
Coexisting Conditions Modifying Phenotypes of Patients with 22q11.2 Deletion Syndrome
 , , , , , , , , and
, , , , , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patients and Deep Phenotyping
2.2. aCGH
2.3. Exome Sequencing
3. Results
3.1. aCGH Analysis
3.2. Exome Sequencing Analysis
3.2.1. Variants in Genes from Remaining 22q11.2 Region
3.2.2. Filtering for SNVs Elsewhere in the Genome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blagojevic, C.; Heung, T.; Theriault, M.; Tomita-Mitchell, A.; Chakraborty, P.; Kernohan, K.; Bulman, D.E.; Bassett, A.S. Estimate of the contemporary live-birth prevalence of recurrent 22q11.2 deletions: A cross-sectional analysis from population-based newborn screening. CMAJ Open 2021, 9, E802–E809. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.M.; Sheppard, S.E.; Crowley, T.B.; McGinn, D.E.; Bailey, A.; McGInn, M.J.; Unolt, M.; Homans, J.F.; Chen, E.Y.; Salmons, H.I.; et al. What is new with 22q? An update from the 22q and You Center at the Children’s Hospital of Philadelphia. Am. J. Med. Genet. A 2018, 176, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Grati, F.R.; Molina Gomes, D.; Ferreira, J.C.P.B.; Dupont, C.; Alesi, V.; Gouas, L.; Horelli-Kuitunen, N.; Choy, K.W.; García-Herrero, S.; De La Vega, A.G.; et al. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat. Diagn. 2015, 35, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Iyengar, S.; Kalyan, A.; Lan, C.; Simon, A.; Stosic, M.; Kobara, K.; Ravi, H.; Truong, T.; Ryan, A.; et al. Clinical experience with a single-nucleotide polymorphism-based non-invasive prenatal test for five clinically significant microdeletions. Clin. Genet. 2017, 93, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Maisenbacher, M.K.; Merrion, K.; Pettersen, B.; Young, M.; Paik, K.; Iyengar, S.; Kareht, S.; Sigurjonsson, S.; Demko, Z.P.; Martin, K.A. Incidence of the 22q11.2 deletion in a large cohort of miscarriage samples. Mol. Cytogenet. 2017, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, T.H.; Kurahashi, H.; Saitta, S.C.; O’Hare, A.M.; Hu, P.; Roe, B.A.; Driscoll, D.A.; McDonald-McGinn, D.M.; Zackai, E.H.; Budarf, M.L.; et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum. Mol. Genet. 2000, 9, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Shprintzen, R.J. Velo-cardio-facial syndrome: 30 Years of study. Dev. Disabil. Res. Rev. 2008, 14, 3–10. [Google Scholar] [CrossRef]
- Monteiro, F.; Vieira, T.P.; Sgardioli, I.C.; Molck, M.C.; Damiano, A.P.; Souza, J.; Monlleo, I.; Fontes, M.I.B.; Fett-Conte, A.C.; Félix, T.M.; et al. Defining new guidelines for screening the 22q11.2 deletion based on a clinical and dysmorphologic evaluation of 194 individuals and review of the literature. Eur. J. Pediatr. 2013, 172, 927–945. [Google Scholar] [CrossRef]
- Desmaze, C.; Prieur, M.; Amblard, F.; Aikem, M.; LeDeist, F.; Demczuk, S.; Zucman, J.; Plougastel, B.; Delattre, O.; Croquette, M.F. Physical mapping by FISH of the DiGeorge critical region (DGCR): Involvement of the region in familial cases. Am. J. Hum. Genet. 1993, 53, 1239–1249. [Google Scholar]
- Scambler, P.J.; Carey, A.H.; Wyse, R.K.; Roach, S.; Dumanski, J.P.; Nordenskjold, M.; Williamson, R. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics 1991, 10, 201–206. [Google Scholar] [CrossRef]
- McDonald-McGinn, D.M.; Tonnesen, M.K.; Laufer-Cahana, A.; Finucane, B.; Driscoll, D.A.; Emanuel, B.S.; Zackai, E.H. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: Cast a wide FISHing net! Genet. Med. 2001, 3, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hemel, J.O.; Schaap, C.; Van Opstal, D.; Mulder, M.P.; Niermeijer, M.F.; Meijers, J.H. Recurrence of DiGeorge syndrome: Prenatal detection by FISH of a molecular 22q11 deletion. J. Med. Genet. 1995, 32, 657–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald-McGinn, D.M.; Kirschner, R.; Goldmuntz, E.; Sullivan, K.; Eicher, P.; Gerdes, M.; Moss, E.; Solot, C.; Wang, P.; Jacobs, I.; et al. The Philadelphia story: The 22q11.2 deletion: Report on 250 patients. Genet. Couns. 1999, 10, 11–24. [Google Scholar] [PubMed]
- McDonald-McGinn, D.M.; Sullivan, K.; Marino, B.; Philip, N.; Swillen, A.; Vortsman, J.; Zackai, E.H.; Emanuel, B.; Vermeesch, J.; Morrow, B.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers. 2015, 19, 15071. [Google Scholar] [CrossRef] [Green Version]
- Swillen, A.; Vogels, A.; Devriendt, K.; Fryns, J.P. Chromosome 22q11 deletion syndrome: Update and review of the clinical features, cognitive-behavioral spectrum, and psychiatric complications. Am. J. Med. Genet. 2000, 97, 128–135. [Google Scholar] [CrossRef]
- Bassett, A.S.; Chow, E.W.C. Schizophrenia and 22q11.2 deletion syndrome. Curr. Psychiatry Rep. 2008, 10, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Bassett, A.S.; Chow, E.W.C.; Husted, J.; Hodgkinson, K.A.; Oechslin, E.; Harris, L.; Silversides, C. Premature death in adults with 22q11.2 deletion syndrome. J. Med. Genet. 2009, 46, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Bertini, V.; Azzarà, A.; Legitimo, A.; Milone, R.; Battini, R.; Consolini, R.; Valetto, A. Deletion Extents Are Not the Cause of Clinical Variability in 22q11.2 Deletion Syndrome: Does the Interaction between DGCR8 and miRNA-CNVs Play a Major Role? Front Genet. 2017, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Rozas, M.F.; Benavides, F.; León, L.; Repetto, G.M. Association between phenotype and deletion size in 22q11.2 microdeletion syndrome: Systematic review and meta-analysis. Orphanet J. Rare Dis. 2019, 14, 195. [Google Scholar] [CrossRef]
- Budarf, M.L.; Konkle, B.A.; Ludlow, L.B.; Michaud, D.; Li, M.; Yamashiro, D.J.; McDonald-McGinn, D.; Zackai, E.H.; Driscoll, D.A. Identification of a patient with Bernard-Soulier syndrome and a deletion in the DiGeorge/velo-cardio-facial chromosomal region in 22q11.2. Hum. Mol. Genet. 1995, 4, 763–766. [Google Scholar] [CrossRef]
- McDonald-McGinn, D.M.; Fahiminiya, S.; Revil, T.; Nowakowska, B.A.; Suhl, J.; Bailey, A.; Mlynarski, E.; Lynch, D.R.; Yan, A.C.; Bilaniuk, L.T.; et al. Hemizygous mutations in SNAP29 unmask autosomal recessive conditions and contribute to atypical findings in patients with 22q11.2DS. J. Med. Genet. 2012, 50, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.J.; van der Smagt, J.J.; Rosenfeld, J.A.; Pagnamenta, A.T.; Alswaid, A.; Baker, E.H.; Blair, E.; Borck, G.; Brinkmann, J.; Craigen, W.; et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet. Med. 2018, 20, 1175–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinton, J.; Huckstadt, V.; Foncuberta, M.E.; Perez, M.M.; Bonetto, M.C.; Gravina, L.P.; Obregon, M.G. Challenges in genetic diagnosis, co-occurrence of 22q11.2 deletion syndrome and Noonan syndrome. Am. J. Med. Genet. Part A 2022, 188, 2505–2508. [Google Scholar] [CrossRef] [PubMed]
- Bedeschi, M.; Colombo, L.; Mari, F.; Hofmann, K.; Rauch, A.; Gentilin, B.; Renieri, A.; Clerici, D. Unmasking of a Recessive SCARF2 Mutation by a 22q11.12 de novo Deletion in a Patient with Van den Ende-Gupta Syndrome. Mol. Syndr. 2010, 1, 239–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenwick, A.L.; Kliszczak, M.; Cooper, F.; Murray, J.; Pulido, L.S.; Twigg, S.; Goriely, A.; McGowan, S.; Miller, K.A.; Taylor, I.B.; et al. Mutations in CDC45, Encoding an Essential Component of the Pre-initiation Complex, Cause Meier-Gorlin Syndrome and Craniosynostosis. Am. J. Hum. Genet. 2016, 99, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Unolt, M.; Kammoun, M.; Nowakowska, B.; Graham, G.E.; Crowley, T.B.; Hestand, M.S.; Demaerel, W.; Geremek, M.; Emanuel, B.S.; Zackai, E.H.; et al. Pathogenic variants in CDC45 on the remaining allele in patients with a chromosome 22q11.2 deletion result in a novel autosomal recessive condition. Genet. Med. 2019, 22, 326–335. [Google Scholar] [CrossRef]
- Hestand, M.S.; Nowakowska, B.A.; Vergaelen, E.; Van Houdt, J.; Dehaspe, L.; Suhl, J.A.; Del-Favero, J.; Mortier, G.; Zackai, E.; Swillen, A.; et al. A catalog of hemizygous variation in 127 22q11 deletion patients. Hum. Genome Var. 2016, 3, 15065. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.L.; Crowley, T.B.; McGinn, D.E.; McDougall, C.; Unolt, M.; Lambert, M.P.; Emanuel, B.S.; Zackai, E.H.; McDonald-McGinn, n.M. 22q and two: 22q11.2 deletion syndrome and coexisting conditions. Am. J. Med. Genet. Part A 2018, 176, 2203–2214. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Gambin, T.; Akdemir, Z.C.; Yuan, B.; Gu, S.; Chiang, T.; Carvalho, C.M.; Shaw, C.; Jhangiani, S.; Boone, P.M.; Eldomery, M.K.; et al. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 2016, 45, 1633–1648. [Google Scholar] [CrossRef]
- Lalani, S.R.; Liu, P.; Rosenfeld, J.A.; Watkin, L.B.; Chiang, T.; Leduc, M.S.; Zhu, W.; Ding, Y.; Pan, S.; Vetrini, F.; et al. Recurrent Muscle Weakness with Rhabdomyolysis, Metabolic Crises, and Cardiac Arrhythmia Due to Bi-allelic TANGO2 Mutations. Am. J. Hum. Genet. 2016, 98, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunishima, S.; Imai, T.; Kobayashi, R.; Kato, M.; Ogawa, S.; Saito, H. Bernard-Soulier syndrome caused by a hemizygous GPIbβ mutation and 22q11.2 deletion. Pediatr Int. 2013, 55, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Diacou, A.; Johnston, H.R.; Musfee, F.I.; McDonald-McGinn, D.M.; McGinn, D.; Crowley, T.B.; Repetto, G.M.; Swillen, A.; Breckpot, J.; et al. Complete Sequence of the 22q11.2 Allele in 1053 Subjects with 22q11.2 Deletion Syndrome Reveals Modifiers of Conotruncal Heart Defects. Am. J. Hum. Genet. 2019, 106, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Dines, J.N.; Golden-Grant, K.; LaCroix, A.; Muir, A.M.; Cintrón, D.L.; McWalter, K.; Cho, M.T.; Sun, A.; Merritt, J.L.; Thies, J.; et al. TANGO2: Expanding the clinical phenotype and spectrum of pathogenic variants. Genet. Med. 2018, 21, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Pedrosa, E.; Hrabovsky, A.; Chen, J.; Puliafito, B.R.; Gilbert, S.R.; Zheng, D.; Lachman, H.M. Integrative transcriptome network analysis of iPSC-derived neurons from schizophrenia and schizoaffective disorder patients with 22q11.2 deletion. BMC Syst. Biol. 2016, 10, 105. [Google Scholar] [CrossRef] [Green Version]
- Ota, V.K.; Bellucco, F.T.; Gadelha, A.; Santoro, M.L.; Noto, C.; Christofolini, D.M.; Assunção, I.B.; Yamada, K.M.; Ribeiro-dos-Santos, A.K.; Santos, S.; et al. PRODH polymorphisms, cortical volumes and thickness in schizophrenia. PLoS One 2014, 9, e87686. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Tang, P.; Yang, C.; Li, R. Proline dehydrogenase gene (PRODH) polymorphisms and schizophrenia susceptibility: A meta-analysis. Metab. Brain Dis. 2017, 33, 89–97. [Google Scholar] [CrossRef]
- Girirajan, S.; Rosenfeld, J.A.; Cooper, G.M.; Antonacci, F.; Siswara, P.; Itsara, A.; Vives, L.; Walsh, T.; McCarthy, S.E.; Baker, C.; et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010, 42, 203–209. [Google Scholar] [CrossRef]
- Li, D.; Tekin, M.; Buch, M.; Fan, Y.S. Co-existence of other copy number variations with 22q11.2 deletion or duplication: A modifier for variable phenotypes of the syndrome? Mol. Cytogenet. 2012, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Bartnik, M.; Nowakowska, B.; Derwińska, K.; Wiśniowiecka-Kowalnik, B.; Kędzior, M.; Bernaciak, J.; Ziemkiewicz, K.; Gambin, T.; Sykulski, M.; Bezniakow, N.; et al. Application of array comparative genomic hybridization in 256 patients with developmental delay or intellectual disability. J. Appl. Genet. 2013, 55, 125–144. [Google Scholar] [CrossRef] [Green Version]
- Mlynarski, E.E.; Sheridan, M.B.; Xie, M.; Guo, T.; Racedo, S.E.; McDonald-McGinn, D.M.; Gai, X.; Chow, E.W.; Vorstman, J.; Swillen, A.; et al. Copy-Number Variation of the Glucose Transporter Gene SLC2A3 and Congenital Heart Defects in the 22q11.2 Deletion Syndrome. Am. J. Hum. Genet. 2015, 96, 753–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlynarski, E.E.; Xie, M.; Taylor, D.; Sheridan, M.B.; Guo, T.; Racedo, S.E.; McDonald-McGinn, D.M.; Chow, E.W.C.; Vorstman, J.; Swillen, A.; et al. Rare copy number variants and congenital heart defects in the 22q11.2 deletion syndrome. Hum. Genet. 2016, 135, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Abdollahi, M.R.; Morrison, E.; Sirey, T.; Molnar, Z.; Hayward, B.E.; Carr, I.M.; Springell, K.; Woods, C.G.; Ahmed, M.; Hattingh, L.; et al. Mutation of the Variant α-Tubulin TUBA8 Results in Polymicrogyria with Optic Nerve Hypoplasia. Am. J. Hum. Genet. 2009, 85, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantas, A.G.; Santoro, M.L.; Nunes, N.; de Mello, C.B.; Pimenta, L.S.E.; Meloni, V.A.; Soares, D.C.Q.; Belangero, S.N.; Carvalheira, G.; Kim, C.A.; et al. Downregulation of genes outside the deleted region in individuals with 22q11.2 deletion syndrome. Hum. Genet. 2019, 138, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Jehee, F.S.; Rosenberg, C.; Krepischi, A.; Kok, F.; Knijnenburg, J.; Froyen, G.; Vianna-Morgante, A.M.; Opitz, J.M.; Passos-Bueno, M.R. An Xq22.3 duplication detected by comparative genomic hybridization microarray (Array-CGH) defines a new locus (FGS5) for FG syndrome. Am. J. Med. Genet. Part A 2005, 139A, 221–226. [Google Scholar] [CrossRef] [Green Version]
- Geetha, T.S.; Michealraj, K.A.; Kabra, M.; Kaur, G.; Juyal, R.C.; Thelma, B. Targeted Deep Resequencing Identifies MID2 Mutation for X-Linked Intellectual Disability with Varied Disease Severity in a Large Kindred from India. Hum. Mutat. 2013, 35, 41–44. [Google Scholar] [CrossRef]
- Chanchani, S.R.; Xie, H.; Sekhon, G.; Melikishvili, A.M.; Moyer Harasink, S.; Pall, H.; Giampietro, P.F. A male infant with Xq22.2q22.3 duplication containing PLP1 and MID2. Mol. Genet. Genom. Med. 2020, 8, e1078. [Google Scholar]


{kind=link}
{kind=link}
| Patient ID | Variant | Sex | Age | Size | Pathogenicity/ CNV Classification | Protein Coding Genes or Known CNV Region | Patients’ Phenotype | 22q11.2 Recurrent Region |
|---|---|---|---|---|---|---|---|---|
| PD5864 | arr[GRCh37] 11q24.2q25(125857822_134868420)x1 | M | 17–18 Hbd | 9.01 Mb | pathogenic | 38 protein coding genes, 11q (Jacobsen syndrome) region | Fetal death, lip and cleft palate, edema. | proximal, A-B |
| arr[hg19] 22q11.1q11.21(16940617_18706059)x1 | 1.77 Mb | likely pathogenic | CCT8L2, XKR3, GAB4, IL17RA, TMEM121B, HDHD5, ADA2, CECR2, SLC25A18, ATP6V1E1, BCL2L13, BID, MICAL3, PEX26, TUBA8, USP18 | |||||
| PD8038 | arr[GRCh37] 22q11.21q11.23(21759521_23822984)x1 | M | 17–18 Hbd | 2.06 Mb | pathogenic | 22q11.2 recurrent region (distal type I, D-E/F) | Symmetrical hypotrophy of the fetus, cardiac defect | proximal, B-D |
| GC034796 | arr[GRCh37] 13q21.32(67204211_67215612)x1 | M | 1 y 3 m | 11 kb | uncertain clinical significance | exon 4 of PCDH9 | Global developmental delay, ventricular septal defect, interrupted aortic arch, bicuspid aortic valve, atrial septal defect, aberrant subclavian, thymic aplasia with absent T cells, hypocalcemia, nasal regurgitation, thyroid hypoplasia, hypothyroidism, growth hormone deficiency, growth delay, dysmorphic features (micrognathia), scoliosis, butterfly vertebrae, additional ribs, hypotonia, ligamentous laxity, delayed dental eruption, chronic constipation | proximal, A-C |
| arr[GRCh37] 16p11.2(29673967_30190593)x1 | 517 kb | pathogenic | 16p11.2 recurrent region (proximal, BP4-BP5) (includes TBX6) | |||||
| arr[GRCh37] 22q11.21(18375151_18661758)x1 | 287 kb | uncertain clinical significance | exons 1–11 of MICAL3, PEX26, TUBA8, USP18 | |||||
| 113787 | arr[GRCh37] 20p12.3(8256689_8558204)x1 | F | 5 y | 301 kb | uncertain clinical significance | exon 3 of PLCB1 | Global developmental delay, delay of speech development, dysmorphic features | proximal, A-B |
| arr[GRCh37] 22q11.1q11.21(16940617_18848020)x3 | 1.91 Mb | pathogenic | 22q11.21 recurrent (Cat eye syndrome) region (includes CECR2) | |||||
| GC034823 | arr[GRCh37] 22q11.1q11.21(17397633_18661758)x1 | F | 25 y | 1.26 Mb | likely pathogenic | GAB4, IL17RA, TMEM121B, HDHD5, ADA2, CECR2, SLC25A18, ATP6V1E1, BCL2L13, BID, MICAL3, PEX26, TUBA8, USP18 | Ventricular septal defect, double outlet right ventricle, pulmonary (valve) stenosis, unilateral cleft palate and lip, bifid uvula, velopharyngeal insufficiency, growth delay, dysmorphic features, myopia, scoliosis, dental problems, conductive hearing loss, nasopharyngeal reflux, chronic constipation, global developmental delay, delayed speech and language development, moderate intellectual disability, mood changes | proximal, A-B |
| arr[GRCh37] 22q13.33(51162483_51178213)x3 | 16 kb | uncertain clinical significance | ACR, exons 1–21 of SHANK3 | |||||
| 111183 | arr[GRCh37] 16p12.2(21959950_22407951)x1 | F | 1 m | 448 kb | likely pathogenic | 16p12.2 recurrent region (proximal) (includes EEF2K, CDR2) | Facial dysmorphic features, cardiac defect, urinary system defect | proximal, A-D |
| 105620 | arr[GRCh37] 12q24.33(132239944_133773393)x1, | M | 6 y | 1.53 Mb | likely pathogenic | SFSWAP, MMP17, ULK1, PUS1, EP400, DDX51, NOC4L, GALNT9, FBRSL1, LRCOL1, P2RX2, POLE, PXMP2, PGAM5, ANKLE2, GOLGA3, CHFR, ZNF605, ZNF26, ZNF84, ZNF140, ZNF891, ZNF10, ZNF268 | Hypotonia (low muscle tone), dysmorphic features, lack of speech development, cardiac defect | proximal, A-B |
| arr[GRCh37] 22q11.1q11.21(17397633_18661758)x1 | 1.26 Mb | likely pathogenic | GAB4, IL17RA, TMEM121B, HDHD5, ADA2, CECR2, SLC25A18, ATP6V1E1, BCL2L13, BID, MICAL3, PEX26, TUBA8, USP18 | |||||
| GC034824 /106502 | arr[GRCh37] Xp11.23(47616035_48204099)x3 | F | 2 y 3 m | 588 kb | uncertain clinical significance | ZNF81, ZNF182, ZNF630, SPACA5, SPACA5B, SSX5, SSX1 | Aberrant subclavian artery, recurrent infections, thymic hypoplasia, hypernasal speech, nasal regurgitation, pronunciation defects, short stature, dysmorphic features (hypotelorism, bitemporal narrowing, micrognathia, retrognathia) long fingers, hypotonia, ligamentous laxity, dental problems, enuresis, gastrointestinal problems, global developmental delay | proximal, A-D |
| 108894 | arr[GRCh37] 1p36.23(8736229_9105539)x3 mat | M | 2 y | 369 kb | uncertain clinical significance | exons 1–2 of RERE, ENO1, CA6, SLC2A7, exons 5–12 of SLC2A5 | Global developmental delay, facial dysmorphic features, hypotonia (low muscle tone) | proximal, A-D |
| 110226 | arr[GRCh37] 4q13.1(65794809_66356976)x1 mat | M | 11 y | 562 kb | uncertain clinical significance | exons 1–2 of EPHA5 | Hypotonia (low muscle tone), delay of speech development | proximal, A-D |
| 111711 | arr[GRCh37] 12q12(44202928_44445117)x1 | M | 1 m | 242 kb | uncertain clinical significance | exons 1–3 of TMEM117 | Facial dysmorphic features, cardiac defect | proximal, A-D |
| 112367 | arr[GRCh37] 22q11.22q11.23(23012069_23648159)x1 pat | M | 9 m | 637 kb | uncertain clinical significance | 22q11.2 recurrent region (distal type II, E-F) | Cardiac defect | proximal, A-D |
| 112587 | arr[GRCh37] 15q13.2q13.3(30419801_32861612)x3 | M | 6 y | 2.44 Mb | uncertain clinical significance | 15q13.3 recurrent region (BP4-BP5) (includes CHRNA7) | No data | proximal, A-D |
| 113324 | arr[GRCh37] 7q21.3q22.1(97939915_98557740)x3 mat | F | 1 y 4 m | 618 kb | uncertain clinical significance | exons 1–8 of BAIAP2L1, NPTX2, TMEM130, exons 1–44 of TRRAP | Defect of the larynx, thymic hypoplasia, dysmorphic features | proximal, A-D |
| arr[GRCh37] 22q11.23(23720181_25066484)x3 mat | 1.35 Mb | uncertain clinical significance | 22q11.2 recurrent region (distal type III, F-H) | |||||
| 116123 | arr[GRCh37] 18p11.31p11.23(7094706_8359012)x3 | F | 1 m | 1.26 Mb | uncertain clinical significance | exon 1 of LAMA1, LRRC30, exons 1–23 of PTPRM | Facial dysmorphic features, defect of the urinary system, cardiac defect (vascular ring), long fingers. | proximal, A-D |
| PD2305 | arr[GRCh37] 7p12.3p12.2(46094932_49190408)x3 | M | 22 Hbd | 3.1 Mb | uncertain clinical significance | TNS3, PKD1L1, HUS1, SUN3, C7orf57, UPP1, ABCA13, CDC14C | Screening test result showing increased risk of chromosomal aberration, fetal cardiac defect | proximal, A-D |
| PD3311 | arr[GRCh37] 22q11.22q11.23(23012069_23648159)x1 pat | M | 21 Hbd | 636 kb | uncertain clinical significance | 22q11.2 recurrent region (distal type II, E-F) | Fetal cardiac defect | proximal, A-D |
| GC034808 | arr[GRCh37] Xq22.3(106791395_107156386)x2 | M | 6y | 465 kb | uncertain clinical significance | FRMPD3, NCBP2L, PRPS1, TSC22D3, exons 1–5 of MID2 | Recurrent infections, acute otitis media, submucosal cleft palate, bifid uvula, secondary hearing loss, pronunciation defects, speech articulation difficulties, short stature, dysmorphic features, hypotonia, ligamentous laxity, feeding difficulties, umbilical hernia, global developmental delay, delayed speech and language development, attention difficulties | proximal, A-D |
| 108366 | arr[GRCh37] 8p12(30498811_30732891)x3 | M | 9 y | 234 kb | uncertain clinical significance | exons 1–2 of GTF2E2, SMIM18, GSR, UBXN8, PPP2CB, exons 2–7 of TEX15 | Global developmental delay, facial dysmorphic features, epilepsy | proximal, A-B |
| GC034815 | arr[GRCh37] 3p26.3(1539221_2206719)x1 | F | 17 y | 667 kb | uncertain clinical significance | exons 1–2 of CNTN4 | Patent foramen ovale, defects in humoral immunity, IgA deficiency, recurrent infections, acute otitis media, chronic sinusitis, T-cell lymphopenia, vitiligo, thrombocytopenia, Hashimoto thyroiditis, velopharyngeal insufficiency, laryngotracheoesophageal anomalies, hypothyroidism, hypoparathyroidism, dysmorphic features, myopia, amblyopia, hypotonia, ligamentous laxity, delayed dental eruption, feeding and swallowing problems, delayed speech and language development, supernumerary spleens | proximal, A-D |
| arr[GRCh37] 7q21.3q22.1(97939915_98557740)x3 | 618 kb | uncertain clinical significance | exons 1–8 of BAIAP2L1, NPTX2, TMEM130, exons 1–44 of TRRAP | |||||
| GC028958 | arr[GRCh37] 8q22.1(96815252_97229772)x1 | F | 2 weeks | 414 kb | uncertain clinical significance | GDF6 | Child succumbed at 14 days of life following her heart repair on day of life 13. Ventricular septal defect, persistent truncus arteriosus, atrial septal defect, bilateral coronal craniosynostosis, diffuse white matter gliosis, reduced cortical thickness, thymic aplasia with absent T cells, hypocalcemia, hypoparathyroidism, intestinal malrotation, anteriorly placed anus, diaphragmatic hernia | proximal, A-D |
| arr[GRCh37] Xq28(151896983_152351582)x3 | 455 kb | uncertain clinical significance | CETN2, CSAG1, MAGEA12, MAGEA2, MAGEA3, NSDHL, PNMA3, PNMA5, PNMA6A, PNMA6C, ZNF185 | |||||
| (8)x2~3 | 146.36 Mpz | pathogenic | mosaic trisomy |
| ID | Sex | Age | Chr | Change | Zyg | Gene | Function | OMIM_Diseases Linked to Gene | Exonic Function | GnomAD Exome ALL | Patient’s Clinical Features Associated with Variant |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GC034931 | M | 7 y | chr2 | NM_000384:c.9115_9119del:p.F3039fs | het | APOB | exonic | Hypercholesterolemia, familial, 2; Hypobetalipoproteinemia | frameshift deletion | 4.071 × 10−6 | NA |
| GC034813 | M | 12 y | chrX | NM_000495:c.G1871A:p.G624D | hom | COL4A5 | exonic | Alport syndrome 1, X-linked | nonsynonymous SNV | 8.97 × 10−5 | NA |
| GC034793 | M | 7 y | chr3 | NM_000094:c.A425G:p.K142R | het | COL7A1 | exonic | EBD inversa; EBD, Bart type; EBD, localisata variant; Epidermolysis bullosa dystrophica, AD; Epidermolysis bullosa dystrophica AR; Epidermolysis bullosa pruriginosa; Epidermolysis bullosa, pretibial; Toenail dystrophy, isolated; Transient bullous of the newborn | nonsynonymous SNV | 4.468 × 10−5 | NA |
| GC034931 | M | 7 y | chr11 | het | F2 | UTR3 | Dysprothrombinemia; Hypoprothrombinemia; Thrombophilia due to thrombin defect | 0 | NA | ||
| GC030951 | M | 6 y | chr17 | NM_000267:c.C5839T:p.R1947X | het | NF1 | exonic | Leukemia, juvenile myelomonocytic; Neurofibromatosis, familial spinal; Neurofibromatosis, type 1; Neurofibromatosis-Noonan syndrome; Watson syndrome | stopgain | 0 | Neurofibromatosis |
| GC034900 | F | 1 y | chr4 | NM_012464.5:c.713T>C:p.p.Val238Ala | het | TLL1 | exonic | Atrial septal defect 6 | nonsynonymous SNV | 2 × 10−4 | Atrial septal defect |
| GC034800 | F | 7 y | chr12 | NM_000552:c.G2561A:p.R854Q | het | VWF | exonic | von Willebrand disease, type 1; von Willebrand disease, type 3; von Willebrand disease, types 2A, 2B, 2M, and 2N | nonsynonymous SNV | 0.0034 | NA |
| Identifier | Sex | Age | Inheritance | Chr | Start | End | Ref | Alt | Zyg | Gene | OMIM Diseases | Patient’s Phenotype Features Correlating with OMIM Phenotype |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GC028956 | F | 13 y | AD | chr9 | 128607934 | 128607934 | A | C | het | SPTAN1 | Developmental and epileptic encephalopathy 5 | Structural CNS anomalies (spina bifida, polyhydramnios), unprovoked seizures, developmental delay |
| GC028956 | F | 13 y | AD | chr18 | 34794223 | 34794223 | T | G | het | DTNA | Left ventricular noncompaction 1, with or without congenital heart defects | Congenital heart defects (VSD) |
| GC030952 | F | 24 y | AD | chr2 | 120982720 | 120982720 | A | C | het | GLI2 | Culler-Jones syndrome; Holoprosencephaly 9 | Palatal abnormalities (bifid uvula), growth delay, delayed psychomotor development |
| GC034768 | M | 6 y | AD | chr19 | 10986246 | 10986246 | G | A | het | SMARCA4 | Coffin-Siris syndrome 4 | Congenital heart defects (VSD, IAA, BAV, ASD), dental problems, hypotonia, global developmental delay, delayed speech and language development, learning disability |
| GC034778 | F | 10 y | AD | chr18 | 44952093 | 44952093 | G | A | het | SETBP1 | Intellectual developmental disorder, autosomal dominant 29; Schinzel–Giedion midface retraction syndrome | Global developmental delay, delayed speech and language development, learning disability |
| GC034779 | F | 7 y | AD | chr2 | 15945723 | 15945723 | A | T | het | MYCN | Feingold syndrome 1 | Asymmetric face, narrow palpebral fissures, epicantic folds, micrognathia, palatal abnormalities (bifid uvula), hearing loss, global developmental delay, delayed speech and language development, learning disability |
| GC034783 | F | 8 y | AD | chr1 | 211082791 | 211082791 | G | A | het | KCNH1 | Temple-Baraitser syndrome; Zimmermann-Laband syndrome 1 | Cardiovascular system defects (ASD, conotruncal cardiac anomaly, aberrant subclavian), hypertelorism, skeletal abnormalities (proximal implantation of thumbs), learning disability |
| GC034785 | M | 19 y | AD | chr8 | 60800422 | 60800422 | G | A | het | CHD7 | CHARGE syndrome; Hypogonadotropic hypogonadism 5 with or without anosmia | Submucosal cleft palate, dysmorphic face (facial asymmetry, hypertelorism, malar flattening, micrognathia, cup ear), hearing loss, skeletal anomalies of limbs, feeding and swallowing problems, intellectual disability, learning disability, thymic hypoplasia, T-cell lymphopenia |
| GC034786 | M | 2 y 6 m | AD | chr1 | 7747776 | 7747776 | A | G | het | CAMTA1 | Cerebellar dysfunction with variable cognitive and behavioral abnormalities | Bulbous, wide nose, low-set ears, delayed speech and language development, hypotonia, dental problems, gastrointestinal problems |
| GC034787 | M | 1 y | AD | chr3 | 119402046 | 119402046 | C | G | het | ARHGAP31 | Adams-Oliver syndrome 1 | Microcephaly, palatal anomalies (bifid uvula), cardiovascular system defects (VSD, PTA, Pulmonary artery stenosis), hypotonia, global developmental delay |
| GC034789 | F | 6 m | XLD | chrX | 41134763 | 41134763 | C | T | het | USP9X | Intellectual developmental disorder, X-linked 99, syndromic, female-restricted | Congenital heart defects (VSD, ASD), dysmorphic face (prominent forehead, bitemporal narrowing, posteriorly rotated ears, broad nasal bridge, bulbous nose), hypotonia, sensory processing problems |
| GC034796 | M | 1 y 3 m | AD | chr1 | 151440971 | 151440971 | G | T | het | POGZ | White-Sutton syndrome | Congenital heart defects (VSD, ASD, IAA, BAV, aberrant subclavian), growth delay, dysmorphic face (low-set ears, posteriorly rotated ears, short philtrum), hypotonia, global developmental delay |
| GC034797 | F | 2 y 2 m | AD | chr2 | 227309265 | 227309265 | C | T | het | COL4A3 | Alport syndrome 3, autosomal dominant; Hematuria, benign familial | Sensorineural hearing loss |
| GC034800 | F | 7 y | AD | chr1 | 27552055 | 27552055 | - | G | het | AHDC1 | Xia-Gibbs syndrome | Low-set ears, global developmental delay, delayed speech and language development, intellectual disability, learning disability |
| GC034800 | F | 7 y | AD | chr6 | 45492054 | 45492054 | C | T | het | RUNX2 | Cleidocranial dysplasia; Cleidocranial dysplasia, forme fruste, dental anomalies only; Cleidocranial dysplasia, forme fruste, with brachydactyly; Metaphyseal dysplasia with maxillary hypoplasia with or without brachydactyly | Prominent forehead, dental problems, high-arched palate, skeletal abnormalities |
| GC034813 | M | 12 y | AD | chr16 | 3769345 | 3769345 | C | A | het | CREBBP | Menke-Hennekam syndrome 1; Rubinstein-Taybi syndrome 1 | Congenital heart defects (Tetralogy of Fallot, VSD, ASD, conotruncal heart defects) submucosal cleft palate, dysmorphic face (prominent forehead, broad nasal bridge, micrognathia, retrognathia, low-set ears), scoliosis, syndactyly, dental problems, cryptorchidism, recurrent infections, hypotonia, developmental delay, delayed speech and language development, intellectual disability |
| GC034813 | M | 12 y | XLD | chrX | 45061396 | 45061396 | T | C | hom | KDM6A | Kabuki syndrome 2 | Congenital heart defects (conotruncal heart defects, Tetralogy of Fallot, VSD, ASD), submucosal cleft palate, cupped ears, dental problems, hypotonia, ligamentous laxity, developmental delay, behavioral difficulties |
| GC034822 | M | 10 y | AD | chr19 | 13300559 | 13300559 | G | C | het | CACNA1A | Developmental and epileptic encephalopathy 42; Episodic ataxia, type 2; Migraine, familial hemiplegic, 1; Migraine, familial hemiplegic, 1, with progressive cerebellar ataxia; Spinocerebellar ataxia 6 | Global developmental delay, hypotonia, intellectual disability (borderline), learning difficulties |
| GC034822 | M | 10 y | XLR | chrX | 70454277 | 70454277 | G | A | hom | DLG3 | Intellectual developmental disorder, X-linked 90 | Global developmental delay, delayed speech development, intellectual disability (borderline), learning difficulties |
| GC034824 | F | 2 y 3 m | AD | chr8 | 143728049 | 143728049 | C | T | het | FAM83H | Amelogenesis imperfecta, type IIIA | Dental problems |
| GC034930 | F | 4 m | AD | chr3 | 111649747 | 111649747 | A | C | het | CD96 | C syndrome | Micrognathia, posteriorly rotated ears, epicantic folds, broad nasal bridge, short nose, congenital heart defect (VSD) |
| GC034930 | F | 4 m | AD | chr1 | 119916604 | 119916604 | C | T | het | NOTCH2 | Alagille syndrome 2; Hajdu-Cheney syndrome | Congenital heart defects (VSD, ASD, PTA), Bulbous nasal tip |
| GC034932 | F | 1 y | AD | chr12 | 115996599 | 115996599 | C | T | het | MED13L | Impaired intellectual development and distinctive facial features with or without cardiac defects | Global developmental delay |
| GC034823 | F | 25 y | AD | chr8 | 105801714 | 105801714 | G | A | het | ZFPM2 | Tetralogy of Fallot | VSD, DORV with pulmonary stenosis |
| GC034789 | F | 6 m | AD | chr5 | 173233189 | 173233189 | C | A | het | NKX2-5 | Atrial septal defect 7, with or without AV conduction defects; Hypoplastic left heart syndrome 2; Hypothyroidism, congenital nongoitrous, 5; Tetralogy of Fallot; Ventricular septal defect 3 | Congenital heart defects (ASD, VSD) |
| GC034808 | M | 6 y | AD | chr6 | 1610202 | 1610202 | C | T | hom | FOXC1 | Axenfeld-Rieger syndrome, type 3 | Secondary hearing loss, redundant periumbilical skin |
| ID | Sex | Age | Reults of HMZdelfinder Analysis | Gene Content | Gene Function |
|---|---|---|---|---|---|
| pathogenic | |||||
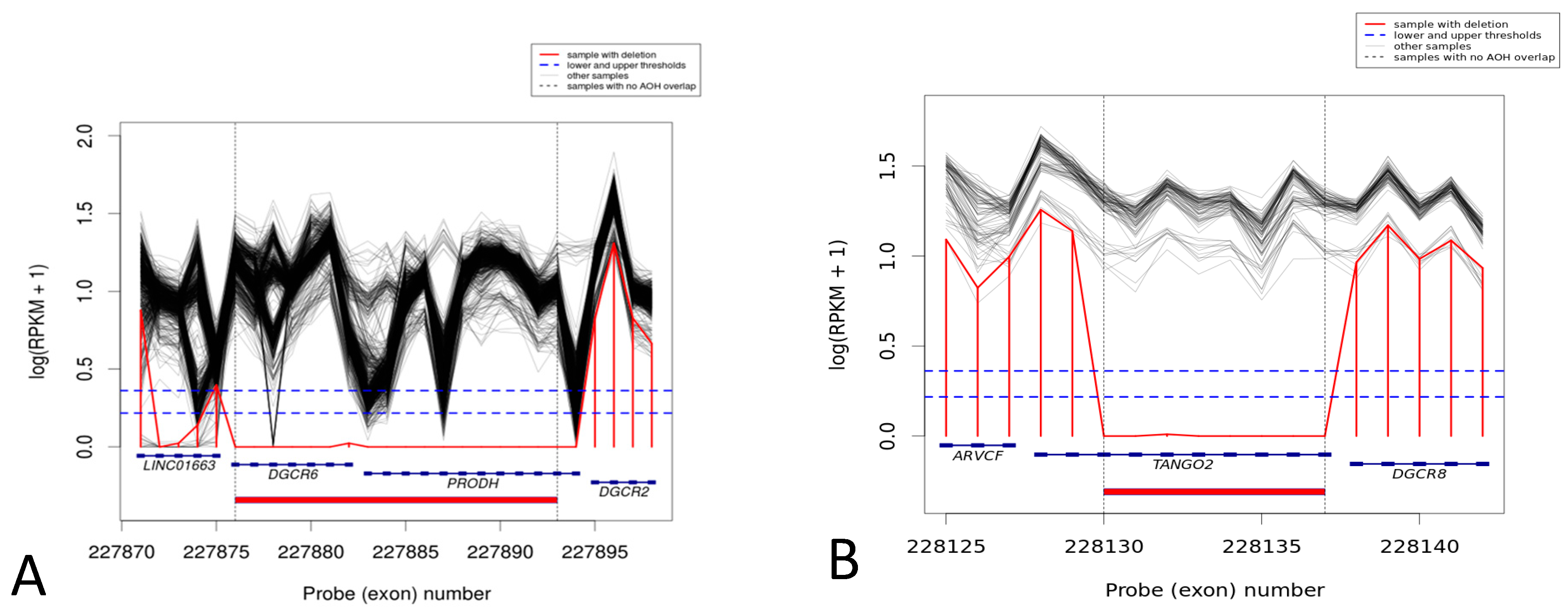
| GC034806 | M | 5 y | chr22_20043265_20064650 | exons 2–9 of TANGO2 | encodes transport and golgi organization 2 homologs; bi-allelic mutations in TANGO2 cause recurrent muscle weakness with rhabdomyolysis, metabolic crises, and cardiac arrhythmia |
| uncertain pathogenicity | |||||
| GC028957 | F | 27 y | chr15_26914781_26914903 | exon 7 of GABRA5 | encodes γ-aminobutyric acid type A receptor alpha5 subunit; restricted expression toward brain; GABA is the major inhibitory neurotransmitter in the mammalian brain where it acts at GABA-A receptors, which are ligand-gated chloride channels |
| GC028958 | F | 2 weeks | chr15_26937134_26937376 | exon 8 of GABRA5 | encodes γ-aminobutyric acid type A receptor alpha5 subunit; restricted expression toward brain; GABA is the major inhibitory neurotransmitter in the mammalian brain where it acts at GABA-A receptors, which are ligand-gated chloride channels |
| GC030952 | F | 24 y | chr16_75635620_75635862 | exon 7 of KARS | encodes lysyl-tRNA synthetase; defects in KARS are associated with the recessive form of Charcot-Mary-Tooth polyneuropathy, the autosomal recessive non-syndromic hearing loss, congenital visual impairment and progressive microcephaly, hypertrophic cardiomyopathy and combined mitochondrial respiratory chain defect |
| chr22_26601827_26608074 | exons 4–5 of CRYBB1 | encodes crystallin β B1; mutations in CRYBB1 are associated with congenital cataract with nystagmus | |||
| GC035414 | M | 9 y | Chr22:18906447-18931248 | DGCR6, PRODH | Proline dehydrogenase is involved in the degradation of the amino acid proline. It catalyzes the conversion of proline to pyrroline-5-carboxylate, or P5C. It is associated with Hyperprolinemia, type I, and susceptibility to schizophrenia |
| GC030953 | M | 3 y | chr12_21568814_21580552 | exons 9–12 of GYS2 | encodes glycogen synthase 2; mutations in this gene cause autosomal recessive glycogen storage disease type 0 (GSD-0)—a rare type of early childhood fasting hypoglycemia with decreased liver glycogen content |
| chr23_55751086_55751208 | exon 7 of RRAGB | encodes Ras related GTP binding protein B | |||
| GC034767 | F | 3 y | chr1_202135375_202154374 | exons 1–5 of ARL8A, exons 6–10 of PTPN7 | ARL8A—encodes ADP ribosylation factor like GTPase 8A; PTPN7—encodes protein tyrosine phosphatase non-receptor type 7 |
| GC034773 | M | 6 y | chr2_3702340_4228633 | exon 12 of ALLC, DCDC2C, LINC01304, ENSG00000215960 | ALLC encodes allantoicase, expressed in testis; DCDC2C encodes doublecortin domain containing 2C, low expression in testis; LINC01304 expressed in testis |
| GC034774 | F | 35 y | chr12_6452218_6452340 | exon 1 of TAPBPL | encodes TAP binding protein like, involved in controlling peptide presentation to the immune system |
| GC034780 | M | 10 y | chr8_3029293_3230255 | exons 28–52 of CSMD1 | encodes CUB and Sushi multiple domains 1, expressed at intermediate level in brain, including cerebellum, substantia nigra, hippocampus and fetal brain; potential suppressor of squamous cell carcinomas; altered expression of the CSMD1 gene in the peripheral blood of schizophrenia patients; association with cognitive function |
| GC034789 | F | 6 m | chr1_145850735_145854625 | exons 8–12 of PIAS3 | encodes protein inhibitor of activated STAT 3, plays a crucial role as a transcriptional coregulation in various cellular pathways, including the STAT pathway and the steroid hormone signaling pathway |
| chr1_145868270_145875066 | exons 8–11 of ANKRD35 | encodes ankyrin repeat domain 35, expression in skin, esophagus, association with chronic lymphocytic leukemia | |||
| GC034850 | M | 3 m | chr1_196902403_196914600 | exons 2–8 of CFHR4 | encodes complement factor H related 4, expressed in liver, can associate with lipoproteins and may play a role in lipid metabolism |
| GC034825 | F | 34 y | chr14_23988867_23990346 | exon 2 of DHRS4L2 | encodes a member of the short chain dehydrogenase reductase family. The encoded protein may be a NADPH-dependent retinol oxidoreductase |
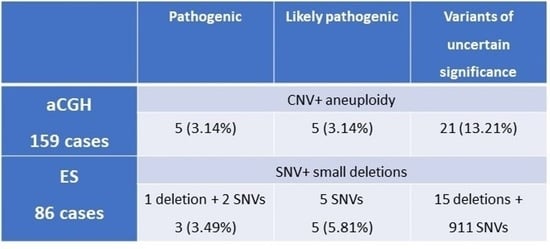
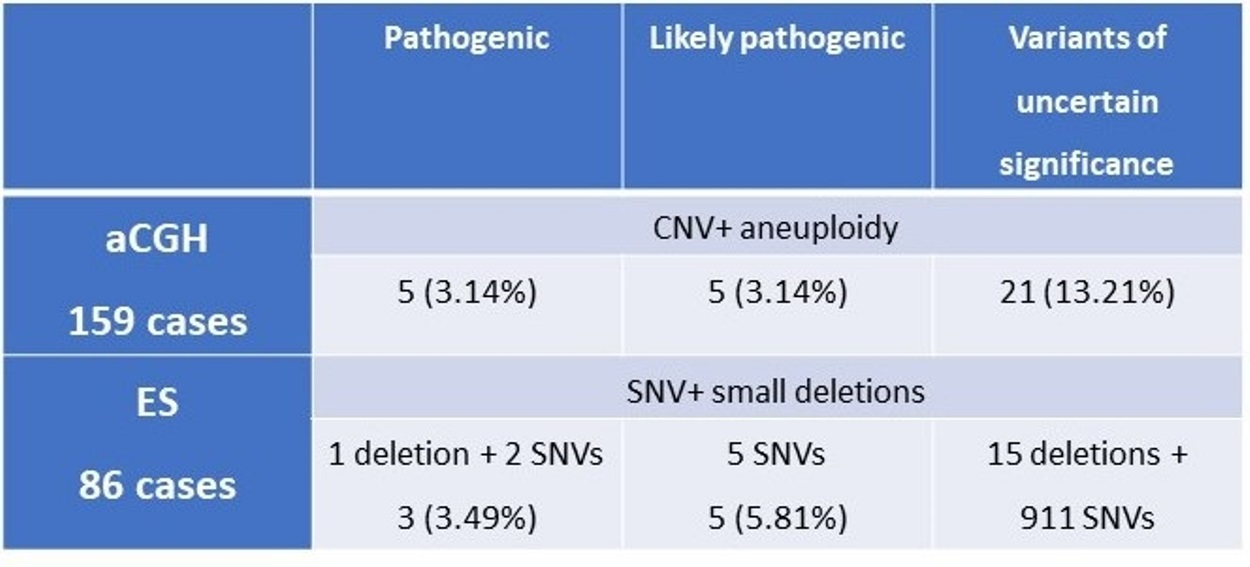
| Pathogenic | Likely Pathogenic | Variants of Uncertain Significance | |
|---|---|---|---|
| aCGH 159 cases | CNV + aneuploidy | ||
| 5 (3.14%) | 5 (3.14%) | 21 (13.21%) | |
| ES 86 cases | SNV + small deletions | ||
| 1 deletion + 2 SNVs 3 (3.49%) | 5 SNVs 5 (5.81%) | 15 deletions + 911 SNVs | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smyk, M.; Geremek, M.; Ziemkiewicz, K.; Gambin, T.; Kutkowska-Kaźmierczak, A.; Kowalczyk, K.; Plaskota, I.; Wiśniowiecka-Kowalnik, B.; Bartnik-Głaska, M.; Niemiec, M.; et al. Coexisting Conditions Modifying Phenotypes of Patients with 22q11.2 Deletion Syndrome. Genes 2023, 14, 680. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14030680
Smyk M, Geremek M, Ziemkiewicz K, Gambin T, Kutkowska-Kaźmierczak A, Kowalczyk K, Plaskota I, Wiśniowiecka-Kowalnik B, Bartnik-Głaska M, Niemiec M, et al. Coexisting Conditions Modifying Phenotypes of Patients with 22q11.2 Deletion Syndrome. Genes. 2023; 14(3):680. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14030680
Chicago/Turabian StyleSmyk, Marta, Maciej Geremek, Kamila Ziemkiewicz, Tomasz Gambin, Anna Kutkowska-Kaźmierczak, Katarzyna Kowalczyk, Izabela Plaskota, Barbara Wiśniowiecka-Kowalnik, Magdalena Bartnik-Głaska, Magdalena Niemiec, and et al. 2023. "Coexisting Conditions Modifying Phenotypes of Patients with 22q11.2 Deletion Syndrome" Genes 14, no. 3: 680. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14030680