
Embryonic Heterogeneity of Smooth Muscle Cells in the Complex Mechanisms of Thoracic Aortic Aneurysms


1
Saha Cardiovascular Research Center, College of Medicine, University of Kentucky, Lexington, KY 40536, USA
2
Saha Aortic Center, College of Medicine, University of Kentucky, Lexington, KY 40536, USA
3
Department of Physiology, College of Medicine, University of Kentucky, Lexington, KY 40536, USA
*
Author to whom correspondence should be addressed.
Genes 2022, 13(9), 1618; https://0-doi-org.brum.beds.ac.uk/10.3390/genes13091618
Submission received: 4 August 2022
/
Revised: 2 September 2022
/
Accepted: 5 September 2022
/
Published: 9 September 2022
(This article belongs to the Special Issue Molecular Mechanisms of Vascular Disease)
Abstract
:Smooth muscle cells (SMCs) are the major cell type of the aortic wall and play a pivotal role in the pathophysiology of thoracic aortic aneurysms (TAAs). TAAs occur in a region-specific manner with the proximal region being a common location. In this region, SMCs are derived embryonically from either the cardiac neural crest or the second heart field. These cells of distinct origins reside in specific locations and exhibit different biological behaviors in the complex mechanism of TAAs. The purpose of this review is to enhance understanding of the embryonic heterogeneity of SMCs in the proximal thoracic aorta and their functions in TAAs.
1. Introduction
Thoracic aortic aneurysms (TAAs) are life-threatening diseases defined as a dilatation of the aortic wall in the thoracic region [1]. TAAs occur either sporadically or in association with a genetic condition, including mutations in FBN1 (encoding fibrillin-1); ACTA2 (encoding α-smooth muscle actin); MYH11 (encoding myosin heavy chain 11); and genes of transforming growth factor (TGF)-β and its receptors [2,3,4,5,6,7,8]. Despite the heterogeneous causes, a common feature of TAAs is the regional specificity that aortic dilatations occur predominantly in the proximal region: the aortic root and ascending aorta [9,10]. For example, patients with Marfan syndrome (MFS) and Ehlers-Danlos syndrome exhibit TAA formation preferentially in the aortic root [11,12,13], and TAAs in Loeys-Dietz syndrome (LDS) and Turner syndrome occur in the aortic root and the ascending aorta [14,15,16,17]. Another example is that bicuspid aortic valve (BAV) leads to TAA formation in the ascending aorta [18]. Multiple mouse models mimic these regional specificities of TAAs. MFS and LDS mouse models have luminal dilatations in the proximal thoracic aorta [19,20,21]. TAAs induced by chronic angiotensin II infusion, representing sporadic TAAs, are located mainly in the ascending aorta [22,23]. Several mechanisms have been reported as a determinant of the regional specificity of TAAs, such as hemodynamic effects due to the complex blood flow [24], the nonuniformity of vascular components across the aorta [25], and embryonic heterogeneity of SMCs [26,27].
SMCs are the most abundant cell type of the aortic wall [28]. Aortic SMCs are derived embryonically from several origins: second heart field (SHF), cardiac neural crest (CNC), somite, and splanchnic mesoderm [29,30]. In the disease-prone proximal thoracic aorta, SMCs are derived from both the SHF and CNC [27,30,31,32,33]. In the past decade, multiple studies have uncovered disparate biological functions of SMCs between their embryonic origins and the pathophysiology of aortic diseases, including TAAs [32,33,34,35,36,37,38,39,40]. This review highlights publications investigating the role of SMC origins and discusses functional divergences of these origins in the development of TAAs.
2. Distributions of CNC- and SHF-Derived SMCs
The CNC is composed of mesenchymal cells derived from the ectoderm [41], which migrates into pharyngeal arches and the outflow tract. The SHF is derived from the mesoderm that forms a part of the cardiac crescent and migrates into the heart tube [42,43]. SHF-derived cells in the heart tube constitute the right ventricle and the proximal thoracic aorta. While selected cells of these origins are differentiated into endothelial cells and fibroblasts, most CNC- and SHF-derived cells in the thoracic aorta are differentiated into SMCs [30].
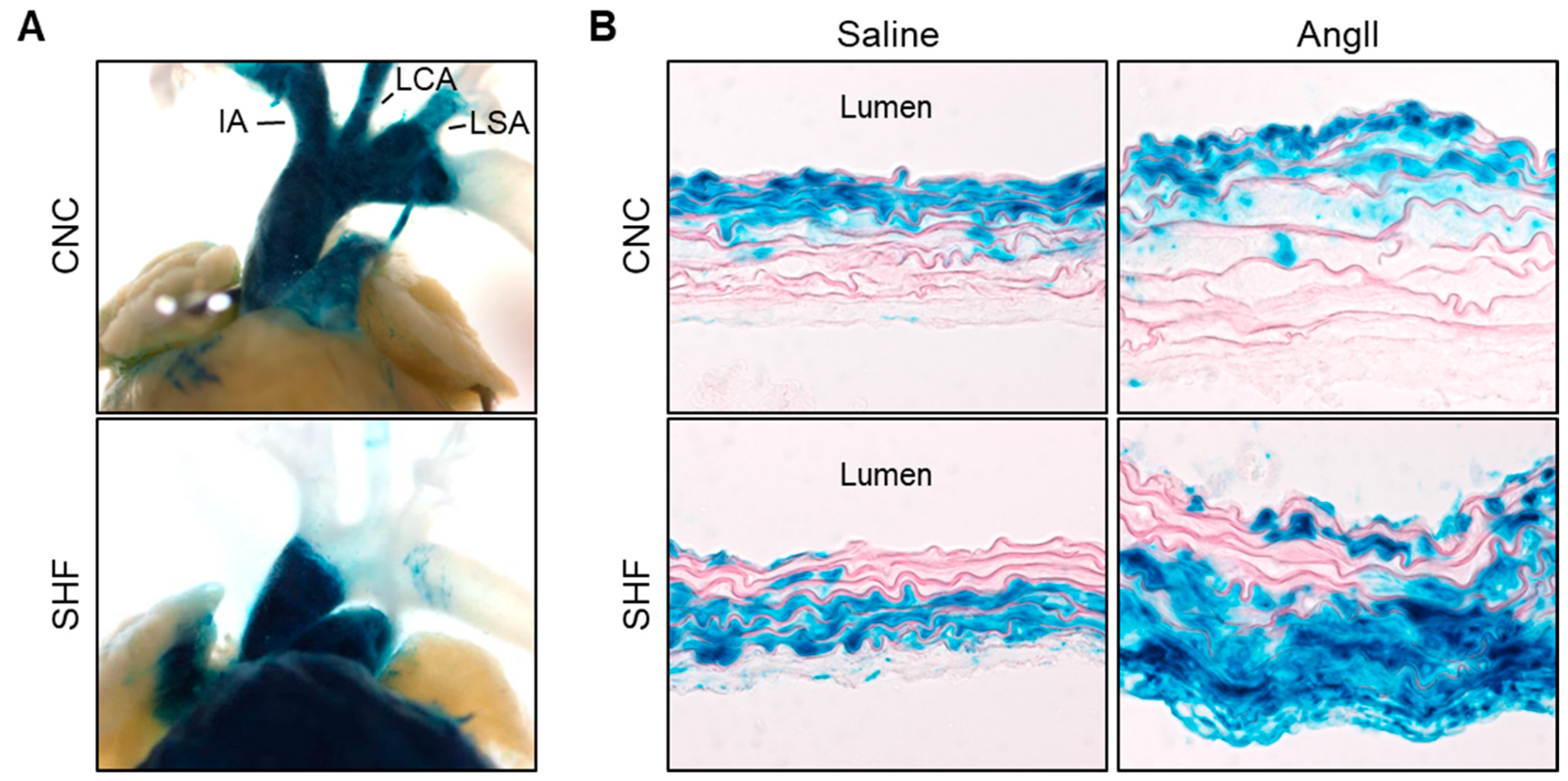
The distribution of CNC-derived SMCs in the proximal thoracic aorta was originally determined by Jiang et al. [44]. A fate-mapping study using mice expressing Cre driven by a Wnt1 promoter revealed that CNC-derived SMCs populate the thoracic aorta from the ascending aorta and throughout the aortic arch (Figure 1A). This distinct distribution has been validated by multiple studies [30,33,45,46,47], and Wnt1-Cre is now a common promoter in studies of CNC-derived SMCs. As shown in cross sections of aortic tissue, CNC-derived SMCs are distributed in the whole media of the posterior curvature of the ascending aorta, but only in the inner media of the anterior curvature (Figure 1B) [30].
SHF-derived cells were initially mapped using avian systems [48]. A fluorescent dye was microinjected into the SHF of chick embryos and the stained cells were tracked. SHF-derived cells migrate into the myocardial outflow myocardium and the outflow tract. These findings were confirmed by subsequent studies using fate mapping in mouse models [30,33,34,49]. Several promoters are available for Cre to track SHF-derived cells in mice: Nkx2.5, Mef2c, and Islet1. Despite some disparities of distributions in the myocardium, these promoters demonstrate consistent distributions in the proximal thoracic aorta that SHF-derived cells populate the aortic root and ascending aorta [30,33,34,49] (Figure 1A). Unlike CNC-derived cells, SHF-derived cells do not extend to the aortic arch. SHF-derived SMCs also have a unique distribution in the media [30]. SHF-derived SMCs are present mainly in the outer media of the ascending aorta (Figure 1B). Thus, the proximal thoracic aorta contains overlapping SMCs from both CNC and SHF origins, and these origins show a spatially distinct distribution.
In humans, aortic medial pathologies, such as a loss of SMCs and collagen deposition, exhibit a gradient across the media that increases from the luminal to the adventitial aspects [23,32]. Aortic dissection occurs preferentially in the outer third of the aortic media [50]. Multiple TAA mouse models also exhibit outer media-dominant pathologies, such as thickening and hemorrhage (Figure 1B) [32,51,52,53]. Thus, medial pathologies show a gradient toward the outer medial aspect in human and mouse TAAs. The gradient of medial pathologies in TAAs corresponds to the distribution of embryologic origins of SMCs that has been shown in mouse studies, indicating that SMCs of different embryonic origins have different functions in the pathophysiology of TAAs.
3. Functional Differences between Embryonic Origins of SMCs in Development of TAAs
In the past decade, multiple studies have uncovered functional differences between CNC- and SHF-derived SMCs in maintaining aortic structure and function (Table 1 and Table 2) [32,33,34,35,36,37,38].
3.1. Marfan Syndrome (MFS)
MFS is a multisystem disorder resulting from mutations in FBN1, encoding fibrillin-1 [11]. TAAs are a devastating manifestation of this syndrome. There is evidence that aortic TGF-β is upregulated in a mode that corresponds with luminal dilatations in MFS [15,55]. The impact of SMC origins on the dysregulation of TGF-β signaling has been investigated using induced pluripotent stem cells (iPSCs) [36]. iPSCs were generated from either patients with MFS or control subjects. Subsequently, iPSCs were differentiated into lateral mesoderm-, paraxial mesoderm-, and neural crest-derived SMCs. Compared to control subjects, the abundance of TGF-β ligands was increased in MFS-SMCs derived from the neural crest, but not from other origins (Table 2). In addition, neural crest-derived MFS-SMCs exhibited severe abnormal organization of extracellular microfibrils. These findings suggest that neural crest-derived SMCs are more susceptible to FBN1 mutations than SMCs from other origins.
The NOTCH1 signaling pathway is important for cardiovascular development and aortic integrity [56,57]. The heterozygous deletion of NOTCH1 in pan-SMCs augmented luminal dilatations in the aortic sinus and disrupted the extracellular matrix in Fbn1 haploinsufficient (Fbn1C1041G/+) mice [37]. Of note, the heterozygous deletion of NOTCH1 in SHF-, but not CNC-, derived cells had a tendency to recapitulate these aortic pathologies (p = 0.08, Table 1). In contrast to the human iPSC data, mouse models revealed a potential role of SHF-derived cells in TAA formation of MFS mice through NOTCH1-mediated mechanisms.
Single-cell RNA sequencing (scRNAseq) using a fate-mapping strategy in mice enables the precise and unbiased determination of biological differences of SMCs between origins. A recent study by Pedroza et al. performed scRNAseq in the proximal thoracic aorta of Fbn1 haploinsufficient mice with tdTomato reporter driven by Nkx2.5 [39]. Cells were selected based on tdTomato signals in SHF-derived cells, and transcriptomes were compared between origins. CNC-derived SMCs displayed a chondrogenic phenotype, whereas SHF-derived SMCs had abundant multiple collagen genes (Table 2). In addition, the transcriptional activity of TWIST1, a mediator of pathologic fibrosis, was enhanced in SHF-derived SMCs compared to CNC-derived SMCs. In MFS, genetic mutations on Fbn1 lead to multiple functional alterations of SMCs in an embryonic origin-specific manner. However, its impact on TAA formation is not fully understood. Further in vivo studies with genetic manipulations in each origin would be helpful to understand the molecular basis of embryonic differences in the pathophysiology of TAAs in MFS.
3.2. Loeys-Dietz Syndrome (LDS)
Patients with LDS have an aggressive TAA formation caused by mutations in genes encoding either type 1 or 2 TGF-β receptors [14] and the downstream pathways. Although TGF-β receptors are obligatory for TGF-β signaling, LDS exhibits characteristics that have been interpreted as overactivated TGF-β pathways including increased SMAD2/3 phosphorylation in the aorta [21]. In vitro experiments using iPSCs from LDS patients with gene mutations on TGFBR1 (TGFBR1A230T) revealed that TGFBR1 mutation downregulates SMAD3 phosphorylation in SHF-derived SMCs, whereas it is not altered in CNC-derived SMCs (Table 2) [38]. The same group also investigated the impact of lineage-specific SMAD3 mutation on aortic TGF-β signaling activity [35]. Aortic SMAD2 phosphorylation was not changed by SMAD3 mutations in SHF-derived SMCs, but SMAD3 mutations increased SMAD2 phosphorylation in CNC-derived SMCs (Table 2). Collectively, CNC- and SHF-derived SMCs demonstrate different responses to different mutations on TGF-β signaling molecules in LDS.
Mice with heterozygous missense mutation on Tgrbr1 (Tgrbr1M318R/+) develop aortic root aneurysms and medial disruptions that mimic many facets of aortic pathologies in patients with LDS [21]. Distinct properties of CNC- and SHF-derived SMCs have also been observed in this LDS mouse model [33]. In vitro experiments defined that SHF-derived SMCs show impaired SMAD2/3 activation in response to TGF-β stimulation and an increased abundance of TGF-β ligands. In contrast, CNC-derived SMCs preserve TGF-β signaling potential without the alteration of TGF-β abundance. Of interest, aortic root dilatations are ameliorated by SMAD2 deletion in cells derived from the CNC, but not SHF in mice (Table 1). These findings indicate a critical role of CNC-derived SMCs in the development of TAAs through TGF-β signaling.
The constitutive deletion of TGFBR2 in pan-SMCs causes cardiovascular defects and embryonic lethality in mice [58]. Consistent with these phenotypes, TGFBR2 deletion in either CNC- or SHF-derived cells also causes vascular malformation (Table 1) [32,54]. CNC-specific TGFBR2 deficient mice die in the early postnatal phase with persistent truncus arteiosus and craniofacial defects [54]. TGFBR2 deletion in SHF-derived cells induces prenatal death around E11.5–12.5 with dilatation of the outflow tract and retroperitoneal hemorrhage [32]. Thus, both CNC and SHF origins play an important role in aortic development through TGFBR2.
There is compelling evidence that the renin-angiotensin system exerts a pivotal role in the development of TAAs [59]. Losartan, an angiotensin receptor blocker, ameliorates aneurysm formation in multiple mouse models, including LDS [21,60,61]. In Tgrbr1M318R/+ LDS mice, mRNA abundance of Agtr1a encoding angiotensin II (AngII) type 1a receptor is increased in SHF-derived SMCs, but not in CNC-derived cells [33]. In agreement, in vitro experiments revealed that AngII stimulation upregulates Tgfb1 and Tgfb3 mRNA in SHF-derived cells, which is suppressed by losartan. However, in vivo studies using LDS mice demonstrated that Agtr1a deletion in SHF-derived cells results in only a modest reduction in aortic dilatations (Table 1) [40]. Since TAA formation in LDS mice is attenuated remarkably by either pharmacological inhibition of AT1 receptors or whole-body Agtr1a genetic deficiency [21,40], it would be interesting to investigate the impact of Agtr1a in other cell types, including CNC-derived SMCs, on AngII-mediated mechanisms of TAAs in LDS.
TAAs are present in both MFS and LDS. However, the region prone to TAA formation differs between the two syndromes. Aortic dilatations in MFS are located primarily in the aortic root, whereas LDS displays aneurysm formation in both the aortic root and the ascending aorta [11,12,14,15]. Of note, the population of SMC origins is different between the aortic root and ascending aorta. The aortic root is predominantly populated with SHF-derived SMCs, while the ascending aorta is composed of both CNC- and SHF-derived SMCs [30]. SMCs show functional differences between origins (Table 1 and Table 2). Thus, the difference in SMC populations may contribute to the regional specificity of TAAs in MFS and LDS.
3.3. Angiotensin II-Mediated TAAs
AngII infusion leads to aortopathies, including luminal dilatation and medial thickening, in the ascending aorta of mice [22,23,32,62,63]. SMC-specific deletion of low-density lipoprotein receptor-related protein 1 (LRP1) that plays a critical role in extracellular matrix maturation augments AngII-induced aortopathies [64,65]. Of note, SHF-specific LRP1 deletion recapitulates the ascending aortic pathologies in AngII-infused mice with LRP1 deletion in pan-SMCs (Table 1) [32]. These data suggest that SHF-derived cells exert a critical role in AngII-induced TAA formation. scRNAseq using Mef2c-Cre ROSA26RmTmG mice demonstrated that a short-interval of AngII infusion decreased mRNA abundance of TGF-β receptors (Tgfbr1, Tgfbr2) in SHF-derived SMCs prior to TAA formation [32]. Thus, AngII compromises the TGF-β signaling pathway in SHF-derived SMCs that is vital in maintaining the aortic integrity. In contrast, there are no publications describing LRP1 in CNC-derived cells. In addition, transcriptomic alteration in CNC-derived cells by AngII infusion has not been determined. Further study, including scRNAseq, is desirable to uncover the role of CNC-derived cells in AngII-mediated aortopathy formation.
AngII-induced medial thickening shows a transmedial gradient that is dominant in the outer media. This pathological gradient is consistent with the distribution of SHF-derived SMCs (Figure 1B) [27,32], suggesting the susceptibility of SHF-derived SMCs to AngII-induced pathologies. Since the gradient of medial thickening is observed in other TAA mouse models and human TAAs [23,32,51,52,53], SHF-derived SMCs may play an important role in the pathophysiology of TAAs. It will be fascinating to investigate the molecular mechanisms of how SHF-derived cells are involved in the transmedial gradient of medial pathologies in TAAs.
3.4. Other Aortic Diseases
Elastin is the major extracellular component of the aorta and a key determinant factor for aortic resilience [28]. Numerous studies have reported elastic fiber disruption as a key structural alteration in TAAs [66]. Nevertheless, genetic deletion of elastin did not cause TAA formation. Whole-body elastin deletion led to aortic stenosis by neointimal hyperplasia of SMCs in the proximal thoracic aorta [31,67,68]. Of note, elastin deletion in cells from either CNC- or SHF-derived cells also developed neointimal SMC hyperplasia (Table 1) [34]. Interestingly, the aortic neointima, despite being adjacent to the CNC-derived cells, is predominantly composed of SHF-lineage cells.
CNC- and SHF-derived cells contribute to aortic valve development in addition to the proximal thoracic aorta [69]. Lineage tracking studies using Wnt1 and Nkx2.5 promotors revealed that right- and left-coronary leaflets are primarily composed of CNC-derived cells, whereas the non-coronary leaflet is derived from the SHF. These origins are associated with the pathophysiology of BAV [70]. CNC-specific Krox20 deletion leads to BAV with the fusion of non- and right-coronary leaflets [71]. Although lesions of valve fusion and the incidence of BAV vary by genes, SHF-specific deficiency of Gata6, Vangl2, Jag1, or Mib1 also displays BAV in mice [72,73,74].
Vascular Ehlers-Danlos syndrome (vEDS) is an autosomal dominant disorder caused by genetic mutations in COL3A1 [75]. Similar to MFS and LDS, vEDS also shows the regional specificity of TAAs that the proximal thoracic region is dominant for aneurysm formation [13,76]. Although preclinical investigation of vEDS was restricted by the lack of animal models, a recent study established a mouse model that mimics multiple facets of vEDS [77]. Thus, it would be interesting to explore the contribution of SMC origins to vEDS-induced TAAs.
4. Summary
CNC- and SHF-derived SMCs reside in distinct locations of the proximal thoracic aorta. Multiple studies have uncovered embryonic origin-specific mechanisms in aortic diseases, including TAAs. However, CNC- and SHF-derived cells demonstrate distinct properties in different regions and diseases, which has painted a complex landscape for origin-specific mechanisms in aortic diseases. Since TAAs are mediated by complex mechanisms, including the alterations in the extracellular matrix, mechano-transduction, and SMC functions [78,79,80,81], it is important to continue efforts to understand the divergent behaviors of embryonic origins in the pathophysiology of TAAs.
Funding
The authors’ aortic aneurysm-related research work is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (R35HL155649) and the American Heart Association SFRN in Vascular Disease (18SFRN33900001). The content in this review is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E., Jr.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. J. Am. Coll. Cardiol. 2010, 55, e27–e129. [Google Scholar] [CrossRef] [PubMed]
- Sakai, L.Y.; Keene, D.R.; Renard, M.; De Backer, J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene 2016, 591, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007, 39, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Vranckx, R.; Khau Van Kien, P.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.M.; Brunotte, F.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Guo, D.C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Bertoli-Avella, A.M.; Gillis, E.; Morisaki, H.; Verhagen, J.M.A.; de Graaf, B.M.; van de Beek, G.; Gallo, E.; Kruithof, B.P.T.; Venselaar, H.; Myers, L.A.; et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015, 65, 1324–1336. [Google Scholar] [CrossRef]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, T.; Collod-Beroud, G.; Akiyama, T.; Abifadel, M.; Harada, N.; Morisaki, T.; Allard, D.; Varret, M.; Claustres, M.; Morisaki, H.; et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet. 2004, 36, 855–860. [Google Scholar] [CrossRef]
- Albornoz, G.; Coady, M.A.; Roberts, M.; Davies, R.R.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Familial thoracic aortic aneurysms and dissections--incidence, modes of inheritance, and phenotypic patterns. Ann. Thorac. Surg. 2006, 82, 1400–1405. [Google Scholar] [CrossRef]
- Isselbacher, E.M. Thoracic and abdominal aortic aneurysms. Circulation 2005, 111, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Dietz, H.C.; Cutting, G.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Dietz, H.C. Marfan’s syndrome. Lancet 2005, 366, 1965–1976. [Google Scholar] [CrossRef]
- Wenstrup, R.J.; Meyer, R.A.; Lyle, J.S.; Hoechstetter, L.; Rose, P.S.; Levy, H.P.; Francomano, C.A. Prevalence of aortic root dilation in the Ehlers-Danlos syndrome. Genet. Med. 2002, 4, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Chen, J.; Neptune, E.R.; Judge, D.P.; Podowski, M.; Holm, T.; Meyers, J.; Leitch, C.C.; Katsanis, N.; Sharifi, N.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005, 37, 275–281. [Google Scholar] [CrossRef]
- Lindsay, M.E.; Dietz, H.C. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 2011, 473, 308–316. [Google Scholar] [CrossRef]
- Ostberg, J.E.; Donald, A.E.; Halcox, J.P.; Storry, C.; McCarthy, C.; Conway, G.S. Vasculopathy in Turner syndrome: Arterial dilatation and intimal thickening without endothelial dysfunction. J. Clin. Endocrinol. Metab. 2005, 90, 5161–5166. [Google Scholar] [CrossRef]
- Cleemann, L.; Mortensen, K.H.; Holm, K.; Smedegaard, H.; Skouby, S.O.; Wieslander, S.B.; Leffers, A.M.; Leth-Espensen, P.; Pedersen, E.M.; Gravholt, C.H. Aortic dimensions in girls and young women with turner syndrome: A magnetic resonance imaging study. Pediatric Cardiol. 2010, 31, 497–504. [Google Scholar] [CrossRef]
- Fazel, S.S.; Mallidi, H.R.; Lee, R.S.; Sheehan, M.P.; Liang, D.; Fleischman, D.; Herfkens, R.; Mitchell, R.S.; Miller, D.C. The aortopathy of bicuspid aortic valve disease has distinctive patterns and usually involves the transverse aortic arch. J. Thorac. Cardiovasc. Surg. 2008, 135, 901–907. [Google Scholar] [CrossRef]
- Judge, D.P.; Biery, N.J.; Keene, D.R.; Geubtner, J.; Myers, L.; Huso, D.L.; Sakai, L.Y.; Dietz, H.C. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Investig. 2004, 114, 172–181. [Google Scholar] [CrossRef]
- Pereira, L.; Lee, S.Y.; Gayraud, B.; Andrikopoulos, K.; Shapiro, S.D.; Bunton, T.; Biery, N.J.; Dietz, H.C.; Sakai, L.Y.; Ramirez, F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc. Natl. Acad. Sci. USA 1999, 96, 3819–3823. [Google Scholar] [CrossRef] [Green Version]
- Gallo, E.M.; Loch, D.C.; Habashi, J.P.; Calderon, J.F.; Chen, Y.; Bedja, D.; van Erp, C.; Gerber, E.E.; Parker, S.J.; Sauls, K.; et al. Angiotensin II-dependent TGF-beta signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J. Clin. Investig. 2014, 124, 448–460. [Google Scholar] [CrossRef]
- Daugherty, A.; Rateri, D.L.; Charo, I.F.; Owens, A.P.; Howatt, D.A.; Cassis, L.A. Angiotensin II infusion promotes ascending aortic aneurysms: Attenuation by CCR2 deficiency in apoE−/− mice. Clin. Sci. 2010, 118, 681–689. [Google Scholar] [CrossRef]
- Rateri, D.L.; Davis, F.M.; Balakrishnan, A.; Howatt, D.A.; Moorleghen, J.J.; O’Connor, W.N.; Charnigo, R.; Cassis, L.A.; Daugherty, A. Angiotensin II induces region-specific medial disruption during evolution of ascending aortic aneurysms. Am. J. Pathol. 2014, 184, 2586–2595. [Google Scholar] [CrossRef]
- den Reijer, P.M.; Sallee, D., 3rd; van der Velden, P.; Zaaijer, E.R.; Parks, W.J.; Ramamurthy, S.; Robbie, T.Q.; Donati, G.; Lamphier, C.; Beekman, R.P.; et al. Hemodynamic predictors of aortic dilatation in bicuspid aortic valve by velocity-encoded cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2010, 12, 4. [Google Scholar] [CrossRef]
- Jana, S.; Hu, M.; Shen, M.; Kassiri, Z. Extracellular matrix, regional heterogeneity of the aorta, and aortic aneurysm. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef]
- Topouzis, S.; Majesky, M.W. Smooth muscle lineage diversity in the chick embryo. Two types of aortic smooth muscle cell differ in growth and receptor-mediated transcriptional responses to transforming growth factor-beta. Dev. Biol. 1996, 178, 430–445. [Google Scholar] [CrossRef]
- Sawada, H.; Chen, J.Z.; Wright, B.C.; Sheppard, M.B.; Lu, H.S.; Daugherty, A. Heterogeneity of aortic smooth muscle cells: A determinant for regional characteristics of thoracic aortic aneurysms? J. Transl. Int. Med. 2018, 6, 93–96. [Google Scholar] [CrossRef]
- Shen, Y.H.; LeMaire, S.A.; Webb, N.R.; Cassis, L.A.; Daugherty, A.; Lu, H.S. Aortic aneurysms and dissections series. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e37–e46. [Google Scholar] [CrossRef]
- Majesky, M.W. Developmental basis of vascular smooth muscle diversity. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1248–1258. [Google Scholar] [CrossRef]
- Sawada, H.; Rateri, D.L.; Moorleghen, J.J.; Majesky, M.W.; Daugherty, A. Smooth muscle cells derived from second heart field and cardiac neural crest reside in spatially distinct domains in the media of the ascending aorta-Brief report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1722–1726. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.J.; Staiculescu, M.C.; Hawes, J.Z.; Cocciolone, A.J.; Hunkins, B.M.; Roth, R.A.; Lin, C.Y.; Mecham, R.P.; Wagenseil, J.E. Heterogeneous cellular contributions to elastic laminae formation in arterial wall development. Circ. Res. 2019, 125, 1006–1018. [Google Scholar] [CrossRef]
- Sawada, H.; Katsumata, Y.; Higashi, H.; Zhang, C.; Li, Y.; Morgan, S.; Lee, L.H.; Singh, S.A.; Chen, J.Z.; Franklin, M.K.; et al. Second heart field-derived cells contribute to angiotensin II-mediated ascending aortopathies. Circulation 2022, 145, 987–1001. [Google Scholar] [CrossRef]
- MacFarlane, E.G.; Parker, S.J.; Shin, J.Y.; Kang, B.E.; Ziegler, S.G.; Creamer, T.J.; Bagirzadeh, R.; Bedja, D.; Chen, Y.; Calderon, J.F.; et al. Lineage-specific events underlie aortic root aneurysm pathogenesis in Loeys-Dietz syndrome. J. Clin. Investig. 2019, 129, 659–675. [Google Scholar] [CrossRef]
- Lin, C.J.; Hunkins, B.M.; Roth, R.A.; Lin, C.Y.; Wagenseil, J.E.; Mecham, R.P. Vascular smooth muscle cell subpopulations and neointimal formation in mouse models of elastin insufficiency. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2890–2905. [Google Scholar] [CrossRef]
- Gong, J.; Zhou, D.; Jiang, L.; Qiu, P.; Milewicz, D.M.; Chen, Y.E.; Yang, B. In vitro lineage-specific differentiation of vascular smooth muscle cells in response to SMAD3 deficiency: Implications for SMAD3-related thoracic aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1651–1663. [Google Scholar] [CrossRef]
- Granata, A.; Serrano, F.; Bernard, W.G.; McNamara, M.; Low, L.; Sastry, P.; Sinha, S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat. Genet. 2017, 49, 97–109. [Google Scholar] [CrossRef]
- Koenig, S.N.; LaHaye, S.; Feller, J.D.; Rowland, P.; Hor, K.N.; Trask, A.J.; Janssen, P.M.; Radtke, F.; Lilly, B.; Garg, V. Notch1 haploinsufficiency causes ascending aortic aneurysms in mice. JCI Insight 2017, 2, e91353. [Google Scholar] [CrossRef]
- Zhou, D.; Feng, H.; Yang, Y.; Huang, T.; Qiu, P.; Zhang, C.; Olsen, T.; Zhang, J.; Chen, Y.E.; Mizrak, D.; et al. hiPSC modeling of lineage-specific smooth muscle cell defects caused by TGFBR1A230T variant, and its therapeutic implications for Loeys-Dietz syndrome. Circulation 2021, 144, 1145–1159. [Google Scholar] [CrossRef]
- Pedroza, A.J.; Dalal, A.R.; Shad, R.; Yokoyama, N.; Nakamura, K.; Cheng, P.; Wirka, R.C.; Mitchel, O.; Baiocchi, M.; Hiesinger, W.; et al. Embryologic origin influences smooth muscle cell phenotypic modulation signatures in murine Marfan syndrome aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1154–1168. [Google Scholar] [CrossRef]
- Bramel, E.E.; Bagirzadeh, R.; Saqib, M.; Creamer, T.J.; Espinoza Camejo, W.A.; Roker, L.A.; Pardo Habashi, J.; Dietz, H.C.; Gallo MacFarlane, E. Distinct contribution of global and regional angiotensin II type 1a receptor inactivation to amelioration of aortopathy in Tgfbr1M318R/+ mice. Front. Cardiovasc. Med. 2022, 9, 936142. [Google Scholar] [CrossRef]
- Hutson, M.R.; Kirby, M.L. Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin. Cell Dev. Biol. 2007, 18, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D. Making or breaking the heart: From lineage determination to morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [CrossRef]
- Lewis, A.E.; Vasudevan, H.N.; O’Neill, A.K.; Soriano, P.; Bush, J.O. The widely used Wnt1-Cre transgene causes developmental phenotypes by ectopic activation of Wnt signaling. Dev. Biol. 2013, 379, 229–234. [Google Scholar] [CrossRef]
- Makki, N.; Capecchi, M.R. Hoxa1 lineage tracing indicates a direct role for Hoxa1 in the development of the inner ear, the heart, and the third rhombomere. Dev. Biol. 2010, 341, 499–509. [Google Scholar] [CrossRef]
- Passman, J.N.; Dong, X.R.; Wu, S.P.; Maguire, C.T.; Hogan, K.A.; Bautch, V.L.; Majesky, M.W. A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1+ smooth muscle progenitor cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9349–9354. [Google Scholar] [CrossRef]
- Waldo, K.L.; Hutson, M.R.; Ward, C.C.; Zdanowicz, M.; Stadt, H.A.; Kumiski, D.; Abu-Issa, R.; Kirby, M.L. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev. Biol. 2005, 281, 78–90. [Google Scholar] [CrossRef]
- Harmon, A.W.; Nakano, A. Nkx2-5 lineage tracing visualizes the distribution of second heart field-derived aortic smooth muscle. Genesis 2013, 51, 862–869. [Google Scholar] [CrossRef]
- Osada, H.; Kyogoku, M.; Ishidou, M.; Morishima, M.; Nakajima, H. Aortic dissection in the outer third of the media: What is the role of the vasa vasorum in the triggering process? Eur. J. Cardiothorac. Surg. 2013, 43, e82–e88. [Google Scholar] [CrossRef] [Green Version]
- Wanga, S.; Hibender, S.; Ridwan, Y.; van Roomen, C.; Vos, M.; van der Made, I.; van Vliet, N.; Franken, R.; van Riel, L.A.; Groenink, M.; et al. Aortic microcalcification is associated with elastin fragmentation in Marfan syndrome. J. Pathol. 2017, 243, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Schmit, B.M.; Fu, C.; DeSart, K.; Oh, S.P.; Berceli, S.A.; Jiang, Z. Smooth muscle cell-specific Tgfbr1 deficiency promotes aortic aneurysm formation by stimulating multiple signaling events. Sci. Rep. 2016, 6, 35444. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Q.; Jiao, Y.; Qin, L.; Ali, R.; Zhou, J.; Ferruzzi, J.; Kim, R.W.; Geirsson, A.; Dietz, H.C.; et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Investig. 2014, 124, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, B.; Ito, Y.; Makita, T.; Sasaki, T.; Chai, Y.; Sucov, H.M. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFbeta receptor (Tgfbr2) mutant mice. Dev. Biol. 2006, 289, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Gerber, E.E.; Dietz, H.C. Matrix-dependent perturbation of TGFbeta signaling and disease. FEBS Lett. 2012, 586, 2003–2015. [Google Scholar] [CrossRef]
- Swiatek, P.J.; Lindsell, C.E.; del Amo, F.F.; Weinmaster, G.; Gridley, T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994, 8, 707–719. [Google Scholar] [CrossRef]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000, 14, 1343–1352. [Google Scholar] [CrossRef]
- Oshima, M.; Oshima, H.; Taketo, M.M. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 1996, 179, 297–302. [Google Scholar] [CrossRef]
- Shen, Y.H.; LeMaire, S.A. Molecular pathogenesis of genetic and sporadic aortic aneurysms and dissections. Curr. Probl. Surg. 2017, 54, 95–155. [Google Scholar] [CrossRef]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Kuang, S.Q.; Geng, L.; Prakash, S.K.; Cao, J.M.; Guo, S.; Villamizar, C.; Kwartler, C.S.; Peters, A.M.; Brasier, A.R.; Milewicz, D.M. Aortic remodeling after transverse aortic constriction in mice is attenuated with AT1 receptor blockade. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2172–2179. [Google Scholar] [CrossRef]
- Owens, A.P., 3rd; Subramanian, V.; Moorleghen, J.J.; Guo, Z.; McNamara, C.A.; Cassis, L.A.; Daugherty, A. Angiotensin II induces a region-specific hyperplasia of the ascending aorta through regulation of inhibitor of differentiation 3. Circ. Res. 2010, 106, 611–619. [Google Scholar] [CrossRef]
- Trachet, B.; Piersigilli, A.; Fraga-Silva, R.A.; Aslanidou, L.; Sordet-Dessimoz, J.; Astolfo, A.; Stampanoni, M.F.; Segers, P.; Stergiopulos, N. Ascending aortic aneurysm in angiotensin II-infused mice: Formation, progression, and the role of focal dissections. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 673–681. [Google Scholar] [CrossRef]
- Davis, F.M.; Rateri, D.L.; Balakrishnan, A.; Howatt, D.A.; Strickland, D.K.; Muratoglu, S.C.; Haggerty, C.M.; Fornwalt, B.K.; Cassis, L.A.; Daugherty, A. Smooth muscle cell deletion of low-density lipoprotein receptor-related protein 1 augments angiotensin II-induced superior mesenteric arterial and ascending aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 155–162. [Google Scholar] [CrossRef]
- Strickland, D.K.; Au, D.T.; Cunfer, P.; Muratoglu, S.C. Low-density lipoprotein receptor-related protein-1: Role in the regulation of vascular integrity. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 487–498. [Google Scholar] [CrossRef]
- Yamashiro, Y.; Yanagisawa, H. Crossing bridges between extra- and intra-cellular events in thoracic aortic aneurysms. J. Atheroscler. Thromb. 2018, 25, 99–110. [Google Scholar] [CrossRef]
- Li, D.Y.; Brooke, B.; Davis, E.C.; Mecham, R.P.; Sorensen, L.K.; Boak, B.B.; Eichwald, E.; Keating, M.T. Elastin is an essential determinant of arterial morphogenesis. Nature 1998, 393, 276–280. [Google Scholar] [CrossRef]
- Wagenseil, J.E.; Ciliberto, C.H.; Knutsen, R.H.; Levy, M.A.; Kovacs, A.; Mecham, R.P. Reduced vessel elasticity alters cardiovascular structure and function in newborn mice. Circ. Res. 2009, 104, 1217–1224. [Google Scholar] [CrossRef]
- Peterson, J.C.; Chughtai, M.; Wisse, L.J.; Gittenberger-de Groot, A.C.; Feng, Q.; Goumans, M.T.H.; VanMunsteren, J.C.; Jongbloed, M.R.M.; DeRuiter, M.C. Bicuspid aortic valve formation: Nos3 mutation leads to abnormal lineage patterning of neural crest cells and the second heart field. Dis. Models Mech. 2018, 11, dmm034637. [Google Scholar] [CrossRef]
- Kern, C.B. Excess Provisional Extracellular Matrix: A Common Factor in Bicuspid Aortic Valve Formation. J. Cardiovasc. Dev. Dis. 2021, 8, 92. [Google Scholar] [CrossRef]
- Odelin, G.; Faure, E.; Coulpier, F.; Di Bonito, M.; Bajolle, F.; Studer, M.; Avierinos, J.F.; Charnay, P.; Topilko, P.; Zaffran, S. Krox20 defines a subpopulation of cardiac neural crest cells contributing to arterial valves and bicuspid aortic valve. Development 2018, 145, dev151944. [Google Scholar] [CrossRef]
- Eley, L.; Alqahtani, A.M.; MacGrogan, D.; Richardson, R.V.; Murphy, L.; Salguero-Jimenez, A.; Sintes Rodriguez San Pedro, M.; Tiurma, S.; McCutcheon, L.; Gilmore, A.; et al. A novel source of arterial valve cells linked to bicuspid aortic valve without raphe in mice. elife 2018, 7, e34110. [Google Scholar] [CrossRef]
- Gharibeh, L.; Komati, H.; Bosse, Y.; Boodhwani, M.; Heydarpour, M.; Fortier, M.; Hassanzadeh, R.; Ngu, J.; Mathieu, P.; Body, S.; et al. GATA6 regulates aortic valve remodeling, and its haploinsufficiency leads to right-left type bicuspid aortic valve. Circulation 2018, 138, 1025–1038. [Google Scholar] [CrossRef]
- MacGrogan, D.; D’Amato, G.; Travisano, S.; Martinez-Poveda, B.; Luxan, G.; Del Monte-Nieto, G.; Papoutsi, T.; Sbroggio, M.; Bou, V.; Gomez-Del Arco, P.; et al. Sequential ligand-dependent Notch signaling activation regulates valve primordium formation and morphogenesis. Circ. Res. 2016, 118, 1480–1497. [Google Scholar] [CrossRef]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef]
- Meester, J.A.N.; Verstraeten, A.; Schepers, D.; Alaerts, M.; Van Laer, L.; Loeys, B.L. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann. Cardiothorac. Surg. 2017, 6, 582–594. [Google Scholar] [CrossRef]
- Bowen, C.J.; Calderon Giadrosic, J.F.; Burger, Z.; Rykiel, G.; Davis, E.C.; Helmers, M.R.; Benke, K.; Gallo MacFarlane, E.; Dietz, H.C. Targetable cellular signaling events mediate vascular pathology in vascular Ehlers-Danlos syndrome. J. Clin. Investig. 2020, 130, 686–698. [Google Scholar] [CrossRef]
- Halushka, M.K.; Angelini, A.; Bartoloni, G.; Basso, C.; Batoroeva, L.; Bruneval, P.; Buja, L.M.; Butany, J.; d’Amati, G.; Fallon, J.T.; et al. Consensus statement on surgical pathology of the aorta from the Society for Cardiovascular Pathology and the Association For European Cardiovascular Pathology: II. Noninflammatory degenerative diseases—Nomenclature and diagnostic criteria. Cardiovasc. Pathol. 2016, 25, 247–257. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Schwartz, M.A.; Tellides, G.; Milewicz, D.M. Role of mechanotransduction in vascular biology: Focus on thoracic aortic aneurysms and dissections. Circ. Res. 2015, 116, 1448–1461. [Google Scholar] [CrossRef]
- Michel, J.B.; Jondeau, G.; Milewicz, D.M. From genetics to response to injury: Vascular smooth muscle cells in aneurysms and dissections of the ascending aorta. Cardiovasc. Res. 2018, 114, 578–589. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Trybus, K.M.; Guo, D.C.; Sweeney, H.L.; Regalado, E.; Kamm, K.; Stull, J.T. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 26–34. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Embryonic origins of SMCs in the ascending aorta. Representative images of X-gal-stained aortic (A) tissues and (B) sections from Wnt1-Cre and Mef2c-Cre ROSA26RLacZ mice. Blue color indicates the distribution of cells driven by either Cre. CNC indicates cardiac neural crest; SHF, second heart field; IA, innominate artery; LCA, left common carotid artery; LSA, left subclavian artery. Images are cited from [30,32] with permission from Wolters Kluwer Health (2022).
Figure 1.
Embryonic origins of SMCs in the ascending aorta. Representative images of X-gal-stained aortic (A) tissues and (B) sections from Wnt1-Cre and Mef2c-Cre ROSA26RLacZ mice. Blue color indicates the distribution of cells driven by either Cre. CNC indicates cardiac neural crest; SHF, second heart field; IA, innominate artery; LCA, left common carotid artery; LSA, left subclavian artery. Images are cited from [30,32] with permission from Wolters Kluwer Health (2022).


{kind=link}
Table 1.
Aortic phenotypes caused by genetic manipulations in either SHF- or CNC-derived cells in mice.
Table 1.
Aortic phenotypes caused by genetic manipulations in either SHF- or CNC-derived cells in mice.
| Gene | Mouse Model | Aortic Phenotypes | Ref. | |
|---|---|---|---|---|
| CNC | SHF | |||
| Notch1 | Fbn1C1041G/+ | TAA ↔ | TAA ↑ (trend, p = 0.08) | [37] |
| Fbn1 | Fbn1C1041G/+ | Chondrogenic | Collagenic | [39] |
| Tgfbr2 | Spontaneous | Persistent truncus arteriosus | Outflow tract dilatation | [32,54] |
| Smad2 | Tgfbr1M318R/+ | TAA ↓ | TAA ↔ | [33] |
| Agtr1a | Tgfbr1M318R/+ | N/D | TAA ↓ (modestly) | [40] |
| Lrp1 | AngII infusion | N/D | TAA ↑ | [32] |
| Eln | Spontaneous | Neointimal hyperplasia | Neointimal hyperplasia | [34] |
N/D indicates not determined; ↑, augmented; ↔, not changed; ↓, suppressed.
Table 2.
TGF-β-related phenotypes in SHF- and CNC-derived SMCs generated from human iPSCs.
| Model | Experiment | Phenotype | Ref. | |
|---|---|---|---|---|
| CNC | SHF | |||
| iPSCs generated from MFS patients | In vitro | TGF-β1 ↑ | TGF-β1 ↔ | [36] |
| iPSCs generated from LDS patients | In vitro | pSMAD3 ↔ | pSMAD3 ↓ | [38] |
| iPSCs with LoF mutations on SMAD3 generated from a healthy donor | In vitro | pSMAD2 ↑ | pSMAD2 ↔ | [35] |
LoF indicates loss of function; ↑, augmented; ↔, not changed; ↓, suppressed.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ito, S.; Lu, H.S.; Daugherty, A.; Sawada, H. Embryonic Heterogeneity of Smooth Muscle Cells in the Complex Mechanisms of Thoracic Aortic Aneurysms. Genes 2022, 13, 1618. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13091618
AMA Style
Ito S, Lu HS, Daugherty A, Sawada H. Embryonic Heterogeneity of Smooth Muscle Cells in the Complex Mechanisms of Thoracic Aortic Aneurysms. Genes. 2022; 13(9):1618. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13091618
Chicago/Turabian StyleIto, Sohei, Hong S. Lu, Alan Daugherty, and Hisashi Sawada. 2022. "Embryonic Heterogeneity of Smooth Muscle Cells in the Complex Mechanisms of Thoracic Aortic Aneurysms" Genes 13, no. 9: 1618. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13091618
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.