
Loss of Function TGFBR2 Variant as a Contributing Factor in Generalized Pustular Psoriasis and Adult-Onset Immunodeficiency
 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients
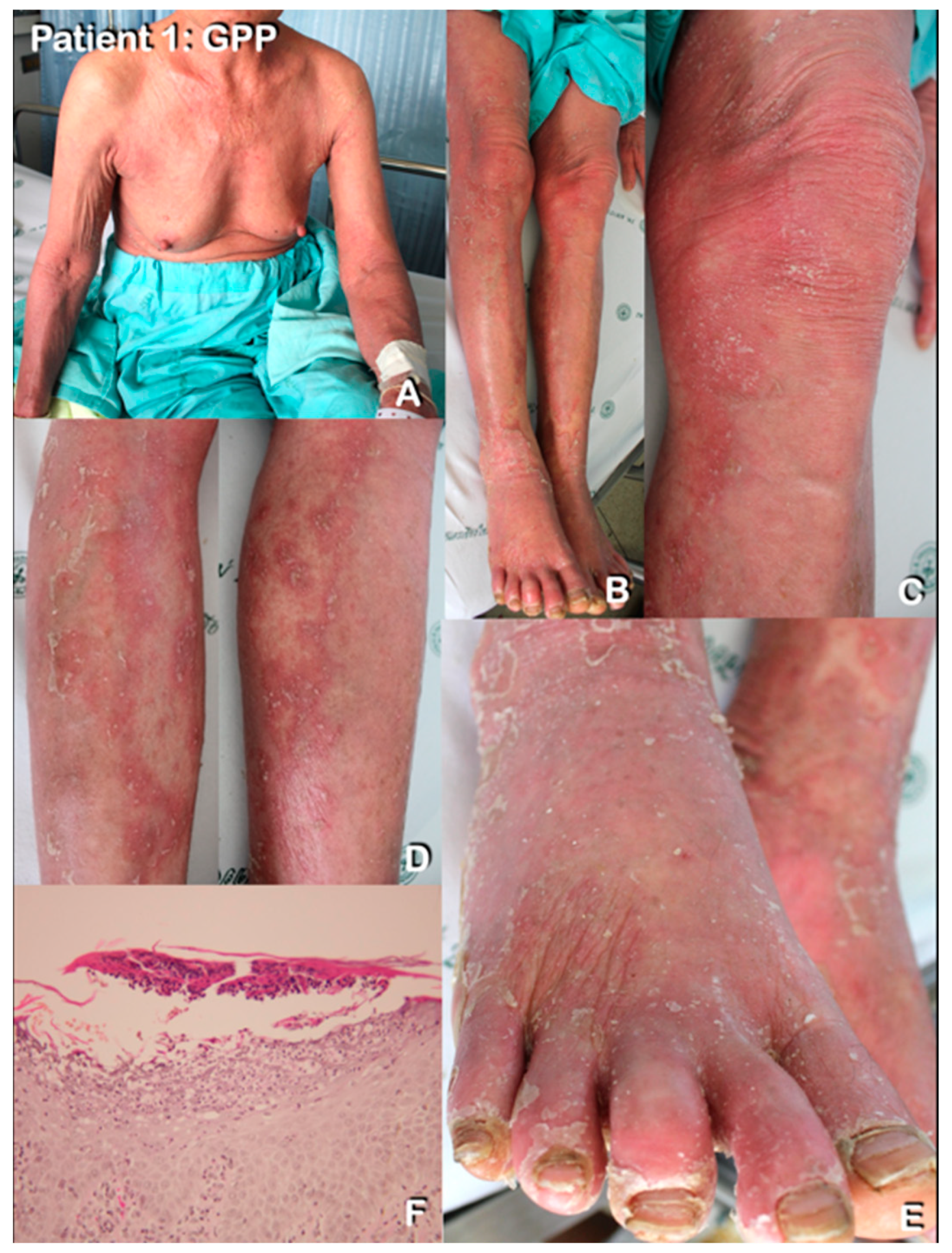
2.1.1. Patient 1
2.1.2. Patient 2
2.1.3. Patient 3
2.1.4. Patient 4
2.2. Whole Exome Sequencing and Mutation Analysis
2.3. Histopathology
2.4. Immunohistochemistry
3. Results
3.1. Whole Exome Sequence Sequencing and Bioinformatic Analysis
3.2. Histopathological Findings
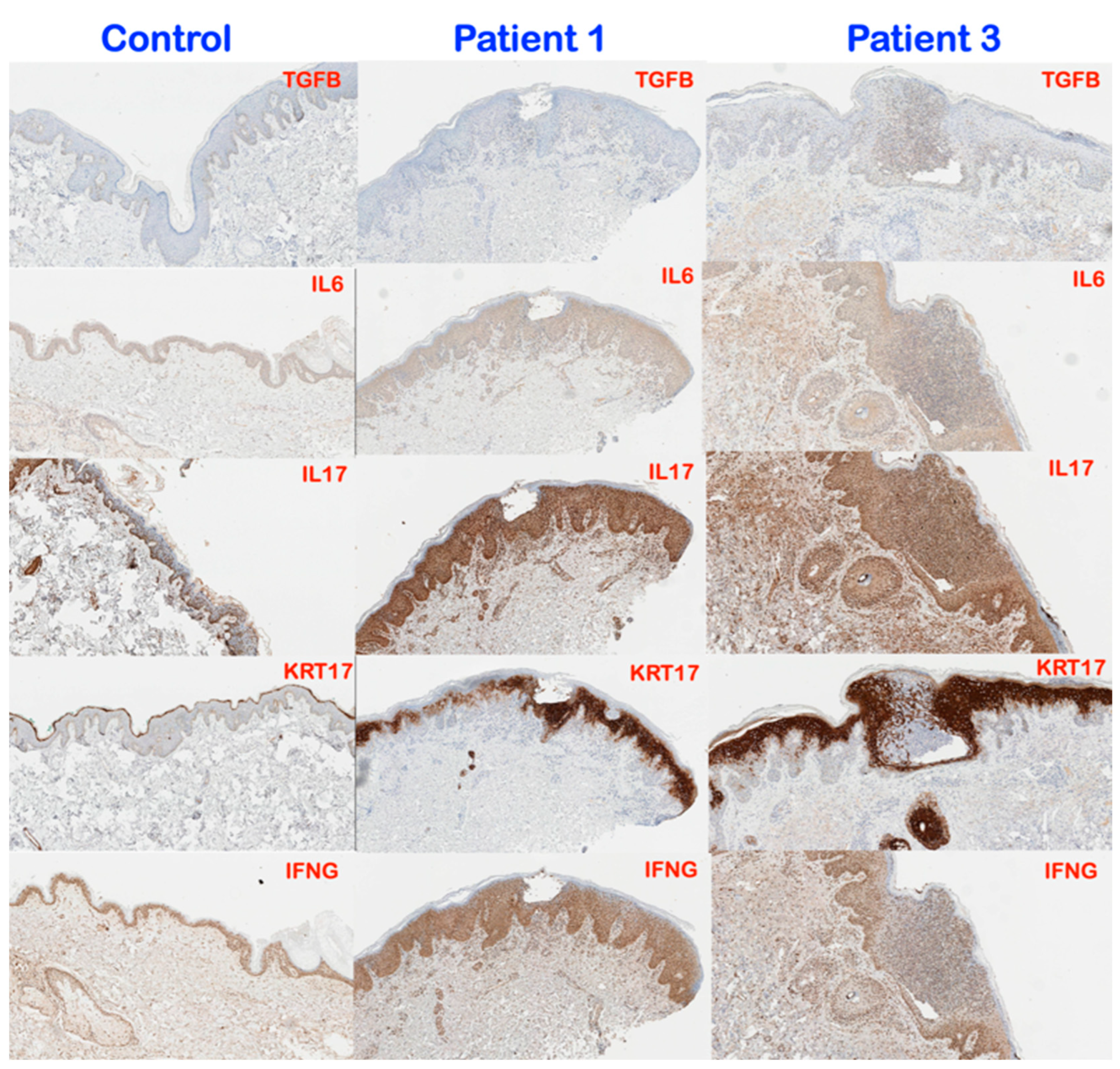
3.3. Immunohistochemical Findings
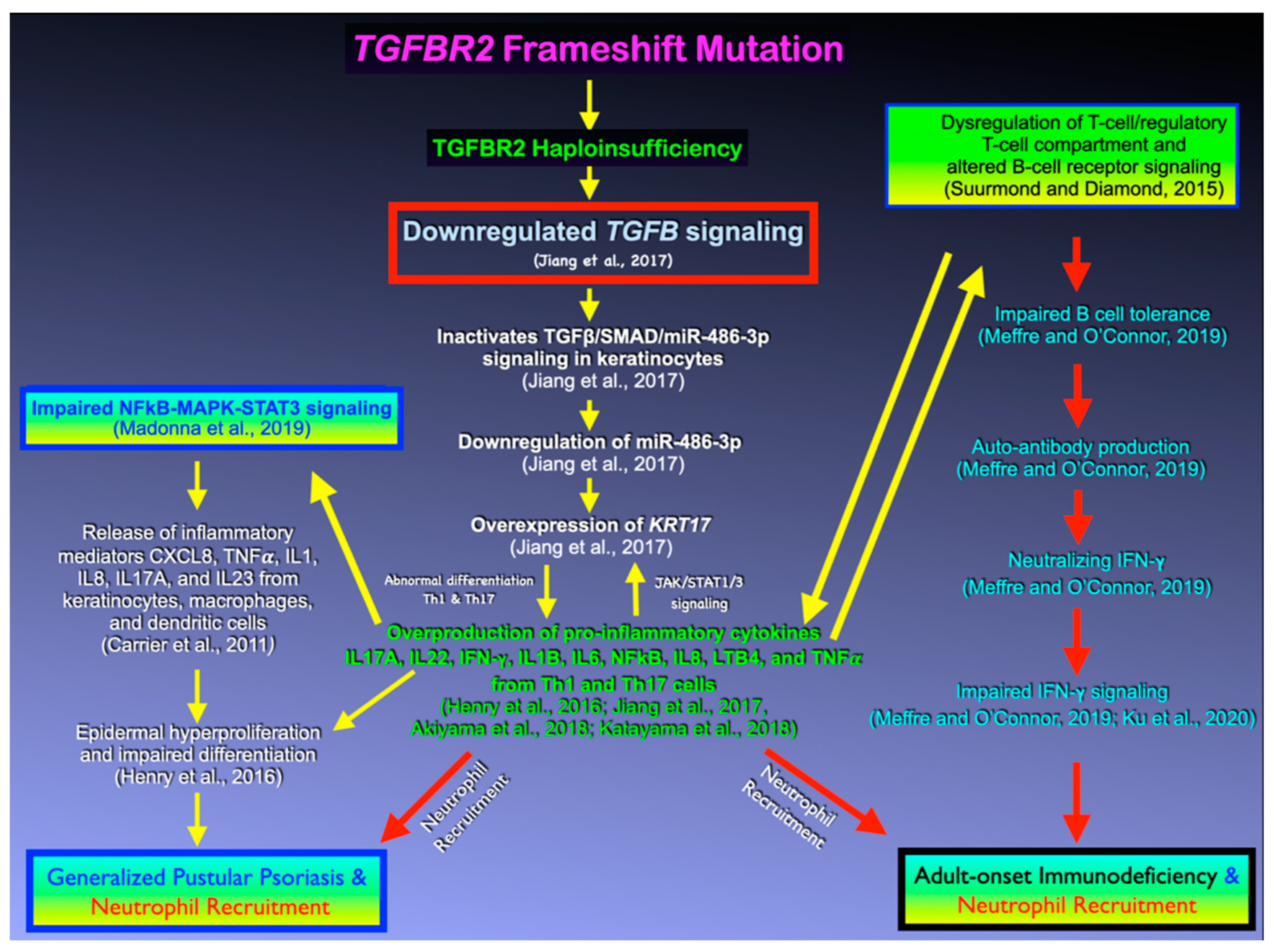
4. Discussion
4.1. TGFB Signaling, Its Antiproliferative Effect, and GPP
4.2. TGFB Signaling, Immunosuppressive Effects, and AOID
4.3. The Effects of Variants in Other Immunodeficiency Genes
5. Conclusions
- A frameshift mutation in TGFBR2 might be associated with GPP and AOID.
- A frameshift mutation in TGFBR2 is associated with overexpression of KRT17 gene expression, a hallmark of psoriatic skin lesion.
- AOID might share pathogenetic mechanisms with GPP.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uppala, R.; Tsoi, L.C.; Harms, P.W.; Wang, B.; Billi, A.C.; Maverakis, E.; Kahlenberg, J.M.; Ward, N.L.; Gudjonsson, J.E. “Autoinflammatory psoriasis”—Genetics and biology of pustular psoriasis. Cell. Mol. Immunol. 2021, 18, 307–317. [Google Scholar] [CrossRef]
- Browne, S.K. Adult-onset immunodeficiency in Thailand and Taiwan. N. Engl. J. Med. 2012, 367, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Ku, C.L.; Chi, C.Y.; von Bernuth, H.; Doffinger, R. Autoantibodies against cytokines: Phenocopies of primary immunodeficiencies? Hum. Genet. 2020, 139, 783–794. [Google Scholar] [CrossRef]
- Pithukpakorn, M.; Roothumnong, E.; Angkasekwinai, N.; Suktitipat, B.; Assawamakin, A.; Luangwedchakarn, V.; Umrod, P.; Thongnoppakhun, W.; Foongladda, S.; Suputtamongkol, Y. HLA-DRB1 and HLA-DQB1 Are Associated with Adult-Onset Immunodeficiency with Acquired Anti-Interferon-γ Autoantibodies. PLoS ONE 2015, 10, e0128481. [Google Scholar] [CrossRef] [PubMed]
- Jutivorakool, K.; Sittiwattanawong, P.; Kantikosum, K.; Hurst, C.; Kumtornrut, C.; Asawanonda, P.; Klaewsongkram, J.; Rerknimitr, P. Skin manifestations in patients with adult-onset Immunodeficiency due to anti-interferon-γ autoantibody: A Relationship with Systemic Infections. Acta Derm. Venereol. 2018, 98, 742–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantaputra, P.N.; Chuamanochan, M.; Kiratikanon, S.; Chiewchanvit, S.; Chaiwarith, R.; Intachai, W.; Quarto, N.; Tongsima, S.; McGrath, J.A.; Ngamphiw, C. A truncating variant in SERPINA3, skin pustules and adult-onset immunodeficiency. J. Dermatol. 2021, 48, e370–e371. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.; Chaowattanapanit, S.; Kiratikanon, S.; Chaiwarith, R.; Choonhakarn, C.; Intachai, W.; Quarto, N.; Tongsima, S.; Cairns, J.R.K.; Ngamphiw, C.; et al. SERPINA1, generalized pustular psoriasis, and adult-onset immunodeficiency. J. Dermatol. 2021, 48, 1597–1601. [Google Scholar] [CrossRef]
- Doi, H.; Shibata, M.A.; Kiyokane, K.; Otsuki, Y. Downregulation of TGF β isoforms and their receptors contributes to keratinocyte hyperproliferation in psoriasis vulgaris. J. Dermatol. Sci. 2003, 33, 7–16. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massagué, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef]
- Robinson, P.N.; Arteaga-Solis, E.; Baldock, C.; Collod-Beroud, G.; Booms, P.; De Paepe, A.; Dietz, H.C.; Guo, G.; Handford, P.A.; Judge, D.; et al. The molecular genetics of Marfan syndrome and related disorders. J. Med. Genet. 2006, 43, 69–787. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef] [PubMed]
- Navarini, A.A.; Burden, A.D.; Capon, F.; Mrowietz, U.; Puig, L.; Köks, S.; Kingo, K.; Smith, C.; Barker, J.N. European consensus statement on phenotypes of pustular psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1792–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.H.; Ebner, R.; Derynck, R. Inactivation of the type II receptor reveals two receptor pathways for the diverse TGF-β activities. Science 1993, 260, 1335–1338. [Google Scholar] [CrossRef]
- Jiang, M.; Sun, Z.; Dang, E.; Li, B.; Fang, H.; Li, J.; Gao, L.; Zhang, K.; Wang, G. TGFβ/SMAD/microRNA-486-3p Signaling Axis Mediates Keratin 17 Expression and Keratinocyte Hyperproliferation in Psoriasis. J. Investig. Dermatol. 2017, 137, 2177–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blauvelt, A. New Concepts in the pathogenesis and treatment of psoriasis: Key roles for IL-23, Il17A, and TGF-β1. Expert Rev. Dermatol. 2007, 2, 69–78. [Google Scholar] [CrossRef]
- Kampitak, T.; Suwanpimolkul, G.; Browne, S.; Suankratay, C. Anti-interferon γ autoantibody and opportunistic infections: Case series and review of the literature. Infection 2011, 39, 65–71. [Google Scholar] [CrossRef]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-β induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef]
- Chiang, C.C.; Cheng, W.J.; Korinek, M.; Lin, C.Y.; Hwang, T.L. Neutrophils in Psoriasis. Front. Immunol. 2019, 10, 2376. [Google Scholar] [CrossRef] [PubMed]
- Meffre, E.; O’Connor, K. C. Impaired B-cell tolerance checkpoints promote the development of autoimmune diseases and pathogenic autoantibodies. Immunol. Rev. 2019, 292, 90–101. [Google Scholar] [CrossRef]
- Akiyama, M.; Takeichi, T.; McGrath, J.A.; Sugiura, K. Autoinflammatory keratinization diseases: An emerging concept encompassing various inflammatory keratinization disorders of the skin. J. Dermatol. Sci. 2018, 90, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrier, Y.; Ma, H.L.; Ramon, H.E.; Napierata, L.; Small, C.; O’Toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M.; et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: Implications in psoriasis pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, H. Development of psoriasis by continuous neutrophil infiltration into the epidermis. Exp. Dermatol. 2018, 27, 1084–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madonna, S.; Girolomoni, G.; Dinarello, C.A.; Albanesi, C. The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis. Int. J. Mol. Sci. 2019, 20, 3318. [Google Scholar] [CrossRef] [Green Version]
- Suurmond, J.; Diamond, B. Autoantibodies in systemic autoimmune diseases: Specificity and pathogenicity. J. Clin. Investig. 2015, 125, 2194–2202. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Gender Age (year) | Diagnosis | TGFBRII Variant 1 | TGFBRII Variant 2 | JAK2 Variant | JAK3 Variant | IL17RA Variant | IL12RB2 Variant | CARD14 Variant | TYK2 Variant |
|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Female (70 Yr ) | GPP | NM_001024847.2: c.458del; NP_001020018.1: p.Lys153SerfsTer35; rs79375991 3-30691880-AA-A (GRCh37) | NM_001289905.1: c.833G>A; NP_001276834.1: p.Arg278His; rs141467790; AF = 0.00128 | NM_001258214.1: c.52T>G; NP_001245143.1: p.Trp18Gly; rs751550346; AF = 0.000003981 | NM_024110.4; c.2473G>A; NP_077015.2; p.Ala825Thr; rs538251591 | ||||
| Patient 2 | Female (24 year) | GPP | NM_001024847.2: c.458del; NP_001020018.1: p.Lys153SerfsTer35; rs79375991 3-30691880-AA-A (GRCh37) | NM_001024847.2: c.1019C>T; NP_001020018.1: p.Thr340Met; rs34833812; AF = 0.001062 Het/Hom 4/0 | NM_003331.5: c.2977C>T; NP_003322.3: p.His993Tyr; rs201397594; AF = 0.0001920 | |||||
| Patient 3 | Male (66 year) | AOID | NM_001024847.2: c.458del; NP_001020018.1: p.Lys153SerfsTer35; rs79375991 3-30691880-AA-A (GRCh37) | NM_000215.3: c.2678C>T; NP_000206.2: p.Pro893Leu; rs772027199; AF = 0.000003978 | NM_001289905.1: c.679T>G; NP_001276834.1: p.Ser227Ala; rs371494126; AF = 0.00001193 | |||||
| Patient 4 | Female (41 year) | AOID | NM_001024847.2: c.458del; NP_001020018.1: p.Lys153SerfsTer35; rs79375991 3-30691880-AA-A (GRCh37) | NM_001322194.1: c.1174G>A; NP_001309123.1: p.Val392Met; rs200018153; AF = 0.0006015 | NM_003331.5:c.2107C>T; NP_003322.3:p.Arg703Trp; rs55882956; AF = 0.006692 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kantaputra, P.; Daroontum, T.; Chuamanochan, M.; Chaowattanapanit, S.; Intachai, W.; Olsen, B.; Sastraruji, T.; Tongsima, S.; Ngamphiw, C.; Kampuansai, J.; et al. Loss of Function TGFBR2 Variant as a Contributing Factor in Generalized Pustular Psoriasis and Adult-Onset Immunodeficiency. Genes 2023, 14, 103. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010103
Kantaputra P, Daroontum T, Chuamanochan M, Chaowattanapanit S, Intachai W, Olsen B, Sastraruji T, Tongsima S, Ngamphiw C, Kampuansai J, et al. Loss of Function TGFBR2 Variant as a Contributing Factor in Generalized Pustular Psoriasis and Adult-Onset Immunodeficiency. Genes. 2023; 14(1):103. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010103
Chicago/Turabian StyleKantaputra, Piranit, Teerada Daroontum, Mati Chuamanochan, Suteeraporn Chaowattanapanit, Worrachet Intachai, Bjorn Olsen, Thanapat Sastraruji, Sissades Tongsima, Chumpol Ngamphiw, Jatupol Kampuansai, and et al. 2023. "Loss of Function TGFBR2 Variant as a Contributing Factor in Generalized Pustular Psoriasis and Adult-Onset Immunodeficiency" Genes 14, no. 1: 103. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010103