
The Role of PARP1 and PAR in ATP-Independent Nucleosome Reorganisation during the DNA Damage Response
Abstract
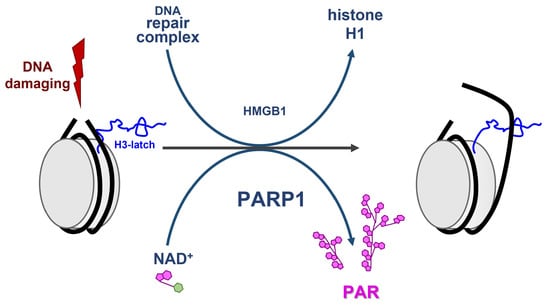
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Structure of the Nucleosome Core Particle (NCP) and Its Subtypes
3. Histone Variants
4. Linker Histone H1 as a Factor Affecting Chromatin Compaction Dynamics
5. HMGB1 as an ATP-Independent Chromatin Remodelling Factor
6. Nuclear Protein Poly(ADP-ribose)polymerase 1 (PARP1): Interaction with the NCP
7. PAR in the DDR
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Noll, M.; Kornberg, R.D. Action of micrococcal nuclease on chromatin and the location of histone H1. J. Mol. Biol. 1977, 109, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Cutter, A.R.; Hayes, J.J. A brief review of nucleosome structure. FEBS Lett. 2015, 589, 2914–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednar, J.; Horowitz, R.A.; Grigoryev, S.A.; Carruthers, L.M.; Hansen, J.C.; Koster, A.J.; Woodcock, C.L. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. USA 1998, 95, 14173–14178. [Google Scholar] [CrossRef] [Green Version]
- Thåström, A.; Lowary, P.; Widlund, H.; Cao, H.; Kubista, M.; Widom, J. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 1999, 288, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Ngo, T.T.; Zhang, Q.; Zhou, R.; Yodh, J.G.; Ha, T. Asymmetric Unwrapping of Nucleosomes under Tension Directed by DNA Local Flexibility. Cell 2015, 160, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Chua, E.Y.D.; Vasudevan, D.; Davey, G.E.; Wu, B.; Davey, C.A. The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 2012, 40, 6338–6352. [Google Scholar] [CrossRef] [Green Version]
- Zhurkin, V.B. Sequence-Dependent Bending of DNA and Phasing of Nucleosomes. J. Biomol. Struct. Dyn. 1985, 2, 785–804. [Google Scholar] [CrossRef]
- Segal, E.; Fondufe-Mittendorf, Y.; Chen, L.; Thåström, A.; Field, Y.; Moore, I.K.; Wang, J.-P.Z.; Widom, J. A genomic code for nucleosome positioning. Nature 2006, 442, 772–778. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Zhao, Y.; Garcia, B.A. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenhard, B.; Sandelin, A.; Carninci, P. Metazoan promoters: Emerging characteristics and insights into transcriptional regulation. Nat. Rev. Genet. 2012, 13, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.C.; Workman, J.L. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol. Cell. Biol. 1995, 15, 1405–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.A.; Widom, J. Collaborative Competition Mechanism for Gene Activation In Vivo. Mol. Cell. Biol. 2003, 23, 1623–1632. [Google Scholar] [CrossRef] [Green Version]
- Gurova, K.; Chang, H.-W.; Valieva, M.E.; Sandlesh, P.; Studitsky, V.M. Structure and function of the histone chaperone—Resolving FACTual issues. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2018, 1861, 892–904. [Google Scholar] [CrossRef]
- Sabantsev, A.; Levendosky, R.F.; Zhuang, X.; Bowman, G.D.; Deindl, S. Direct observation of coordinated DNA movements on the nucleosome during chromatin remodelling. Nat. Commun. 2019, 10, 1720. [Google Scholar] [CrossRef] [Green Version]
- Andonegui-Elguera, M.A.; Cáceres-Gutiérrez, R.E.; López-Saavedra, A.; Cisneros-Soberanis, F.; Justo-Garrido, M.; Díaz-Chávez, J.; Herrera, L.A. The Roles of Histone Post-Translational Modifications in the Formation and Function of a Mitotic Chromosome. Int. J. Mol. Sci. 2022, 23, 8704. [Google Scholar] [CrossRef]
- White, C.L.; Suto, R.K.; Luger, K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001, 20, 5207–5218. [Google Scholar] [CrossRef]
- Kornberg, R.D.; Lorch, Y. Twenty-Five Years of the Nucleosome, Fundamental Particle of the Eukaryote Chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- McGinty, R.K.; Tan, S. Nucleosome Structure and Function. Chem. Rev. 2014, 115, 2255–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luger, K.; Richmond, T.J. DNA binding within the nucleosome core. Curr. Opin. Struct. Biol. 1998, 8, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Armeev, G.A.; Kniazeva, A.S.; Komarova, G.A.; Kirpichnikov, M.P.; Shaytan, A.K. Histone dynamics mediate DNA unwrapping and sliding in nucleosomes. Nat. Commun. 2021, 12, 2387. [Google Scholar] [CrossRef]
- Beato, M.; Eisfeld, K. Transcription factor access to chromatin. Nucleic Acids Res. 1997, 25, 3559–3563. [Google Scholar] [CrossRef] [Green Version]
- Imbalzano, A.N.; Kwon, H.; Green, M.R.; Kingston, R.E. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature 1994, 370, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, D.S.; Papoulas, O.; Dang, C.V.; Kingston, R.E. Differential binding of c-Myc and Max to nucleosomal DNA. Mol. Cell. Biol. 1994, 14, 4097–4107. [Google Scholar] [CrossRef] [Green Version]
- Boyes, J.; Omichinski, J.; Clark, D.; Pikaart, M.; Felsenfeld, G. Perturbation of nucleosome structure by the erythroid transcription factor GATA-1. J. Mol. Biol. 1998, 279, 529–544. [Google Scholar] [CrossRef]
- Hayes, J.J.; Clark, D.J.; Wolffe, A.P. Histone contributions to the structure of DNA in the nucleosome. Proc. Natl. Acad. Sci. USA 1991, 88, 6829–6833. [Google Scholar] [CrossRef] [Green Version]
- Luzete-Monteiro, E.; Zaret, K.S. Structures and consequences of pioneer factor binding to nucleosomes. Curr. Opin. Struct. Biol. 2022, 75, 102425. [Google Scholar] [CrossRef]
- Lowary, P.; Widom, J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998, 276, 19–42. [Google Scholar] [CrossRef]
- Li, G.; Levitus, M.; Bustamante, C.; Widom, J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2005, 12, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Tillo, D.; Hughes, T.R. G+C content dominates intrinsic nucleosome occupancy. BMC Bioinform. 2009, 10, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richmond, T.J.; Davey, C.A. The structure of DNA in the nucleosome core. Nature 2003, 423, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Widom, J. Role of DNA sequence in nucleosome stability and dynamics. Q. Rev. Biophys. 2001, 34, 269–324. [Google Scholar] [CrossRef]
- van Holde, K.E. Chromatin; Springer: New York, NY, USA, 2012; Available online: https://0-link-springer-com.brum.beds.ac.uk/book/10.1007/978-1-4612-3490-6 (accessed on 6 December 2012).
- Satchwell, S.C.; Drew, H.R.; Travers, A.A. Sequence periodicities in chicken nucleosome core DNA. J. Mol. Biol. 1986, 191, 659–675. [Google Scholar] [CrossRef]
- Winogradoff, D.; Aksimentiev, A. Molecular Mechanism of Spontaneous Nucleosome Unraveling. J. Mol. Biol. 2018, 431, 323–335. [Google Scholar] [CrossRef]
- Hall, M.A.; Shundrovsky, A.; Bai, L.; Fulbright, R.M.; Lis, J.T.; Wang, M.D. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 2009, 16, 124–129. [Google Scholar] [CrossRef]
- Gagniuc, P.; Ionescu-Tirgoviste, C. Eukaryotic genomes may exhibit up to 10 generic classes of gene promoters. BMC Genom. 2012, 13, 512. [Google Scholar] [CrossRef] [Green Version]
- Brower-Toland, B.D.; Smith, C.L.; Yeh, R.C.; Lis, J.T.; Peterson, C.L.; Wang, M.D. Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 1960–1965. [Google Scholar] [CrossRef] [Green Version]
- Polach, K.; Widom, J. Mechanism of Protein Access to Specific DNA Sequences in Chromatin: A Dynamic Equilibrium Model for Gene Regulation. J. Mol. Biol. 1995, 254, 130–149. [Google Scholar] [CrossRef]
- Sinha, K.K.; Gross, J.D.; Narlikar, G.J. Distortion of histone octamer core promotes nucleosome mobilization by a chromatin remodeler. Science 2017, 355, eaaa3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilokapic, S.; Strauss, M.; Halic, M. Structural rearrangements of the histone octamer translocate DNA. Nat. Commun. 2018, 9, 1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagi, A.; Ando, T.; Lyubchenko, Y.L. Dynamics of Nucleosomes Assessed with Time-Lapse High-Speed Atomic Force Microscopy. Biochemistry 2011, 50, 7901–7908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyubchenko, Y.L. Nanoscale nucleosome dynamics assessed with time-lapse AFM. Biophys. Rev. 2013, 6, 181–190. [Google Scholar] [CrossRef]
- Wei, S.; Falk, S.J.; Black, B.E.; Lee, T.-H. A novel hybrid single molecule approach reveals spontaneous DNA motion in the nucleosome. Nucleic Acids Res. 2015, 43, e111. [Google Scholar] [CrossRef] [Green Version]
- Gansen, A.; Felekyan, S.; Kühnemuth, R.; Lehmann, K.; Tóth, K.; Seidel, C.A.M.; Langowski, J. High precision FRET studies reveal reversible transitions in nucleosomes between microseconds and minutes. Nat. Commun. 2018, 9, 4628. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, H.; Somers, J.; Webster, R.; Flaus, A.; Owen-Hughes, T. Histone Tails and the H3 αN Helix Regulate Nucleosome Mobility and Stability. Mol. Cell. Biol. 2007, 27, 4037–4048. [Google Scholar] [CrossRef] [Green Version]
- Stormberg, T.; Stumme-Diers, M.; Lyubchenko, Y.L. Sequence-dependent nucleosome nanoscale structure characterized by atomic force microscopy. FASEB J. 2019, 33, 10916–10923. [Google Scholar] [CrossRef] [Green Version]
- Edayathumangalam, R.S.; Weyermann, P.; Dervan, P.B.; Gottesfeld, J.M.; Luger, K. Nucleosomes in Solution Exist as a Mixture of Twist-defect States. J. Mol. Biol. 2005, 345, 103–114. [Google Scholar] [CrossRef]
- Shukla, M.S.; Syed, S.H.; Montel, F.; Faivre-Moskalenko, C.; Bednar, J.; Travers, A.; Angelov, D.; Dimitrov, S. Remosomes: RSC generated non-mobilized particles with approximately 180 bp DNA loosely associated with the histone octamer. Proc. Natl. Acad. Sci. USA 2010, 107, 1936–1941. [Google Scholar] [CrossRef]
- Erler, J.; Zhang, R.; Petridis, L.; Cheng, X.; Smith, J.C.; Langowski, J. The Role of Histone Tails in the Nucleosome: A Computational Study. Biophys. J. 2014, 107, 2911–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmanian, N.; Shokrzadeh, M.; Eskandani, M. Recent advances in γH2AX biomarker-based genotoxicity assays: A marker of DNA damage and repair. DNA Repair 2021, 108, 103243. [Google Scholar] [CrossRef] [PubMed]
- Wootton, J.; Soutoglou, E. Chromatin and Nuclear Dynamics in the Maintenance of Replication Fork Integrity. Front. Genet. 2021, 12, 773426. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kono, H. Distinct Roles of Histone H3 and H2A Tails in Nucleosome Stability. Sci. Rep. 2016, 6, 31437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, G.-L.; Bleakley, M.; Bradley, R.K.; Malik, H.S.; Henikoff, S.; Molaro, A.; Sarthy, J. Short H2A histone variants are expressed in cancer. Nat. Commun. 2021, 12, 490. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Konesky, K.; Park, Y.-J.; Rosu, S.; Dyer, P.N.; Rangasamy, D.; Tremethick, D.; Laybourn, P.J.; Luger, K. Nucleosomes containing the histone variant H2A.Bbd organize only 118 base pairs of DNA. EMBO J. 2004, 23, 3314–3324. [Google Scholar] [CrossRef] [Green Version]
- Tolstorukov, M.Y.; Goldman, J.; Gilbert, C.; Ogryzko, V.; Kingston, R.E.; Park, P.J. Histone Variant H2A.Bbd Is Associated with Active Transcription and mRNA Processing in Human Cells. Mol. Cell 2012, 47, 596–607. [Google Scholar] [CrossRef] [Green Version]
- Oberdoerffer, P.; Miller, K.M. Histone H2A variants: Diversifying chromatin to ensure genome integrity. Semin. Cell Dev. Biol. 2022, 135, 59–72. [Google Scholar] [CrossRef]
- Xu, Y.; Ayrapetov, M.K.; Xu, C.; Gursoy-Yuzugullu, O.; Hu, Y.; Price, B.D. Histone H2A.Z Controls a Critical Chromatin Remodeling Step Required for DNA Double-Strand Break Repair. Mol. Cell 2012, 48, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Delaney, S. Histone H2A Variants Enhance the Initiation of Base Excision Repair in Nucleosomes. ACS Chem. Biol. 2019, 14, 1041–1050. [Google Scholar] [CrossRef]
- Yu, Y.; Deng, Y.; Reed, S.; Millar, C. Waters Histone variant Htz1 promotes histone H3 acetylation to enhance nucleotide excision repair in Htz1 nucleosomes. Nucleic Acids Res. 2013, 41, 9006–9019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bewersdorf, J.; Bennett, B.T.; Knight, K.L. H2AX chromatin structures and their response to DNA damage revealed by 4Pi microscopy. Proc. Natl. Acad. Sci. USA 2006, 103, 18137–18142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK Function Redundantly to Phosphorylate H2AX after Exposure to Ionizing Radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; De Falco, L.; Padavattan, S.; Rao, C.; Geifman-Shochat, S.; Liu, C.-F.; Davey, C.A. PARP1 exhibits enhanced association and catalytic efficiency with γH2A.X-nucleosome. Nat. Commun. 2019, 10, 5751. [Google Scholar] [CrossRef] [Green Version]
- Karras, G.; Kustatscher, G.; Buhecha, H.R.; Allen, M.D.; Pugieux, C.; Sait, F.; Bycroft, M.; Ladurner, A.G. The macro domain is an ADP-ribose binding module. EMBO J. 2005, 24, 1911–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Xu, Y.; Gursoy-Yuzugullu, O.; Price, B.D. The histone variant macroH2A1.1 is recruited to DSBs through a mechanism involving PARP1. FEBS Lett. 2012, 586, 3920–3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timinszky, G.; Till, S.; Hassa, P.O.; Hothorn, M.; Kustatscher, G.; Nijmeijer-Winter, B.; Colombelli, J.; Altmeyer, M.; Stelzer, E.H.; Scheffzek, K.; et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009, 16, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.D.; Hamilton, G.A.; Park, J.W.; Gamble, M.J. MacroH2A1 Regulation of Poly(ADP-Ribose) Synthesis and Stability Prevents Necrosis and Promotes DNA Repair. Mol. Cell. Biol. 2019, 40, e00230-19. [Google Scholar] [CrossRef] [Green Version]
- Kozlowski, M.; Corujo, D.; Hothorn, M.; Guberovic, I.; Mandemaker, I.; Blessing, C.; Sporn, J.; Gutierrez-Triana, A.; Smith, R.; Portmann, T.; et al. MacroH2A histone variants limit chromatin plasticity through two distinct mechanisms. EMBO Rep. 2018, 19, e44445. [Google Scholar] [CrossRef]
- Khurana, S.; Kruhlak, M.J.; Kim, J.; Tran, A.D.; Liu, J.; Nyswaner, K.; Shi, L.; Jailwala, P.; Sung, M.-H.; Hakim, O.; et al. A Macrohistone Variant Links Dynamic Chromatin Compaction to BRCA1-Dependent Genome Maintenance. Cell Rep. 2014, 8, 1049–1062. [Google Scholar] [CrossRef]
- Sebastian, R.; Hosogane, E.K.; Sun, E.G.; Tran, A.D.; Reinhold, W.C.; Burkett, S.; Sturgill, D.M.; Gudla, P.R.; Pommier, Y.; Aladjem, M.I.; et al. Epigenetic Regulation of DNA Repair Pathway Choice by MacroH2A1 Splice Variants Ensures Genome Stability. Mol. Cell 2020, 79, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Allis, C.D.; Nussenzweig, A. Phosphorylation of Histone H2B at DNA Double-Strand Breaks. J. Exp. Med. 2004, 199, 1671–1677. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Kato, A.; Kobayashi, J.; Yanagihara, H.; Sakamoto, S.; Oliveira, D.V.; Shimada, M.; Tauchi, H.; Suzuki, H.; Tashiro, S.; et al. Regulation of Homologous Recombination by RNF20-Dependent H2B Ubiquitination. Mol. Cell 2011, 41, 515–528. [Google Scholar] [CrossRef]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.-Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-Dependent Monoubiquitylation of Histone H2B for Timely Repair of DNA Double-Strand Breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef]
- Suraweera, A.; Gandhi, N.S.; Beard, S.; Burgess, J.T.; Croft, L.V.; Bolderson, E.; Naqi, A.; Ashton, N.W.; Adams, M.N.; Savage, K.I.; et al. COMMD4 functions with the histone H2A-H2B dimer for the timely repair of DNA double-strand breaks. Commun. Biol. 2021, 4, 484. [Google Scholar] [CrossRef]
- Verreault, A.; Kaufman, P.D.; Kobayashi, R.; Stillman, B. Nucleosome Assembly by a Complex of CAF-1 and Acetylated Histones H3/H4. Cell 1996, 87, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Yoda, K.; Ando, S.; Morishita, S.; Houmura, K.; Hashimoto, K.; Takeyasu, K.; Okazaki, T. Human centromere protein A (CENP-A) can replace histone H3 in nucleosome reconstitution in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 7266–7271. [Google Scholar] [CrossRef] [Green Version]
- Luijsterburg, M.S.; de Krijger, I.; Wiegant, W.W.; Shah, R.G.; Smeenk, G.; de Groot, A.J.L.; Pines, A.; Vertegaal, A.C.O.; Jacobs, J.J.L.; Shah, G.M.; et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell 2016, 61, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.L.; Portoso, M.; Mata, J.; Bähler, J.; Allshire, R.C.; Kouzarides, T. Methylation of Histone H4 Lysine 20 Controls Recruitment of Crb2 to Sites of DNA Damage. Cell 2004, 119, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.-M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef]
- Ge, Z.; Nair, D.; Guan, X.; Rastogi, N.; Freitas, M.A.; Parthun, M.R. Sites of Acetylation on Newly Synthesized Histone H4 Are Required for Chromatin Assembly and DNA Damage Response Signaling. Mol. Cell. Biol. 2013, 33, 3286–3298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, S.; Gursoy-Yuzugullu, O.; Parasuram, R.; Price, B.D. The tale of a tail: Histone H4 acetylation and the repair of DNA breaks. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muyldermans, S.; Travers, A.A. DNA Sequence Organization in Chromatosomes. J. Mol. Biol. 1994, 235, 855–870. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Gu, L.; Ye, Y.; Zheng, M.; Xu, Z.; Lin, J.; Du, Y.; Tian, M.; Luo, L.; Wang, B.; et al. Dynamic placement of the linker histone H1 associated with nucleosome arrangement and gene transcription in early Drosophila embryonic development. Cell Death Dis. 2018, 9, 765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Happel, N.; Doenecke, D. Histone H1 and its isoforms: Contribution to chromatin structure and function. Gene 2009, 431, 1–12. [Google Scholar] [CrossRef]
- Izzo, A.; Schneider, R. The role of linker histone H1 modifications in the regulation of gene expression and chromatin dynamics. Biochim. Biophys. Acta 2016, 1859, 486–495. [Google Scholar] [CrossRef]
- Liao, R.; Mizzen, C.A. Interphase H1 phosphorylation: Regulation and functions in chromatin. Biochim. Biophys. Acta 2016, 1859, 476–485. [Google Scholar] [CrossRef]
- Bowman, G.D.; Poirier, M.G. Post-Translational Modifications of Histones That Influence Nucleosome Dynamics. Chem. Rev. 2014, 115, 2274–2295. [Google Scholar] [CrossRef] [Green Version]
- Liao, R.; Mizzen, C.A. Site-specific regulation of histone H1 phosphorylation in pluripotent cell differentiation. Epigenetics Chromatin 2017, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Noberini, R.; Torres, C.M.; Savoia, E.O.; Brandini, S.; Jodice, M.G.; Bertalot, G.; Bonizzi, G.; Capra, M.; Diaferia, G.; Scaffidi, P.; et al. Label-Free Mass Spectrometry-Based Quantification of Linker Histone H1 Variants in Clinical Samples. Int. J. Mol. Sci. 2020, 21, 7330. [Google Scholar] [CrossRef]
- Woodcock, C.L.; Skoultchi, A.I.; Fan, Y. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosom. Res. 2006, 14, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Perišić, O.; Portillo-Ledesma, S.; Schlick, T. Sensitive effect of linker histone binding mode and subtype on chromatin condensation. Nucleic Acids Res. 2019, 47, 4948–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misteli, T.; Gunjan, A.; Hock, R.; Bustin, M.; Brown, D.T. Dynamic binding of histone H1 to chromatin in living cells. Nature 2000, 408, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Chapman, G.E.; Hartman, P.G.; Bradbury, E.M. Studies on the Role and Mode of Operation of the Very-Lysine-Rich Histone H1 in Eukaryote Chromatin. The Isolation of the Globular and Non-Globular Regions of the Histone H1 Molecule. JBIC J. Biol. Inorg. Chem. 1976, 61, 69–75. [Google Scholar] [CrossRef]
- Ma, C.; Malessa, A.; Boersma, A.J.; Liu, K.; Herrmann, A. Supercharged Proteins and Polypeptides. Adv. Mater. 2020, 32, e1905309. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hoover, M.E.; Dang, X.; Shomo, A.A.; Guan, X.; Marshall, A.G.; Freitas, M.A.; Young, N.L. Quantitative Mass Spectrometry Reveals that Intact Histone H1 Phosphorylations are Variant Specific and Exhibit Single Molecule Hierarchical Dependence. Mol. Cell. Proteom. 2016, 15, 818–833. [Google Scholar] [CrossRef] [Green Version]
- Hendzel, M.J.; Lever, M.A.; Crawford, E.; Th’Ng, J.P.H. The C-terminal Domain Is the Primary Determinant of Histone H1 Binding to Chromatin in Vivo. J. Biol. Chem. 2004, 279, 20028–20034. [Google Scholar] [CrossRef] [Green Version]
- Th’Ng, J.P.H.; Sung, R.; Ye, M.; Hendzel, M. H1 Family Histones in the Nucleus: Control of binding and localization by the C-terminal domain. J. Biol. Chem. 2005, 280, 27809–27814. [Google Scholar] [CrossRef] [Green Version]
- Öztürk, M.A.; Cojocaru, V.; Wade, R.C. Toward an Ensemble View of Chromatosome Structure: A Paradigm Shift from One to Many. Structure 2018, 26, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Saez, I.; Menoni, H.; Boopathi, R.; Shukla, M.S.; Soueidan, L.; Noirclerc-Savoye, M.; Le Roy, A.; Skoufias, D.; Bednar, J.; Hamiche, A.; et al. Structure of an H1-Bound 6-Nucleosome Array Reveals an Untwisted Two-Start Chromatin Fiber Conformation. Mol. Cell 2018, 72, 902–915.e7. [Google Scholar] [CrossRef]
- Zhou, B.-R.; Jiang, J.; Feng, H.; Ghirlando, R.; Xiao, T.S.; Bai, Y. Structural Mechanisms of Nucleosome Recognition by Linker Histones. Mol. Cell 2015, 59, 628–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutter, A.R.; Hayes, J.J. Linker histones: Novel insights into structure-specific recognition of the nucleosome. Biochem. Cell Biol. 2017, 95, 171–178. [Google Scholar] [CrossRef]
- Dombrowski, M.; Engeholm, M.; Dienemann, C.; Dodonova, S.; Cramer, P. Histone H1 binding to nucleosome arrays depends on linker DNA length and trajectory. Nat. Struct. Mol. Biol. 2022, 29, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.-R.; Feng, H.; Ghirlando, R.; Li, S.; Schwieters, C.D.; Bai, Y. A Small Number of Residues Can Determine if Linker Histones Are Bound On or Off Dyad in the Chromatosome. J. Mol. Biol. 2016, 428, 3948–3959. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Z.; Dong, L.; Tang, M.; Zhang, P.; Zhang, C.; Cao, Z.; Zhu, Q.; Chen, Y.; Wang, H.; et al. Histone H1 acetylation at lysine 85 regulates chromatin condensation and genome stability upon DNA damage. Nucleic Acids Res. 2018, 46, 7716–7730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stützer, A.; Liokatis, S.; Kiesel, A.; Schwarzer, D.; Sprangers, R.; Söding, J.; Selenko, P.; Fischle, W. Modulations of DNA Contacts by Linker Histones and Post-translational Modifications Determine the Mobility and Modifiability of Nucleosomal H3 Tails. Mol. Cell 2016, 61, 247–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, D.C.; Wereszczynski, J. Elucidating the influence of linker histone variants on chromatosome dynamics and energetics. Nucleic Acids Res. 2020, 48, 3591–3604. [Google Scholar] [CrossRef] [Green Version]
- Braunschweig, U.; Hogan, G.J.; Pagie, L.; Van Steensel, B. Histone H1 binding is inhibited by histone variant H3.3. EMBO J. 2009, 28, 3635–3645. [Google Scholar] [CrossRef]
- Lever, M.A.; Th’Ng, J.P.H.; Sun, X.; Hendzel, M.J. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature 2000, 408, 873–876. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Gamble, M.J.; Frizzell, K.M.; Berrocal, J.G.; Kininis, M.; Kraus, W.L. Reciprocal Binding of PARP-1 and Histone H1 at Promoters Specifies Transcriptional Outcomes. Science 2008, 319, 819–821. [Google Scholar] [CrossRef]
- Nalabothula, N.; McVicker, G.; Maiorano, J.; Martin, R.; Pritchard, J.K.; Fondufe-Mittendorf, Y.N. The chromatin architectural proteins HMGD1 and H1 bind reciprocally and have opposite effects on chromatin structure and gene regulation. BMC Genom. 2014, 15, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catez, F.; Ueda, T.; Bustin, M. Determinants of histone H1 mobility and chromatin binding in living cells. Nat. Struct. Mol. Biol. 2006, 13, 305–310. [Google Scholar] [CrossRef] [PubMed]
- McBryant, S.J.; Lu, X.; Hansen, J.C. Multifunctionality of the linker histones: An emerging role for protein-protein interactions. Cell Res. 2010, 20, 519–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalashnikova, A.A.; Winkler, D.D.; McBryant, S.J.; Henderson, R.K.; Herman, J.; DeLuca, J.G.; Luger, K.; Prenni, J.; Hansen, J.C. Linker histone H1.0 interacts with an extensive network of proteins found in the nucleolus. Nucleic Acids Res. 2013, 41, 4026–4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Choi, J.; Heo, K.; Kim, H.; Levens, D.; Kohno, K.; Johnson, E.M.; Brock, H.W.; An, W. Isolation and Characterization of a Novel H1.2 Complex That Acts as a Repressor of p53-mediated Transcription. J. Biol. Chem. 2008, 283, 9113–9126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szerlong, H.J.; Herman, J.A.; Krause, C.M.; DeLuca, J.G.; Skoultchi, A.; Winger, Q.A.; Prenni, J.E.; Hansen, J.C. Proteomic Characterization of the Nucleolar Linker Histone H1 Interaction Network. J. Mol. Biol. 2015, 427, 2056–2071. [Google Scholar] [CrossRef] [Green Version]
- Kraus, W.L. Transcriptional control by PARP-1: Chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef] [Green Version]
- Kalashnikova, A.A.; Rogge, R.A.; Hansen, J.C. Linker histone H1 and protein–protein interactions. Biochim. Biophys. Acta (BBA) 2016, 1859, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Bustin, M. Regulation of DNA-Dependent Activities by the Functional Motifs of the High-Mobility-Group Chromosomal Proteins. Mol. Cell. Biol. 1999, 19, 5237–5246. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, G.; Johns, E. Are the high mobility group non-histone chromosomal proteins associated with ‘active’ chromatin? Biochim. Biophys. Acta (BBA) 1978, 519, 279–284. [Google Scholar] [CrossRef]
- Catez, F.; Yang, H.; Tracey, K.J.; Reeves, R.; Misteli, T.; Bustin, M. Network of Dynamic Interactions between Histone H1 and High-Mobility-Group Proteins in Chromatin. Mol. Cell. Biol. 2004, 24, 4321–4328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, S.S.; Vasquez, K.M. HMGB1: The jack-of-all-trades protein is a master DNA repair mechanic. Mol. Carcinog. 2009, 48, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agresti, A.; Bianchi, M.E. HMGB proteins and gene expression. Curr. Opin. Genet. Dev. 2003, 13, 170–178. [Google Scholar] [CrossRef]
- Calogero, S.; Grassi, F.; Aguzzi, A.; Voigtländer, T.; Ferrier, P.; Ferrari, S.; Bianchi, M.E. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 1999, 22, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, H.; Tsuda, K.; Yoshida, M. Primary structure of non-histone chromosomal protein HMG2 revealed by the nucleotide sequence. Biochemistry 1990, 29, 4419–4423. [Google Scholar] [CrossRef]
- Ohndorf, U.-M.; Rould, M.A.; He, Q.; Pabo, C.O.; Lippard, S.J. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature 1999, 399, 708–712. [Google Scholar] [CrossRef]
- Li, J.; Kokkola, R.; Tabibzadeh, S.; Yang, R.; Ochani, M.; Qiang, X.; Harris, H.; Czura, C.; Wang, H.; Ulloa, L.; et al. Yang Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol. Med. 2003, 9, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Ugrinova, I.; Zlateva, S.; Pashev, I.G.; Pasheva, E.A. Native HMGB1 protein inhibits repair of cisplatin-damaged nucleosomes in vitro. Int. J. Biochem. Cell Biol. 2009, 41, 1556–1562. [Google Scholar] [CrossRef]
- Mitkova, E.; Ugrinova, I.; Pashev, I.G.; Pasheva, E.A. The Inhibitory Effect of HMGB-1 Protein on the Repair of Cisplatin-Damaged DNA Is Accomplished through the Acidic Domain. Biochemistry 2005, 44, 5893–5898. [Google Scholar] [CrossRef]
- Watson, M.; Stott, K.; Thomas, J.O. Mapping Intramolecular Interactions between Domains in HMGB1 using a Tail-truncation Approach. J. Mol. Biol. 2007, 374, 1286–1297. [Google Scholar] [CrossRef]
- Stott, K.; Watson, M.; Howe, F.S.; Grossmann, J.G.; Thomas, J.O. Tail-Mediated Collapse of HMGB1 Is Dynamic and Occurs via Differential Binding of the Acidic Tail to the A and B Domains. J. Mol. Biol. 2010, 403, 706–722. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zeng, M.; Wang, W.; Tang, J. The HMGB1 acidic tail regulates HMGB1 DNA binding specificity by a unique mechanism. Biochem. Biophys. Res. Commun. 2007, 360, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, E. Chromatin Structure and Dynamics: State-of-the-Art. Mol. Cell 2002, 10, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Ragab, A.; Travers, A. HMG-D and histone H1 alter the local accessibility of nucleosomal DNA. Nucleic Acids Res. 2003, 31, 7083–7089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, D.; Peterson, R.C.; Scovell, W.M. High Mobility Group B Proteins Facilitate Strong Estrogen Receptor Binding to Classical and Half-Site Estrogen Response Elements and Relax Binding Selectivity. Mol. Endocrinol. 2004, 18, 2616–2632. [Google Scholar] [CrossRef] [Green Version]
- Balliano, A.; Hao, F.; Njeri, C.; Balakrishnan, L.; Hayes, J.J. HMGB1 Stimulates Activity of Polymerase β on Nucleosome Substrates. Biochemistry 2017, 56, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.R.; Sarpong, Y.C.; Peterson, R.C.; Scovell, W.M. Nucleosome dynamics: HMGB1 relaxes canonical nucleosome structure to facilitate estrogen receptor binding. Nucleic Acids Res. 2012, 40, 10161–10171. [Google Scholar] [CrossRef] [Green Version]
- Scovell, W.M. High mobility group protein 1: A collaborator in nucleosome dynamics and estrogen-responsive gene expression. World J. Biol. Chem. 2016, 7, 206–222. [Google Scholar] [CrossRef]
- Heo, K.; Kim, B.; Kim, K.; Choi, J.; Kim, H.; Zhan, Y.; Ranish, J.A.; An, W. Isolation and Characterization of Proteins Associated with Histone H3 Tails in Vivo. J. Biol. Chem. 2007, 282, 15476–15483. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.; Stott, K.; Fischl, H.; Cato, L.; Thomas, J.O. Characterization of the interaction between HMGB1 and H3--a possible means of positioning HMGB1 in chromatin. Nucleic Acids Res. 2013, 42, 848–859. [Google Scholar] [CrossRef]
- Phair, R.D.; Scaffidi, P.; Elbi, C.; Vecerová, J.; Dey, A.; Ozato, K.; Brown, D.T.; Hager, G.; Bustin, M.; Misteli, T. Global Nature of Dynamic Protein-Chromatin Interactions In Vivo: Three-Dimensional Genome Scanning and Dynamic Interaction Networks of Chromatin Proteins. Mol. Cell. Biol. 2004, 24, 6393–6402. [Google Scholar] [CrossRef] [Green Version]
- Giese, K.; Cox, J.; Grosschedl, R. The HMG domain of lymphoid enhancer factor 1 bends DNA and facilitates assembly of functional nucleoprotein structures. Cell 1992, 69, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Travers, A.A. Priming the nucleosome: A role for HMGB proteins? EMBO Rep. 2003, 4, 131–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, S.S.; Mitchell, D.L.; Vasquez, K.M. High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 10320–10325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, R. High mobility group (HMG) proteins: Modulators of chromatin structure and DNA repair in mammalian cells. DNA Repair 2015, 36, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Liu, Y.; Deterding, L.J.; Poltoratsky, V.P.; Kedar, P.S.; Horton, J.K.; Kanno, S.-I.; Asagoshi, K.; Hou, E.W.; Khodyreva, S.N.; et al. HMGB1 Is a Cofactor in Mammalian Base Excision Repair. Mol. Cell 2007, 27, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krynetskaia, N.; Xie, H.M.; Vucetic, S.; Obradovic, Z.; Krynetskiy, E. High Mobility Group Protein B1 Is an Activator of Apoptotic Response to Antimetabolite Drugs. Mol. Pharmacol. 2007, 73, 260–269. [Google Scholar] [CrossRef]
- Yuan, F.; Gu, L.; Guo, S.; Wang, C.; Li, G.-M. Evidence for Involvement of HMGB1 Protein in Human DNA Mismatch Repair. J. Biol. Chem. 2004, 279, 20935–20940. [Google Scholar] [CrossRef] [Green Version]
- Genschel, J.; Modrich, P. Functions of MutLα, Replication Protein A (RPA), and HMGB1 in 5′-Directed Mismatch Repair. J. Biol. Chem. 2009, 284, 21536–21544. [Google Scholar] [CrossRef]
- Ueda, T.; Shirakawa, H.; Yoshida, M. Involvement of HMGB1 and HMGB2 proteins in exogenous DNA integration reaction into the genome of HeLa S3 cells. Biochim. Biophys. Acta 2002, 1593, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Mansure, J.J.; Almajed, W.; Cury, F.; Ferbeyre, G.; Popovic, M.; Seuntjens, J.; Kassouf, W. The Role of HMGB1 in Radioresistance of Bladder Cancer. Mol. Cancer Ther. 2016, 15, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Celona, B.; Weiner, A.; Di Felice, F.; Mancuso, F.M.; Cesarini, E.; Rossi, R.L.; Gregory, L.; Baban, D.; Rossetti, G.; Grianti, P.; et al. Substantial Histone Reduction Modulates Genomewide Nucleosomal Occupancy and Global Transcriptional Output. PLoS Biol. 2011, 9, e1001086. [Google Scholar] [CrossRef]
- Cato, L.; Stott, K.; Watson, M.; Thomas, J.O. The Interaction of HMGB1 and Linker Histones Occurs Through their Acidic and Basic Tails. J. Mol. Biol. 2008, 384, 1262–1272. [Google Scholar] [CrossRef]
- Lüscher, B.; Ahel, I.; Altmeyer, M.; Ashworth, A.; Bai, P.; Chang, P.; Cohen, M.; Corda, D.; Dantzer, F.; Daugherty, M.D.; et al. ADP-ribosyltransferases, an update on function and nomenclature. FEBS J. 2021, 289, 7399–7410. [Google Scholar] [CrossRef]
- Eisemann, T.; Pascal, J.M. Poly(ADP-ribose) polymerase enzymes and the maintenance of genome integrity. Cell. Mol. Life Sci. CMLS 2020, 77, 19–33. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Aleksandrov, R.; Dotchev, A.; Poser, I.; Krastev, D.; Georgiev, G.; Panova, G.; Babukov, Y.; Danovski, G.; Dyankova, T.; Hubatsch, L.; et al. Protein Dynamics in Complex DNA Lesions. Mol. Cell 2018, 69, 1046–1061.e5. [Google Scholar] [CrossRef] [Green Version]
- D’Silva, I.; Pelletier, J.D.; Lagueux, J.; D’Amours, D.; Chaudhry, M.; Weinfeld, M.; Lees-Miller, S.P.; Poirier, G.G. Relative affinities of poly(ADP-ribose) polymerase and DNA-dependent protein kinase for DNA strand interruptions. Biochim. Biophys. Acta 1999, 1430, 119–126. [Google Scholar] [CrossRef]
- Khodyreva, S.N.; Lavrik, O.I. Poly(ADP-Ribose) polymerase 1 as a key regulator of DNA repair. Mol. Biol. 2016, 50, 580–595. [Google Scholar] [CrossRef]
- Sukhanova, M.V.; Abrakhi, S.; Joshi, V.; Pastre, D.; Kutuzov, M.M.; Anarbaev, R.O.; Curmi, P.A.; Hamon, L.; Lavrik, O.I. Single molecule detection of PARP1 and PARP2 interaction with DNA strand breaks and their poly(ADP-ribosyl)ation using high-resolution AFM imaging. Nucleic Acids Res. 2015, 44, e60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maluchenko, N.V.; Nilov, D.K.; Pushkarev, S.V.; Kotova, E.Y.; Gerasimova, N.S.; Kirpichnikov, M.P.; Langelier, M.-F.; Pascal, J.M.; Akhtar, S.; Feofanov, A.V.; et al. Mechanisms of Nucleosome Reorganization by PARP1. Int. J. Mol. Sci. 2021, 22, 12127. [Google Scholar] [CrossRef] [PubMed]
- Ukraintsev, A.A.; Belousova, E.A.; Kutuzov, M.M.; Lavrik, O.I. Study of Interaction of the PARP Family DNA-Dependent Proteins with Nucleosomes Containing DNA Intermediates of the Initial Stages of BER Process. Biochemistry 2022, 87, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Muthurajan, U.M.; Hepler, M.R.D.; Hieb, A.R.; Clark, N.J.; Kramer, M.; Yao, T.; Luger, K. Automodification switches PARP-1 function from chromatin architectural protein to histone chaperone. Proc. Natl. Acad. Sci. USA 2014, 111, 12752–12757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, N.J.; Kramer, M.; Muthurajan, U.M.; Luger, K. Alternative Modes of Binding of Poly(ADP-ribose) Polymerase 1 to Free DNA and Nucleosomes. J. Biol. Chem. 2012, 287, 32430–32439. [Google Scholar] [CrossRef] [Green Version]
- Sultanov, D.; Gerasimova, N.; Kudryashova, K.; Maluchenko, N.; Kotova, E.; Langelier, M.-F.; Pascal, J.; Kirpichnikov, M.; Feofanov, A.; Studitsky, V. Unfolding of core nucleosomes by PARP-1 revealed by spFRET microscopy. AIMS Genet. 2017, 4, 21–31. [Google Scholar] [CrossRef]
- Kotova, E.Y.; Hsieh, F.-K.; Chang, H.-W.; Maluchenko, N.V.; Langelier, M.-F.; Pascal, J.M.; Luse, D.S.; Feofanov, A.V.; Studitsky, V.M. Human PARP1 Facilitates Transcription through a Nucleosome and Histone Displacement by Pol II In Vitro. Int. J. Mol. Sci. 2022, 23, 7107. [Google Scholar] [CrossRef]
- Haince, J.-F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.-Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent Kinetics of Recruitment of MRE11 and NBS1 Proteins to Multiple DNA Damage Sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T.; Satoh, M.S.; Poirier, G.G.; Klungland, A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995, 20, 405–411. [Google Scholar] [CrossRef]
- Martello, R.; Leutert, M.; Jungmichel, S.; Bilan, V.; Larsen, S.C.; Young, C.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat. Commun. 2016, 7, 12917. [Google Scholar] [CrossRef]
- Messner, S.; Hottiger, M.O. Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 2011, 21, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Karch, K.R.; Langelier, M.-F.; Pascal, J.M.; Garcia, B.A. The nucleosomal surface is the main target of histone ADP-ribosylation in response to DNA damage. Mol. Biosyst. 2017, 13, 2660–2671. [Google Scholar] [CrossRef] [PubMed]
- Gibbs-Seymour, I.; Fontana, P.; Rack, J.; Ahel, I. HPF1/C4orf27 Is a PARP-1-Interacting Protein that Regulates PARP-1 ADP-Ribosylation Activity. Mol. Cell 2016, 62, 432–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suskiewicz, M.J.; Zobel, F.; Ogden, T.E.H.; Fontana, P.; Ariza, A.; Yang, J.-C.; Zhu, K.; Bracken, L.; Hawthorne, W.J.; Ahel, D.; et al. HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 2020, 579, 598–602. [Google Scholar] [CrossRef]
- Krüger, A.; Bürkle, A.; Hauser, K.; Mangerich, A. Real-time monitoring of PARP1-dependent PARylation by ATR-FTIR spectroscopy. Nat. Commun. 2020, 11, 2174. [Google Scholar] [CrossRef]
- Schuhwerk, H.; Bruhn, C.; Siniuk, K.; Min, W.; Erener, S.; Grigaravicius, P.; Krüger, A.; Ferrari, E.; Zubel, T.; Lazaro, D.; et al. Kinetics of poly(ADP-ribosyl)ation, but not PARP1 itself, determines the cell fate in response to DNA damage in vitro and in vivo. Nucleic Acids Res. 2017, 45, 11174–11192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.-W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of Poly(ADP-Ribose) Polymerase-1-Dependent Cell Death by Apoptosis-Inducing Factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Singatulina, A.S.; Hamon, L.; Sukhanova, M.V.; Desforges, B.; Joshi, V.; Bouhss, A.; Lavrik, O.I.; Pastré, D. PARP-1 Activation Directs FUS to DNA Damage Sites to Form PARG-Reversible Compartments Enriched in Damaged DNA. Cell Rep. 2019, 27, 1809–1821.e5. [Google Scholar] [CrossRef] [Green Version]
- Alemasova, E.E.; Lavrik, O.I. A sePARate phase? Poly(ADP-ribose) versus RNA in the organization of biomolecular condensates. Nucleic Acids Res. 2022, 50, 10817–10838. [Google Scholar] [CrossRef]
- Shieh, W.M.; Amé, J.-C.; Wilson, M.V.; Wang, Z.-Q.; Koh, D.W.; Jacobson, M.K.; Jacobson, E.L. Poly(ADP-ribose) Polymerase Null Mouse Cells Synthesize ADP-ribose Polymers. J. Biol. Chem. 1998, 273, 30069–30072. [Google Scholar] [CrossRef]
- Amé, J.-C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Höger, T.; Ménissier-De Murcia, J.; de Murcia, G. PARP-2, A Novel Mammalian DNA Damage-dependent Poly(ADP-ribose) Polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [Green Version]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, J.; Ding, M.; Yu, Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat. Methods 2013, 10, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Jungmichel, S.; Rosenthal, F.; Altmeyer, M.; Lukas, J.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide Identification of Poly(ADP-Ribosyl)ation Targets in Different Genotoxic Stress Responses. Mol. Cell 2013, 52, 272–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, B.A.; Zhang, Y.; Jiang, H.; Hussey, K.M.; Shrimp, J.H.; Lin, H.; Schwede, F.; Yu, Y.; Kraus, W.L. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 2016, 353, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; Larsen, S.C.; Nielsen, M.L. An Advanced Strategy for Comprehensive Profiling of ADP-ribosylation Sites Using Mass Spectrometry-based Proteomics. Mol. Cell. Proteom. 2019, 18, 1010–1026. [Google Scholar] [CrossRef]
- Bonfiglio, J.J.; Fontana, P.; Zhang, Q.; Colby, T.; Gibbs-Seymour, I.; Atanassov, I.; Bartlett, E.; Zaja, R.; Ahel, I.; Matic, I. Serine ADP-Ribosylation Depends on HPF1. Mol. Cell 2017, 65, 932–940.e6. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; Buch-Larsen, S.C.; Prokhorova, E.; Elsborg, J.D.; Rebak, A.K.; Zhu, K.; Ahel, D.; Lukas, C.; Ahel, I.; Nielsen, M.L. The regulatory landscape of the human HPF1- and ARH3-dependent ADP-ribosylome. Nat. Commun. 2021, 12, 5893. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.C.; Hendriks, I.A.; Lyon, D.; Jensen, L.J.; Nielsen, M.L. Systems-wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep. 2018, 24, 2493–2505.e4. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Yu, X. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinform. 2016, 14, 131–139. [Google Scholar] [CrossRef]
- Wang, Z.; Michaud, G.A.; Cheng, Z.; Zhang, Y.; Hinds, T.R.; Fan, E.; Cong, F.; Xu, W. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 2012, 26, 235–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahel, I.; Ahel, D.; Matsusaka, T.; Clark, A.J.; Pines, J.; Boulton, S.J.; West, S.C. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 2008, 451, 81–85. [Google Scholar] [CrossRef]
- Li, M.; Lu, L.-Y.; Yang, C.-Y.; Wang, S.; Yu, X. The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev. 2013, 27, 1752–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Tanaka, M.; Shimada, T.; Miwa, M.; Sugimura, T. Size and shape of poly(ADP-ribose): Examination by gel filtration, gel electrophoresis and electron microscopy. Biochem. Biophys. Res. Commun. 1983, 112, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Reber, J.M.; Mangerich, A. Why structure and chain length matter: On the biological significance underlying the structural heterogeneity of poly(ADP-ribose). Nucleic Acids Res. 2021, 49, 8432–8448. [Google Scholar] [CrossRef]
- Aberle, L.; Krüger, A.; Reber, J.M.; Lippmann, M.; Hufnagel, M.; Schmalz, M.; Trussina, I.R.E.A.; Schlesiger, S.; Zubel, T.; Schütz, K.; et al. PARP1 catalytic variants reveal branching and chain length-specific functions of poly(ADP-ribose) in cellular physiology and stress response. Nucleic Acids Res. 2020, 48, 10015–10033. [Google Scholar] [CrossRef]
- Naumenko, K.N.; Sukhanova, M.V.; Hamon, L.; Kurgina, T.A.; Anarbaev, R.O.; Mangerich, A.; Pastré, D.; Lavrik, O.I. The C-terminal Domain of Y-Box Binding Protein 1 Exhibits Structure-Specific Binding to Poly(ADP-Ribose), Which Regulates PARP1 Activity. Front. Cell Dev. Biol. 2022, 10, 831741. [Google Scholar] [CrossRef]
- Erijman, A.; Rosenthal, E.; Shifman, J.M. How Structure Defines Affinity in Protein-Protein Interactions. PLoS ONE 2014, 9, e110085. [Google Scholar] [CrossRef] [Green Version]
- Miwa, M.; Saikawa, N.; Yamaizumi, Z.; Nishimura, S.; Sugimura, T. Structure of poly(adenosine diphosphate ribose): Identification of 2′-[1″-ribosyl-2″-(or 3″-)(1‴-ribosyl)]adenosine-5′,5″,5‴-tris(phosphate) as a branch linkage. Proc. Natl. Acad. Sci. USA 1979, 76, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Gonzalez, R.; Jacobson, M.K. Characterization of polymers of adenosine diphosphate ribose generated in vitro and in vivo. Biochemistry 1987, 26, 3218–3224. [Google Scholar] [CrossRef]
- Fouquerel, E.; Goellner, E.M.; Yu, Z.; Gagné, J.-P.; de Moura, M.B.; Feinstein, T.; Wheeler, D.; Redpath, P.; Li, J.; Romero, G.; et al. ARTD1/PARP1 Negatively Regulates Glycolysis by Inhibiting Hexokinase 1 Independent of NAD + Depletion. Cell Rep. 2014, 8, 1819–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrer, J.; Kranaster, R.; Altmeyer, M.; Marx, A.; Bürkle, A. Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res. 2007, 35, e143. [Google Scholar] [CrossRef] [PubMed]
- Maltseva, E.A.; Rechkunova, N.I.; Sukhanova, M.V.; Lavrik, O.I. Poly(ADP-ribose) Polymerase 1 Modulates Interaction of the Nucleotide Excision Repair Factor XPC-RAD23B with DNA via Poly(ADP-ribosyl)ation. J. Biol. Chem. 2015, 290, 21811–21820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moor, N.A.; Vasil’Eva, I.A.; Kuznetsov, N.A.; Lavrik, O.I. Human apurinic/apyrimidinic endonuclease 1 is modified in vitro by poly(ADP-ribose) polymerase 1 under control of the structure of damaged DNA. Biochimie 2019, 168, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Panzeter, P.L.; Realini, C.A.; Althaus, F.R. Noncovalent interactions of poly(adenosine diphosphate ribose) with histones. Biochemistry 1992, 31, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kassab, M.A.; Dantzer, F.; Yu, X. PARP2 mediates branched poly ADP-ribosylation in response to DNA damage. Nat. Commun. 2018, 9, 3233. [Google Scholar] [CrossRef]
- Maltseva, E.A.; Krasikova, Y.S.; Sukhanova, M.V.; Rechkunova, N.I.; Lavrik, O.I. Replication protein A as a modulator of the poly(ADP-ribose)polymerase 1 activity. DNA Repair 2018, 72, 28–38. [Google Scholar] [CrossRef]
- Dasovich, M.; Beckett, M.Q.; Bailey, S.; Ong, S.-E.; Greenberg, M.M.; Leung, A.K.L. Identifying Poly(ADP-ribose)-Binding Proteins with Photoaffinity-Based Proteomics. J. Am. Chem. Soc. 2021, 143, 3037–3042. [Google Scholar] [CrossRef]
- Kutuzov, M.M.; Belousova, E.A.; Kurgina, T.A.; Ukraintsev, A.A.; Vasil’Eva, I.A.; Khodyreva, S.N.; Lavrik, O.I. The contribution of PARP1, PARP2 and poly(ADP-ribosyl)ation to base excision repair in the nucleosomal context. Sci. Rep. 2021, 11, 4849. [Google Scholar] [CrossRef]
- Schreiber, V.; Amé, J.-C.; Dollé, P.; Schultz, I.; Rinaldi, B.; Fraulob, V.; Ménissier-De Murcia, J.; de Murcia, G. Poly(ADP-ribose) Polymerase-2 (PARP-2) Is Required for Efficient Base Excision DNA Repair in Association with PARP-1 and XRCC1. J. Biol. Chem. 2002, 277, 23028–23036. [Google Scholar] [CrossRef]
- Naumenko, K.N.; Sukhanova, M.V.; Hamon, L.; Kurgina, T.A.; Alemasova, E.E.; Kutuzov, M.M.; Pastré, D.; Lavrik, O.I. Regulation of Poly(ADP-Ribose) Polymerase 1 Activity by Y-Box-Binding Protein 1. Biomolecules 2020, 10, 1325. [Google Scholar] [CrossRef]
- Kurgina, T.A.; Moor, N.A.; Kutuzov, M.M.; Naumenko, K.N.; Ukraintsev, A.A.; Lavrik, O.I. Dual function of HPF1 in the modulation of PARP1 and PARP2 activities. Commun. Biol. 2021, 4, 1259. [Google Scholar] [CrossRef]
- Rack, J.G.M.; Palazzo, L.; Ahel, I. (ADP-ribosyl)hydrolases: Structure, function, and biology. Genes Dev. 2020, 34, 263–284. [Google Scholar] [CrossRef]
- Slade, D.; Dunstan, M.S.; Barkauskaite, E.; Weston, R.; Lafite, P.; Dixon, N.; Ahel, M.; Leys, D.; Ahel, I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 2011, 477, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.; Kato, J.; Moss, J. Identification and Characterization of a Mammalian 39-kDa Poly(ADP-ribose) Glycohydrolase. J. Biol. Chem. 2006, 281, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Ono, T.; Kasamatsu, A.; Oka, S.; Moss, J. The 39-kDa poly(ADP-ribose) glycohydrolase ARH3 hydrolyzes O- acetyl-ADP-ribose, a product of the Sir2 family of acetyl-histone deacetylases. Proc. Natl. Acad. Sci. USA 2006, 103, 16687–16691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, P.; Bonfiglio, J.J.; Palazzo, L.; Bartlett, E.; Matic, I.; Ahel, I. Serine ADP-ribosylation reversal by the hydrolase ARH3. eLife 2017, 6, e28533. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.; Ferreira, M.T.; Gagné, J.-P.; Sharma, A.K.; Hendzel, M.J.; Masson, J.-Y.; Poirier, G.G. Emerging roles of eraser enzymes in the dynamic control of protein ADP-ribosylation. Nat. Commun. 2019, 10, 1182. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, K.; Nemoto, Y.; Ueda, K.; Hayaishi, O. Purification and characterization of poly(ADP-ribose) glycohydrolase. Different modes of action on large and small poly(ADP-ribose). J. Biol. Chem. 1986, 261, 14902–14911. [Google Scholar] [CrossRef]
- Braun, S.A.; Panzeter, P.L.; Collinge, M.A.; Althaus, F.R. Endoglycosidic cleavage of branched polymers by poly(ADP-ribose) glycohydrolase. JBIC J. Biol. Inorg. Chem. 1994, 220, 369–375. [Google Scholar] [CrossRef]
- Rack, J.G.M.; Liu, Q.; Zorzini, V.; Voorneveld, J.; Ariza, A.; Ebrahimi, K.H.; Reber, J.M.; Krassnig, S.C.; Ahel, D.; van der Marel, G.A.; et al. Mechanistic insights into the three steps of poly(ADP-ribosylation) reversal. Nat. Commun. 2021, 12, 4581. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K. Poly(ADP-ribose): A Dynamic Trigger for Biomolecular Condensate Formation. Trends Cell Biol. 2020, 30, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Štros, M. HMGB proteins: Interactions with DNA and chromatin. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2010, 1799, 101–113. [Google Scholar] [CrossRef]
- Ju, B.G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.-F.; Rouleau, M.; Poirier, G.G. Gene Expression Needs a Break to Unwind Before Carrying On. Science 2006, 312, 1752–1753. [Google Scholar] [CrossRef]
- Pinnola, A.; Naumova, N.; Shah, M.; Tulin, A.V. Nucleosomal Core Histones Mediate Dynamic Regulation of Poly(ADP-ribose) Polymerase 1 Protein Binding to Chromatin and Induction of Its Enzymatic Activity. J. Biol. Chem. 2007, 282, 32511–32519. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Mauro, S.; Gévry, N.; Lis, J.T.; Kraus, W. NAD+-Dependent Modulation of Chromatin Structure and Transcription by Nucleosome Binding Properties of PARP-1. Cell 2004, 119, 803–814. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belousova, E.A.; Lavrik, O.I. The Role of PARP1 and PAR in ATP-Independent Nucleosome Reorganisation during the DNA Damage Response. Genes 2023, 14, 112. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010112
Belousova EA, Lavrik OI. The Role of PARP1 and PAR in ATP-Independent Nucleosome Reorganisation during the DNA Damage Response. Genes. 2023; 14(1):112. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010112
Chicago/Turabian StyleBelousova, Ekaterina A., and Olga I. Lavrik. 2023. "The Role of PARP1 and PAR in ATP-Independent Nucleosome Reorganisation during the DNA Damage Response" Genes 14, no. 1: 112. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14010112