
Transferability of Human and Environmental Microbiome on Clothes as a Tool for Forensic Investigations

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Collection
2.2. Microbiome Extraction
2.3. Sequencing and Data Analysis
2.4. Human STRs Profiles
3. Results
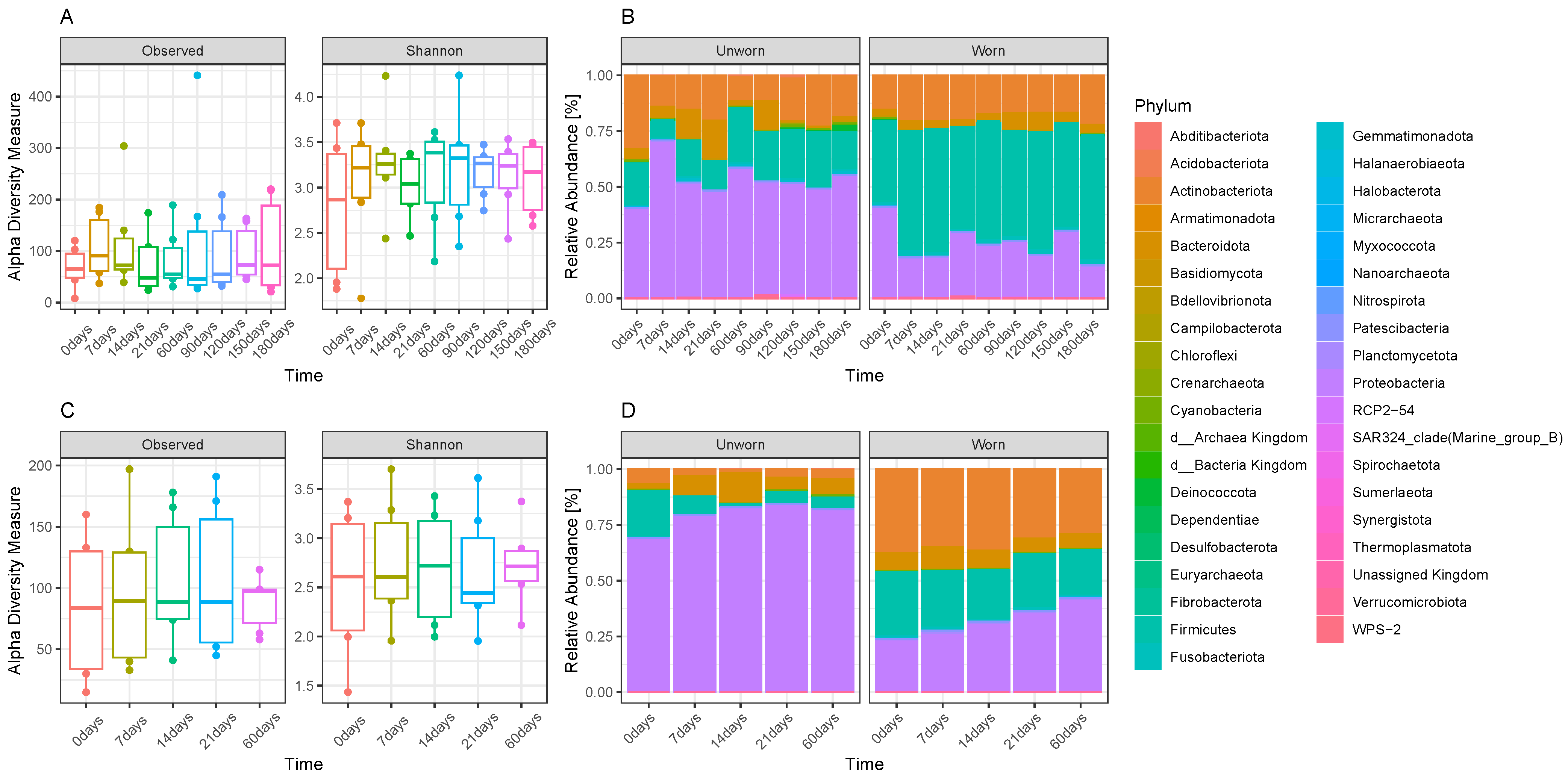
3.1. Microbiome Transfer from Skin to Clothes
3.2. Stability of the Microbiome Transferred on Clothes and Transferability between Adjacent Garments
3.3. Evaluation on Microbial Transferability and Inter-Individual Variations
3.4. Comparison of Efficiency between Three Extraction Kits
3.5. Human STR Profile
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Assenmacher, D.M.; Fields, S.D.; Crupper, S.S. Comparison of Commercial Kits for Recovery and Analysis of Bacterial DNA from Fingerprints. J. Forensic Sci. 2020, 65, 1310–1314. [Google Scholar] [CrossRef]
- Procopio, N.; Lovisolo, F.; Sguazzi, G.; Ghignone, S.; Voyron, S.; Migliario, M.; Renò, F.; Sellitto, F.; D’Angiolella, G.; Tozzo, P.; et al. “Touch microbiome” as a potential tool for forensic investigation: A pilot study. J. Forensic Leg. Med. 2021, 82, 102223. [Google Scholar] [CrossRef]
- The Threat of an Anthrax Attack; CDC: Atlanta, GA, USA, 2020.
- Bock, E. 2001 Anthrax Attacks Revealed Need to Develop Countermeasures Against Biological Threats. NIH Rec. 2002, LXXIV, 2. [Google Scholar]
- Budowle, B.; Schutzer, S.E.; Einseln, A.; Kelley, L.C.; Walsh, A.C.; Smith, J.A.; Marrone, B.L.; Robertson, J.; Campos, J. Building microbial forensics as a response to bioterrorism. Science 2003, 301, 1852–1853. [Google Scholar] [CrossRef] [PubMed]
- Habtom, H.; Pasternak, Z.; Matan, O.; Azulay, C.; Gafny, R.; Jurkevitch, E. Applying microbial biogeography in soil forensics. Forensic Sci. Int. Genet. 2019, 38, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Tozzo, P.; D’angiolella, G.; Brun, P.; Castagliuolo, I.; Gino, S.; Caenazzo, L. Skin Microbiome Analysis for Forensic Human Identification: What Do We Know So Far? Microorganisms 2020, 8, 873. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Lauber, C.L.; Zhou, N.; McDonald, D.; Costello, E.K.; Knight, R. Forensic identification using skin bacterial communities. Proc. Natl. Acad. Sci. USA 2010, 107, 6477–6481. [Google Scholar] [CrossRef] [PubMed]
- Schmedes, S.E.; Woerner, A.E.; Budowle, B. Forensic Human Identification Using Skin Microbiomes. Appl. Environ. Microbiol. 2017, 83, e01672-17. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480. [Google Scholar] [CrossRef] [PubMed]
- Phan, K.; Barash, M.; Spindler, X.; Gunn, P.; Roux, C. Retrieving forensic information about the donor through bacterial profiling. Int. J. Leg. Med. 2020, 134, 21–29. [Google Scholar] [CrossRef]
- Huang, S.; Haiminen, N.; Carrieri, A.P.; Hu, R.; Jiang, L.; Parida, L.; Russell, B.; Allaband, C.; Zarrinpar, A.; Vázquez-Baeza, Y.; et al. Human Skin, Oral, and Gut Microbiomes Predict Chronological Age. mSystems 2020, 5, 10–128. [Google Scholar] [CrossRef]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; Green, E.D.; et al. Topographical and Temporal Diversity of the Human Skin Microbiome. Science 2009, 324, 1190. [Google Scholar] [CrossRef]
- Huse, S.M.; Ye, Y.; Zhou, Y.; Fodor, A.A. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS ONE 2012, 7, e34242. [Google Scholar] [CrossRef]
- Lee, S.Y.; Woo, S.K.; Lee, S.M.; Eom, Y.B. Forensic analysis using microbial community between skin bacteria and fabrics. Toxicol. Environ. Health Sci. 2016, 8, 263–270. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Robeson, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management. PLoS Comput. Biol. 2021, 17, e1009581. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, D.; Dong, Q. microbiomeMarker an R package for micro-biome marker identification and visualiza-tion 2022. Bioinformatics 2022, 38, 4027–4029. [Google Scholar] [CrossRef]
- Whitehead, K.; Eppinger, J.; Srinivasan, V.; Ijaz, M.K.; Nims, R.W.; McKinney, J. Microbiome of Clothing Items Worn for a Single Day in a Non-Healthcare Setting. Microbiol. Res. 2023, 14, 948–958. [Google Scholar] [CrossRef]
- Sterndorff, E.B.; Russel, J.; Jakobsen, J.; Mortensen, M.S.; Gori, K.; Herschend, J.; Burmølle, M. The T-shirt microbiome is distinct between individuals and shaped by washing and fabric type. Environ. Res. 2020, 185, 109449. [Google Scholar] [CrossRef]
- Teufel, L.; Pipal, A.; Schuster, K.C.; Staudinger, T.; Redl, B. Material-dependent growth of human skin bacteria on textiles investigated using challenge tests and DNA genotyping. J. Appl. Microbiol. 2010, 108, 450–461. [Google Scholar] [CrossRef]
- Gattlen, J.; Amberg, C.; Zinn, M.; Mauclaire, L. Biofilms isolated from washing machines from three continents and their tolerance to a standard detergent. Biofouling 2010, 26, 873–882. [Google Scholar] [CrossRef]
- Jacksch, S.; Kaiser, D.; Weis, S.; Weide, M.; Ratering, S.; Schnell, S.; Egert, M. Influence of Sampling Site and other Environmental Factors on the Bacterial Community Composition of Domestic Washing Machines. Microorganisms 2020, 8, 30. [Google Scholar] [CrossRef]
- Saferstein, R. Criminalistics: An Introduction to Forensic Science; Pearson: London, UK, 2018; Volume 3. [Google Scholar]
- Vilhena, L.; Ramalho, A. Friction of human skin against different fabrics for medical use. Lubricants 2016, 4, 6. [Google Scholar] [CrossRef]
- Sanders, D.; Grunden, A.; Dunn, R.R. A review of clothing microbiology: The history of clothing and the role of microbes in textiles: A Review of Clothing Microbiology. Biol. Lett. 2021, 17, 20200700. [Google Scholar] [CrossRef]
- Zhai, H.; Maibach, H.I. Occlusion vs. skin barrier function. Skin Res. Technol. 2002, 8, 1–6. [Google Scholar] [CrossRef]
- Callewaert, C.; Maeseneire, E.D.; Kerckhof, F.M.; Verliefde, A.; de Wiele, T.V.; Boon, N. Microbial odor profile of polyester and cotton clothes after a fitness session. Appl. Environ. Microbiol. 2014, 80, 6611–6619. [Google Scholar] [CrossRef]
- Honisch, M.; Stamminger, R.; Bockmühl, D.P. Impact of wash cycle time, temperature and detergent formulation on the hygiene effectiveness of domestic laundering. J. Appl. Microbiol. 2014, 117, 1787–1797. [Google Scholar] [CrossRef]
- Yan, H.; Ren, Y.; Zhou, B.; Ye, F.; Wu, Z. Microbial profile of T-shirts after a fitness session of Chinese students. Heliyon 2022, 8, e12379. [Google Scholar] [CrossRef]
- Perez, G.I.; Gao, Z.; Jourdain, R.; Ramirez, J.; Gany, F.; Clavaud, C.; Demaude, J.; Breton, L.; Blaser, M.J. Body site is a more determinant factor than human population diversity in the healthy skin microbiome. PLoS ONE 2016, 11, 1–15. [Google Scholar] [CrossRef]
- Oh, J.; Byrd, A.L.; Park, M.; Kong, H.H.; Segre, J.A. Temporal Stability of the Human Skin Microbiome. Cell 2016, 165, 854–866. [Google Scholar] [CrossRef]
- Delanghe, L.; Spacova, I.; Van Malderen, J.; Oerlemans, E.; Claes, I.; Lebeer, S. The role of lactobacilli in inhibiting skin pathogens. Biochem. Soc. Trans. 2021, 49, 617–627. [Google Scholar] [CrossRef]
- Carmona-Cruz, S.; Orozco-Covarrubias, L.; Sáez-de Ocariz, M. The Human Skin Microbiome in Selected Cutaneous Diseases. Front. Cell. Infect. Microbiol. 2022, 12, 834135. [Google Scholar] [CrossRef]
- Rapin, A.; Rehbinder, E.M.; Macowan, M.; Pattaroni, C.; Lødrup Carlsen, K.C.; Harris, N.L.; Jonassen, C.M.; Landrø, L.; Lossius, A.H.; Nordlund, B.; et al. The skin microbiome in the first year of life and its association with atopic dermatitis. Allergy 2023, 78, 1949–1963. [Google Scholar] [CrossRef]
- Kloos, W.E.; Musselwhite, M.S. Distribution and Persistence of Staphylococcus and Micrococcus Species and Other Aerobic Bacteria on Human Skin. Appl. Microbiol. 1975, 30, 381–395. [Google Scholar] [CrossRef]
- Erbasan, F. Brain abscess caused by Micrococcus luteus in a patient with systemic lupus erythematosus: Case-based review. Rheumatol. Int. 2018, 38, 2323–2328. [Google Scholar] [CrossRef]
- Bien, J.; Palagani, V.; Bozko, P. The intestinal microbiota dysbiosis and Clostridium difficile infection: Is there a relationship with inflammatory bowel disease? Ther. Adv. Gastroenterol. 2012, 6, 53–68. [Google Scholar] [CrossRef]
- Figueiredo, G.G.O.; Lopes, V.R.; Romano, T.; Camara, M.C. Clostridium. In Beneficial Microbes in Agro-Ecology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 477–491. [Google Scholar] [CrossRef]
- Bobulsky, G.S.; Al-Nassir, W.N.; Riggs, M.M.; Sethi, A.K.; Donskey, C.J. Clostridium difficile Skin Contamination in Patients with C. difficile-Associated Disease. Clin. Infect. Dis. 2008, 46, 447–450. [Google Scholar] [CrossRef]
- Callewaert, C.; Nevel, S.V.; Kerckhof, F.M.; Granitsiotis, M.S.; Boon, N. Bacterial Exchange in Household Washing Machines. Front. Microbiol. 2015, 6, 1681. [Google Scholar] [CrossRef]
- Callewaert, C.; Kerckhof, F.M.; Granitsiotis, M.S.; van Gele, M.; van de Wiele, T.; Boon, N. Characterization of Staphylococcus and Corynebacterium Clusters in the Human Axillary Region. PLoS ONE 2013, 8, e70538. [Google Scholar] [CrossRef]
- Kubota, H.; Mitani, A.; Niwano, Y.; Takeuchi, K.; Tanaka, A.; Yamaguchi, N.; Kawamura, Y.; Hitomi, J. Moraxella species are primarily responsible for generating malodor in laundry. Appl. Environ. Microbiol. 2012, 78, 3317–3324. [Google Scholar] [CrossRef]
- Kwon, S.; Moon, E.; Kim, T.S.; Hong, S.; Park, H.D. Pyrosequencing Demonstrated Complex Microbial Communities in a Membrane Filtration System for a Drinking Water Treatment Plant. Microbes Environ. 2011, 26, 149–155. [Google Scholar] [CrossRef]
- Yoon, E.J.; Goussard, S.; Touchon, M.; Krizova, L.; Cerqueira, G.; Murphy, C.; Lambert, T.; Grillot-Courvalin, C.; Nemec, A.; Courvalin, P. Origin in acinetobacter guillouiae and dissemination of the aminoglycoside-modifying enzyme Aph(3’)-VI. mBio 2014, 5, e01972-14. [Google Scholar] [CrossRef]
- Nemec, A.; Musílek, M.; Šedo, O.; Baere, T.D.; Maixnerová, M.; Reijden, T.J.; Zdráhal, Z.; Vaneechoutte, M.; Dijkshoorn, L. Acinetobacter bereziniae sp. nov. and Acinetobacter guillouiae sp. nov., to accommodate Acinetobacter genomic species 10 and 11, respectively. Int. J. Syst. Evol. Microbiol. 2010, 60, 896–903. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244. [Google Scholar] [CrossRef]
- Skowron, K.; Bauza-kaszewska, J.; Kraszewska, Z.; Wiktorczyk-kapischke, N.; Grudlewska-buda, K.; Kwiecińska-piróg, J.; Wałecka-zacharska, E.; Radtke, L.; Gospodarek-komkowska, E. Human skin microbiome: Impact of intrinsic and extrinsic factors on skin microbiota. Microorganisms 2021, 9, 543. [Google Scholar] [CrossRef]
- Ying, S.; Zeng, D.N.; Chi, L.; Tan, Y.; Galzote, C.; Cardona, C.; Lax, S.; Gilbert, J.; Quan, Z.X. The Influence of Age and Gender on Skin-Associated Microbial Communities in Urban and Rural Human Populations. PLoS ONE 2015, 10, e0141842. [Google Scholar] [CrossRef]
- Ehlers, C.; Ivens, U.I.; Møller, M.L.; Senderovitz, T.; Serup, J. Females have lower skin surface pH than men. Ski. Res. Technol. 2001, 7, 90–94. [Google Scholar] [CrossRef]
- Levy, G.; Solt, I. The Human Microbiome and Gender Medicine. Gend. Genome 2018, 2, 123–127. [Google Scholar] [CrossRef]
- Steglińska, A.; Jachowicz, A.; Szulc, J.; Adamiak, J.; Otlewska, A.; Pielech-Przybylska, K.; Gutarowska, B. Factors Influencing Microbiological Biodiversity of Human Foot Skin. Int. J. Environ. Res. Public Health 2019, 16, 3503. [Google Scholar] [CrossRef]
- Robert, C.; Cascella, F.; Mellai, M.; Barizzone, N.; Mignone, F.; Massa, N.; Nobile, V.; Bona, E. Influence of Sex on the Microbiota of the Human Face. Microorganisms 2022, 10, 2470. [Google Scholar] [CrossRef]
- Fierer, N.; Hamady, M.; Lauber, C.L.; Knight, R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 17994–17999. [Google Scholar] [CrossRef] [PubMed]
- Haindl, R.; Schick, S.; Kulozik, U. Influence of Cultivation pH on Composition, Diversity, and Metabolic Production in an In Vitro Human Intestinal Microbiota. Fermentation 2021, 7, 156. [Google Scholar] [CrossRef]
- Sguazzi, G.; Mickleburgh, H.L.; Ghignone, S.; Voyron, S.; Renò, F.; Migliario, M.; Sellitto, F.; Lovisolo, F.; Camurani, G.; Ogbanga, N.; et al. Microbial DNA in human nucleic acid extracts: Recoverability of the microbiome in DNA extracts stored frozen long-term and its potential and ethical implications for forensic investigation. Forensic Sci. Int. Genet. 2022, 59, 102686. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Time | Label | From |
|---|---|---|---|
| M03_B_N | Week 0 | Control | Swab |
| M03_A_N | Week 0 | Control | Swab |
| M03_SN | Week 0 | Control | Fabric |
| M3_SN_W0_W | Week 0 | Worn | Fabric |
| M3_SN_W1_W | Week 1 | Worn | Fabric |
| M3_SN_W1_Q | Week 1 | Unworn | Fabric |
| M3_SN_W2_W | Week 2 | Worn | Fabric |
| M3_SN_W2_Q | Week 2 | Unworn | Fabric |
| M3_SN_W3_W | Week 3 | Worn | Fabric |
| M3_SN_W3_Q | Week 3 | Unworn | Fabric |
| M3_SN_M2_W | Month 2 | Worn | Fabric |
| M3_SN_M2_Q | Month 2 | Unworn | Fabric |
| M3_SN_M3_W | Month 3 | Worn | Fabric |
| M3_SN_M3_Q | Month 3 | Unworn | Fabric |
| M3_SN_M4_W | Month 4 | Worn | Fabric |
| M3_SN_M4_Q | Month 4 | Unworn | Fabric |
| M3_SN_M5_W | Month 5 | Worn | Fabric |
| M3_SN_M5_Q | Month 5 | Unworn | Fabric |
| M3_SN_M6_W | Month 6 | Worn | Fabric |
| M3_SN_M6_Q | Month 6 | Unworn | Fabric |
| Sample Name | Time | Label | From |
|---|---|---|---|
| F01_SN | 0 | N/A | Swab |
| F1_SN_W0_W | 0 | Worn | Fabric |
| F1_SN_W1_W | Week 1 | Worn | Fabric |
| F1_SN_W1_Q | Week 1 | Unworn | Fabric |
| F1_SN_W2_W | Week 2 | Worn | Fabric |
| F1_SN_W2_Q | Week 2 | Unworn | Fabric |
| F1_SN_W3_W | Week 3 | Worn | Fabric |
| F1_SN_W3_Q | Week 3 | Unworn | Fabric |
| F1_SN_M2_W | Month 2 | Worn | Fabric |
| F1_SN_M2_Q | Month 2 | Unworn | Fabric |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Procopio, N.; Sguazzi, G.; Eriksson, E.V.; Ogbanga, N.; McKell, F.C.; Newton, E.P.; Magni, P.A.; Bonicelli, A.; Gino, S. Transferability of Human and Environmental Microbiome on Clothes as a Tool for Forensic Investigations. Genes 2024, 15, 375. https://0-doi-org.brum.beds.ac.uk/10.3390/genes15030375
Procopio N, Sguazzi G, Eriksson EV, Ogbanga N, McKell FC, Newton EP, Magni PA, Bonicelli A, Gino S. Transferability of Human and Environmental Microbiome on Clothes as a Tool for Forensic Investigations. Genes. 2024; 15(3):375. https://0-doi-org.brum.beds.ac.uk/10.3390/genes15030375
Chicago/Turabian StyleProcopio, Noemi, Giulia Sguazzi, Emma V. Eriksson, Nengi Ogbanga, Frazer C. McKell, Eleanor P. Newton, Paola A. Magni, Andrea Bonicelli, and Sarah Gino. 2024. "Transferability of Human and Environmental Microbiome on Clothes as a Tool for Forensic Investigations" Genes 15, no. 3: 375. https://0-doi-org.brum.beds.ac.uk/10.3390/genes15030375