.; Martel, S.; Piccart, M.; de Azambuja, E.; Debiasi, M.; Lambertini, M. Endocrine therapy-based treatments in hormone receptor-positive/HER2-negative advanced breast cancer: Systematic review and network meta-analysis. ESMO Open 2020, 5, e000842. [Google Scholar] [CrossRef]
- O’Leary, B.; Cutts, R.J.; Liu, Y.; Hrebien, S.; Huang, X.; Fenwick, K.; Andre, F.; Loibl, S.; Loi, S.; Garcia-Murillas, I.; et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 2018, 8, 1390–1403. [Google Scholar] [CrossRef]
- Oser, M.G.; Fonseca, R.; Chakraborty, A.A.; Brough, R.; Spektor, A.; Jennings, R.B.; Flaifel, A.; Novak, J.S.; Gulati, A.; Buss, E.; et al. Cells Lacking the RB1 Tumor Suppressor Gene Are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov. 2019, 9, 230–247. [Google Scholar] [CrossRef]
- Alves, C.L.; Ehmsen, S.; Terp, M.H.; Portman, N.; Tuttolomondo, M.; Gammelgaard, O.L.; Hundebol, M.F.; Kaminska, K.; Johansen, L.E.; Bak, M.; et al. Co-targeting CDK4/6 and AKT with endocrine therapy prevents progression in CDK4/6 inhibitor and endocrine therapy-resistant breast cancer. Nat. Commun. 2021, 12, 5112. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef]
- Zheng, Z.-Y.; Anurag, M.; Lei, J.T.; Cao, J.; Singh, P.; Peng, J.; Kennedy, H.; Nguyen, N.-C.; Chen, Y.; Lavere, P.; et al. Neurofibromin is an estrogen receptor-α transcriptional co-repressor in breast cancer. Cancer Cell 2020, 37, 387–402. [Google Scholar] [CrossRef]
- Tao, J.; Sun, D.; Dong, L.; Zhu, H.; Hou, H. Advancement in research and therapy of NF1 mutant malignant tumors. Cancer Cell Int. 2020, 20, 492. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schelssinger, J. Cell signaling by receptor-tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kim, E.; Wang, X.; Novitch, B.G.; Yoshikawa, K.; Chang, L.-S.; Zhu, Y. ERK inhibition rescues defects in fate specification of Nf1-deficient neural progenitors and brain abnormalities. Cell 2012, 150, 816–830. [Google Scholar] [CrossRef]
- Bok, S.; Shin, D.Y.; Yallowitz, A.R.; Eiseman, M.; Cung, M.; Xu, R.; Li, N.; Sun, J.; Williams, A.L.; Scott, J.E.; et al. MEKK2 mediates aberrant ERK activation in neurofibromatosis type 1. Nat. Commun. 2020, 11, 5704. [Google Scholar] [CrossRef]
- Plimack, E.R.; Kantarjian, H.M.; Issa, J.P. Decitabine and its role in the treatment of hematopoietic malignancies. Leuk Lymphoma 2007, 48, 1472–1481. [Google Scholar] [CrossRef]
- Issa, J.P. The myelodysplastic syndrome as a prototypical epigenetic disease. Blood 2013, 121, 3811–3817. [Google Scholar] [CrossRef]
- Navada, S.C.; Steinmann, J.; Lubbert, M.; Silverman, L.R. Clinical development of demethylating agents in hematology. J. Clin. Investig. 2014, 124, 40–46. [Google Scholar] [CrossRef]
- Pappalardi, M.B.; Keenan, K.; Cockerill, M.; Kellner, W.A.; Stowell, A.; Sherk, C.; Wong, K.; Pathuri, S.; Briand, J.; Steidel, M.; et al. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat. Cancer 2021, 2, 1002–1017. [Google Scholar] [CrossRef]
- Matei, D.E.; Nephew, K.P. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol. Oncol. 2010, 116, 195–201. [Google Scholar] [CrossRef]
- Juo, Y.Y.; Gong, X.J.; Mishra, A.; Cui, X.; Baylin, S.B.; Azad, N.S.; Ahuja, N. Epigenetic therapy for solid tumors: From bench science to clinical trials. Epigenomics 2015, 7, 215–235. [Google Scholar] [CrossRef]
- Parker, W.B.; Thottassery, J.V. 5-Aza-4′-thio-2′-deoxycytidine, a New Orally Bioavailable Nontoxic “Best-in-Class”: DNA Methyltransferase 1-Depleting Agent in Clinical Development. J. Pharmacol. Exp. Ther. 2021, 379, 211–222. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef]
- Mehdipour, P.; Marhon, S.A.; Ettayebi, I.; Chakravarthy, A.; Hosseini, A.; Wang, Y.; de Castro, F.A.; Loo Yau, H.; Ishak, C.; Abelson, S.; et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 2020, 588, 169–173. [Google Scholar] [CrossRef]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef]
- Bowling, E.A.; Wang, J.H.; Gong, F.; Wu, W.; Neill, N.J.; Kim, I.S.; Tyagi, S.; Orellana, M.; Kurley, S.J.; Dominguez-Vidana, R.; et al. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell 2021, 184, 384–403.e21. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study | Patients | Samples | Subtypes | Sequencing Approach |
|---|---|---|---|---|
| Roy-Chowdhuri et al. [104] | 354 | 305 primary 110 recurrent/metastatic | 224 ER/PR+ HER2- 33 ER/PR+ HER2+ 8 ER/PR- HER2+ 89 ER/PR- HER2- | A 46 cancer-related gene panel |
| Yates et al. [105] | 170 | 148 locoregional 79 metastatic 72 unknown | 87 ER+/HER2- 34 HER2+ 37 TNBC 12 unknown | A 365 gene panel |
| Lefebvre et al. [106] | 216 | Paired metastatic and blood samples | 143 HR+/HER2- 51 HR-/HER2- 14 HER2+ | WES |
| Ng et al. [108] | 9 | Paired primary and synchronous metastatic samples | 3 HR+/HER2+ 2 HR+/HER2- 2 HR-/HER2+ 2 TNBC | WES |
| Angus et al. [14] | 442 | Metastatic samples | 279 ER+/HER2- 49 ER+/HER2+ 28 ER-/HER2+ 58 TNBC 28 unknown | WES |
| Bertucci et al. [15] | 617 | 543 metastatic samples 74 breast tumors | 381 ER+/HER2- 30 HER2+ 182 TNBC 24 unknown | WES |
| Paul et al. [16] | 66 | 28 paired primary and metastatic samples 38 unpaired | 3 HR+/HER2+ 43 HR+/HER2- 5 HR-/HER2+ 12 TNBC 3 unknown | WES and WGS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Lu, T.; Weisenberger, D.J.; Liang, G. The Multi-Omic Landscape of Primary Breast Tumors and Their Metastases: Expanding the Efficacy of Actionable Therapeutic Targets. Genes 2022, 13, 1555. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13091555
Yang G, Lu T, Weisenberger DJ, Liang G. The Multi-Omic Landscape of Primary Breast Tumors and Their Metastases: Expanding the Efficacy of Actionable Therapeutic Targets. Genes. 2022; 13(9):1555. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13091555
Chicago/Turabian StyleYang, Guang, Tao Lu, Daniel J. Weisenberger, and Gangning Liang. 2022. "The Multi-Omic Landscape of Primary Breast Tumors and Their Metastases: Expanding the Efficacy of Actionable Therapeutic Targets" Genes 13, no. 9: 1555. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13091555