
The Evolutionary History of a DNA Methylase Reveals Frequent Horizontal Transfer and Within-Gene Recombination

 , ,
, ,
Abstract
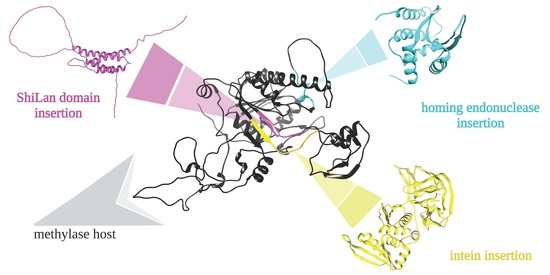
:
1. Introduction
2. Materials and Methods
2.1. Intein Discovery and Dataset Construction
2.2. Sequence Alignments
2.3. Phylogenetic Tree Construction and Divergence Comparison
2.4. Approximately Unbiased (AU) Test
2.5. Protein Structure Prediction
3. Results
3.1. Distribution of Methylases Similar to Dorothy_75
3.2. Alignments of Phage Encoded Methylase Genes and Their Insertion Sequences
- The intein is present in only a fraction of the sequences;
- It is inserted in a conserved region of the methylases (Figure 2A);
- Results from an HHPred search show homology to inteins over the whole length of the insertion (Figure 3);
- The intein sequences have a phylogeny different from the remainder of the methylase (see below).
3.3. Methylase Phylogeny
3.3.1. Methylases Do Not Group According to the Cluster to Which the Phages Belong
- The sequences from phage clusters DC, CV, FL, AR, and AS together with two sequences from separate singletons form a well-supported clan in both phylogenies (a clan is a group of tips that group together in an unrooted phylogeny, corresponding to a clade in a rooted phylogeny);
- The sequences from clusters F, P, E, A, and J do not form clans.
- Details of the branching order are not consistent between the two topologies;
- The two phylogenies have different estimated branch lengths.
3.3.2. Intein and ShiLan Domain-Containing Methylases Do Not Form Clans
3.3.3. Comparison of Phylogenies for the Inserted Elements and the Methylases That Harbor These Elements
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- SEA-PHAGES|Home. Available online: https://seaphages.org/ (accessed on 4 December 2022).
- Liu, X.Q. Protein-Splicing Intein: Genetic Mobility, Origin, and Evolution. Annu. Rev. Genet. 2000, 34, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Pietrokovski, S. Intein Spread and Extinction in Evolution. Trends Genet. 2001, 17, 465–472. [Google Scholar] [CrossRef]
- Perler, F.B.; Olsen, G.J.; Adam, E. Compilation and Analysis of Intein Sequences. Nucleic Acids Res. 1997, 25, 1087–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogarten, J.P.; Senejani, A.G.; Zhaxybayeva, O.; Olendzenski, L.; Hilario, E. Inteins: Structure, Function, and Evolution. Annu. Rev. Microbiol. 2002, 56, 263–287. [Google Scholar] [CrossRef] [Green Version]
- Perler, F.B. Protein Splicing of Inteins and Hedgehog Autoproteolysis: Structure, Function, and Evolution. Cell 1998, 92, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimble, F.S.; Thorner, J. Homing of a DNA Endonuclease Gene by Meiotic Gene Conversion in Saccharomyces Cerevisiae. Nature 1992, 357, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Naor, A.; Altman-Price, N.; Soucy, S.M.; Green, A.G.; Mitiagina, Y.; Turgeman-Grotta, I.; Davidovich, N.; Gogarten, J.P.; Gophna, U. Impact of a Homing Intein on Recombination Frequency and Organismal Fitness. Proc. Natl. Acad. Sci. USA 2016, 113, E4654–E4661. [Google Scholar] [CrossRef] [Green Version]
- Swithers, K.S.; Senejani, A.G.; Fournier, G.P.; Gogarten, J.P. Conservation of Intron and Intein Insertion Sites: Implications for Life Histories of Parasitic Genetic Elements. BMC Evol. Biol. 2009, 9, 303. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.S.; Lennon, C.W.; Belfort, M.; Novikova, O. Mycobacteriophages as Incubators for Intein Dissemination and Evolution. Mbio 2016, 7, e01537-16. [Google Scholar] [CrossRef] [Green Version]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Klenk, H.-P.; Clément, C.; Ouhdouch, Y.; van Wezel, G.P. Taxonomy, Physiology, and Natural Products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2015, 80, 1–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Bustamante, C.A.; Dedrick, R.M.; Garlena, R.A.; Russell, D.A.; Hatfull, G.F. Toward a Phage Cocktail for Tuberculosis: Susceptibility and Tuberculocidal Action of Mycobacteriophages against Diverse Mycobacterium Tuberculosis Strains. Mbio 2021, 12, e00973-21. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F.; Dedrick, R.M.; Schooley, R.T. Phage Therapy for Antibiotic-Resistant Bacterial Infections. Annu. Rev. Med. 2022, 73, 197–211. [Google Scholar] [CrossRef]
- Fruciano, E.; Bourne, S. Phage as an Antimicrobial Agent: D’Herelle’s Heretical Theories and Their Role in the Decline of Phage Prophylaxis in the West. Can. J. Infect. Dis. Med. Microbiol. 2007, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.A.; Hatfull, G.F. PhagesDB: The Actinobacteriophage Database. Bioinformatics 2017, 33, 784–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Actinobacteriophage Database|Home. Available online: https://phagesdb.org/ (accessed on 6 December 2022).
- Pope, W.H.; Bowman, C.A.; Russell, D.A.; Jacobs-Sera, D.; Asai, D.J.; Cresawn, S.G.; Jacobs, W.R.; Hendrix, R.W.; Lawrence, J.G.; Hatfull, G.F. Whole Genome Comparison of a Large Collection of Mycobacteriophages Reveals a Continuum of Phage Genetic Diversity. Elife 2015, 4, e06416. [Google Scholar] [CrossRef] [PubMed]
- Løbner-Olesen, A.; Skovgaard, O.; Marinus, M.G. Dam Methylation: Coordinating Cellular Processes. Curr. Opin. Microbiol. 2005, 8, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T. Initiation of DNA Replication at the Chromosomal Origin of E. Coli, OriC. Adv. Exp. Med. Biol. 2017, 1042, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Mruk, I.; Kobayashi, I. To Be or Not to Be: Regulation of Restriction-Modification Systems and Other Toxin-Antitoxin Systems. Nucleic Acids Res. 2014, 42, 70–86. [Google Scholar] [CrossRef]
- Kobayashi, I. Behavior of Restriction-Modification Systems as Selfish Mobile Elements and Their Impact on Genome Evolution. Nucleic Acids Res. 2001, 29, 3742–3756. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Ma, J.H.; Warren, K.; Tsang, R.S.W.; Low, D.E.; Jamieson, F.B.; Alexander, D.C.; Hao, W. Homologous Recombination Drives Both Sequence Diversity and Gene Content Variation in Neisseria Meningitidis. Genome Biol. Evol. 2013, 5, 1611–1627. [Google Scholar] [CrossRef] [Green Version]
- Fullmer, M.S.; Ouellette, M.; Louyakis, A.S.; Papke, R.T.; Gogarten, J.P. The Patchy Distribution of Restriction–Modification System Genes and the Conservation of Orphan Methyltransferases in Halobacteria. Genes 2019, 10, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, G.G.; Murray, N.E. Restriction and Modification Systems. Annu. Rev. Genet. 1991, 25, 585–627. [Google Scholar] [CrossRef]
- Smith, R.M.; Pernstich, C.; Halford, S.E. TstI, a Type II Restriction-Modification Protein with DNA Recognition, Cleavage and Methylation Functions in a Single Polypeptide. Nucleic Acids Res. 2014, 42, 5809–5822. [Google Scholar] [CrossRef] [Green Version]
- Mavrich, T.N.; Hatfull, G.F. Bacteriophage Evolution Differs by Host, Lifestyle and Genome. Nat. Microbiol. 2017, 2, 17112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cresawn, S.G.; Bogel, M.; Day, N.; Jacobs-Sera, D.; Hendrix, R.W.; Hatfull, G.F. Phamerator: A Bioinformatic Tool for Comparative Bacteriophage Genomics. BMC Bioinform. 2011, 12, 395. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouy, M.; Tannier, E.; Comte, N.; Parsons, D.P. Seaview Version 5: A Multiplatform Software for Multiple Sequence Alignment, Molecular Phylogenetic Analyses, and Tree Reconciliation. Methods Mol. Biol. 2021, 2231, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for Integrated Sequence-Structure Analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339. [Google Scholar] [CrossRef] [Green Version]
- UVA FASTA Server. Available online: https://fastademo.bioch.virginia.edu/fasta_www2/fasta_list2.shtml (accessed on 7 December 2022).
- Kishino, H.; Hasegawa, M. Evaluation of the Maximum Likelihood Estimate of the Evolutionary Tree Topologies from DNA Sequence Data, and the Branching Order in Hominoidea. J. Mol. Evol. 1989, 29, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Hasegawa, M. Multiple Comparisons of Log-Likelihoods with Applications to Phylogenetic Inference. Mol. Biol. Evol. 1999, 16, 1114. [Google Scholar] [CrossRef] [Green Version]
- Shimodaira, H. An Approximately Unbiased Test of Phylogenetic Tree Selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage Resistance Mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Bellas, C.M.; Schroeder, D.C.; Edwards, A.; Barker, G.; Anesio, A.M. Flexible Genes Establish Widespread Bacteriophage Pan-Genomes in Cryoconite Hole Ecosystems. Nat. Commun. 2020, 11, 4403. [Google Scholar] [CrossRef]
- HERSHEY, A.D.; CHASE, M. Independent Functions of Viral Protein and Nucleic Acid in Growth of Bacteriophage. J. Gen. Physiol. 1952, 36, 39–56. [Google Scholar] [CrossRef]
- Luria, S.E. Reactivation of Irradiated Bacteriophage by Transfer of Self-Reproducing Units. Proc. Natl. Acad. Sci. USA 1947, 33, 253–264. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alignment | Best Fitting Model $$ |
|---|---|
| Exteins $ compact alignment | VT + F + R7 |
| Exteins $ gappy alignment | WAG + R5 |
| Inteins (compact alignment) | WAG + F + I |
| Exteins $ (intein containing, compact alignment) | Blosum62 + F + G4 |
| ShiLan domain (compact alignment) | HIVb + F + I |
| Exteins (ShiLan domain containing, compact alignment) | WAG + G4 |
| Second endonuclease domain | Q.pfam |
| Exteins (Second endonuclease domain containing, compact alignment) | WAG + G4 |
| Tree | p-KH [36] | p-SH [37] | p-AU [38] |
|---|---|---|---|
| ml tree | 0.319 | 0.84 | 0.372 |
| all clusters constrained | 0.0006 | 0.001 | 5.41 × 10−5 |
| cluster A constrained | 0.0947 | 0.325 | 0.0529 |
| cluster AS constrained | 0.46 | 0.928 | 0.612 |
| cluster AR constrained | 0.363 | 0.939 | 0.454 |
| cluster E constrained | 0.54 | 1 | 0.618 |
| cluster F constrained | 0.0013 | 0.0027 | 0.00012 |
| cluster E constrained | 0.23 | 0.609 | 0.171 |
| cluster J constrained | 0.299 | 0.81 | 0.335 |
| cluster I constrained | 0.418 | 0.921 | 0.484 |
| all intein-containing seq. | 0 | 0 | 8.05 × 10−54 |
| all ShiLan domain-containing seq. | 0 | 0 | 3.11 × 10−6 |
| all endonucl. domain-containing seq. | 0.501 | 1 | 0.549 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gosselin, S.P.; Arsenault, D.R.; Jennings, C.A.; Gogarten, J.P. The Evolutionary History of a DNA Methylase Reveals Frequent Horizontal Transfer and Within-Gene Recombination. Genes 2023, 14, 288. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14020288
Gosselin SP, Arsenault DR, Jennings CA, Gogarten JP. The Evolutionary History of a DNA Methylase Reveals Frequent Horizontal Transfer and Within-Gene Recombination. Genes. 2023; 14(2):288. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14020288
Chicago/Turabian StyleGosselin, Sophia P., Danielle R. Arsenault, Catherine A. Jennings, and Johann Peter Gogarten. 2023. "The Evolutionary History of a DNA Methylase Reveals Frequent Horizontal Transfer and Within-Gene Recombination" Genes 14, no. 2: 288. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14020288