Genes Involved in Type 1 Diabetes: An Update

1
Center for Applied Genomics, Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA
2
Department of Pediatrics, The Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA
*
Author to whom correspondence should be addressed.
†
Those authors contributed equally to this work.
Genes 2013, 4(3), 499-521; https://0-doi-org.brum.beds.ac.uk/10.3390/genes4030499
Submission received: 31 July 2013
/
Revised: 26 August 2013
/
Accepted: 5 September 2013
/
Published: 16 September 2013
(This article belongs to the Special Issue Genetics of Diabetes)
Abstract
:Type 1 Diabetes (T1D) is a chronic multifactorial disease with a strong genetic component, which, through interactions with specific environmental factors, triggers disease onset. T1D typically manifests in early to mid childhood through the autoimmune destruction of pancreatic β cells resulting in a lack of insulin production. Historically, prior to genome-wide association studies (GWAS), six loci in the genome were fully established to be associated with T1D. With the advent of high-throughput single nucleotide polymorphism (SNP) genotyping array technologies, enabling investigators to perform high-density GWAS, many additional T1D susceptibility genes have been discovered. Indeed, recent meta-analyses of multiple datasets from independent investigators have brought the tally of well-validated T1D disease genes to almost 60. In this mini-review, we address recent advances in the genetics of T1D and provide an update on the latest susceptibility loci added to the list of genes involved in the pathogenesis of T1D.
1. Introduction
Type 1 Diabetes (T1D) is a chronic multifactorial disease with a strong genetic component. It arises as a consequence of autoimmune destruction of pancreatic β-cells, resulting in insufficient insulin production. The prevalence of diabetes is increasing worldwide [1]. According to the International Diabetes Federation (IDF), the worldwide prevalence of diabetes mellitus in 2011 was 366 million, and is predicted to reach 552 million by 2030 [2]. T1D represents approximately 10% of these patients and is most prevalent in populations of European ancestry [3,4]. There is about 3% increase in the incidence of T1D annually, lending further support to complex gene environment interactions in the pathogenesis of T1D [3]. While cumulative evidence supports a strong genetic component associated with T1D, epidemiological data show wide differences in geographic prevalence with populations of European ancestry having the highest presentation rate. T1D also has high concordance among monozygotic twins (33% to 42%) [5], and the disease runs strongly in families with siblings risk being approximately 10 times greater than in the general population [6]; this is in clear contrast to the “less genetic” type 2 diabetes, where the sibling risk ratio is relatively modest at 3.5 [7].
T1D develops at all ages and occurs through the autoimmune destruction of pancreatic β-cells with resulting lack of insulin production. The immune system participates in β-cell destruction through several of its components including CD4+ and CD8+ T cells, natural killer (NK) cells, B lymphocytes, macrophages, dendritic cells (DC), and antigen-presenting cells (APCs). Studies in human and animal models have shown that both innate and adaptive immune responses participate in disease pathogenesis, possibly reflecting the multifactorial nature of this autoimmune disorder.
In this review, we will provide an updated summary of genome-wide association studies (GWAS) including recent meta-analyses and discuss the latest associated regions added to the growing repertoire of gene networks predisposing to T1D.
2. Genetic Component in Type 1 Diabetes
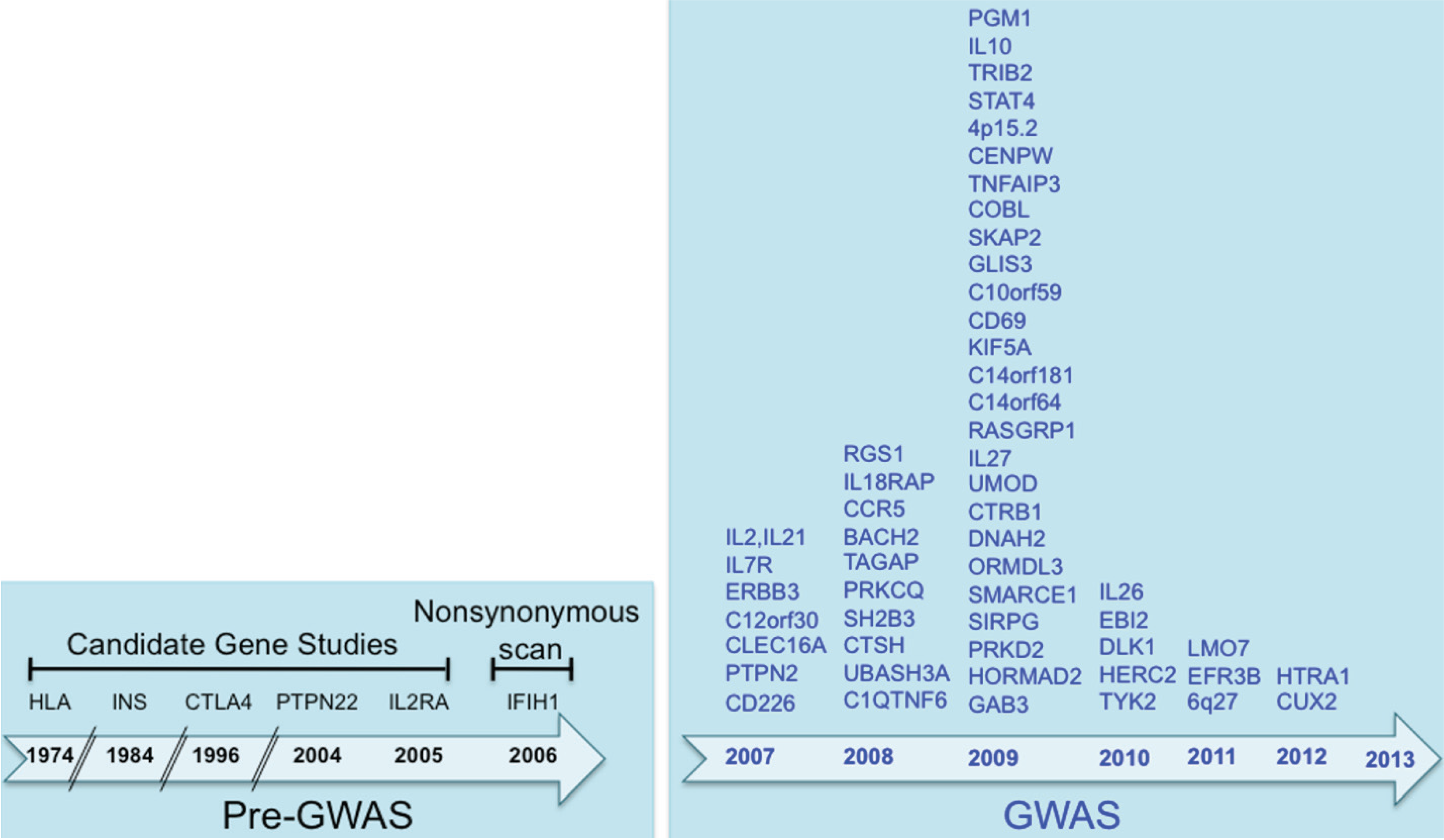
The risk of developing T1D is determined by a complex interaction between multiple genes and environmental factors. The discovery of T1D susceptibility genes started as early as 1974 with five genes discovered, using family and candidate gene approaches. The advent of GWAS led to flurry of novel genes associated with T1D reaching the excess of 40 by 2006, and 60 by 2012 as depicted in Figure 1. It is clear now that many of these genes are novel and were not on any investigators’ radars when they were designing candidate gene studies in the past.
2.1. Before Genome-Wide Association Studies
Historically, prior to GWAS, only six loci were fully established to be associated with T1D. The human leukocyte antigen (HLA) region on chromosome 6p21 was the first known candidate to be strongly associated with T1D in the 1970s [8,9,10]. This cluster of homologous cell-surface proteins is divided into class I (A, B, C) and class II (DP, DQ, RD). The HLA genes encode highly polymorphic proteins, which are essential in self- versus non self-immune recognition. The class I molecules are ubiquitously expressed and present intracellular antigen to CD8+ T cells. Class II molecules are expressed mainly on professional APCs: DCs, macrophages, B lymphocytes and thymus epithelium. Class II molecules are composed of A and B chains, and present antigens to CD4+ T cells, which promote inflammation by secreting cytokines upon recognition of their specific targets. Approximately half of the genetic risk for T1D is conferred by the genomic region harboring the HLA class II genes (primarily HLA-DRB1, -DQA1 and -DQB1 genes). In 1984, insulin (INS) gene encoded on chromosome 11p15 was identified as the second loci linked with T1D [11]. In 1996, the cytotoxic T-lymphocyte-associated protein 4 (CTLA4) gene encoded on chromosome 2q33 was recognized as the third loci [12]. Another case-control study in 2004 reported a protein tyrosine phosphatase, non-receptor type 22 (PTPN22), gene encoded on chromosome 1p13 to be associated with susceptibility to T1D [13]. Vella et al., 2005 reported interleukin 2 receptor alpha (IL2RA) gene as the fifth T1D loci on chromosome 10p15 [14]. In 2006, Smyth et al. identified the interferon-induced with helicase C domain 1 (IFIH1) gene on chromosome 2q24.3 as the sixth candidate to be strongly associated with T1D through genotyping of only 6,500 non-synonymous SNPs genome wide [15]. This study was a precursor to the first GWAS approach.
Figure 1.
The Type 1 Diabetes loci described to date: a timeline. The susceptibility loci are presented by the year they were first implicated in T1D.
Figure 1.
The Type 1 Diabetes loci described to date: a timeline. The susceptibility loci are presented by the year they were first implicated in T1D.

2.2. GWAS of T1D
The advent of GWAS in the mid-2000s has changed the situation dramatically, increasing the pace and efficiency of discovery for the T1D associated loci by a factor of ten. The critical platform for this work was laid by the HapMap project [16,17]. The GWAS approach was made possible by the development of high-density genotyping arrays. It has been shown that the genome is laid out in discrete linkage disequilibrium (LD) blocks, with limited haplotype diversity within each of these blocks. Therefore, a minimal set of single nucleotide polymorphisms (SNPs) can detect almost all common haplotypes present, thus improving genotyping accuracy and reducing the cost. As a result, these technologies enable us and others to perform GWA studies in search of the remaining T1D loci, the outcomes of which are outlined below.
The first full-scale GWAS for T1D were published simultaneously by our group [18] and by the Wellcome Trust Case-Control Consortium (WTCCC) [19]. We examined a large pediatric cohort of European descent using the Illumina HumanHap 550 BeadChip platform. The design involved 561 cases, 1,143 controls, and 467 triads in the discovery stage, followed by a replication effort in 939 nuclear families. In addition to finding the “usual” suspects, including an impressive 392 SNPs capturing the very strong association across the major histocompatibility complex (MHC), we identified significant association with variation at the KIAA0350 gene, which we replicated in an additional cohort. The WTCCC study investigated seven common complex diseases including T1D by genotyping 2,000 cases and 3,000 controls with ~500,000 SNPs using the Affymetrix GeneChip, and reported a number of novel T1D loci, including the KIAA0350 genomic region [19]. Todd et al., 2007 replication study confirmed these findings in 4,000 cases, 5,000 controls, and 3,000 T1D families [20]. In a separate replication effort we elected to fast-track 24 SNPs at 23 distinct loci and established association to the 12q13 region with a combined p-value of 9.13 × 10−10 [21], previously reported by the WTCCC [19] and Todd et al. [20]. The 250-kb LD block on 12q13 region harbors several genes, including ERBB3, RAB5B, SUOX, RPS26, and CDK2. Additional laboratory studies are needed to identify both the corresponding genes and the causative variants for this locus. Later the same year, Concannon et al. reported an association between SNP at the UBASH3A locus on 21q22.3 and T1D by using SNP genotyping data from a linkage study of affected sib pairs in nearly 2,500 multiplex families [22]. UBASH3A (previously known as T-cell ubiquitin ligand [TULA] and suppressor of T-cell signaling 2 [Sts-2]) is expressed predominantly in T cells. It interacts with c-CBL through its SH3 domain and binds to ubiquitin and ubiquitylated proteins via its UBA domain [23]. UBASH3A protein product similar to PTPN22 interacts with c-CBL, but UBASH3A directly downregulates some of the same protein tyrosine kinases by dephosphorylation [24]. Follow-up of 1715 SNPs from the WTCCC genome-wide association study in T1D families confirmed UBASH3A as a susceptibility gene [25]. A recent study reported UBASH3A to be an independent predictor of persistent islet autoimmunity and T1D in children, including those free of family history of T1D but carrying the HLA-DR3/4, DQB1*0302 genotype. UBASH3A may prove useful in T1D risk prediction and pre-screening of the general population children for clinical trials [26].
2.3. Meta-Analyses of T1D GWAS Datasets
In order to get the most from GWAS and to increase the statistical power researchers carried out meta-analyses using datasets from different investigative groups. First meta-analysis was performed by combining the T1D datasets from the Wellcome Trust Case Control Consortium [19] and the Genetics of Kidneys in Diabetes (GoKind) study [27,28], plus control data derived from the National Institute of Mental Health. This study confirmed associations for PTPN22, CTLA4, MHC, IL2RA, 12q13, 12q24, CLEC16A, and PTPN2 [29]. The SNPs with lowest nominal p-values were taken forward for further genotyping in an additional British cohort of approximately 6,000 cases, 7,000 controls, and 2,800 families. As a result, the IL2-IL21 association strengthened further and they found strong evidence for four additional loci: a 6q15 region harboring BACH2; a 10p15 region harboring the protein kinase C, theta gene (PRKCQ); a 15q24 region harboring nine genes including cathepsin H (CTSH) and 22q13 harboring the C1q and tumor necrosis factor-related protein 6 (C1QTNF6) and somatostatin receptor 3 (SSTR3) genes [29]. Study of polymorphisms in 4q27, 10p15, and 22q13 regions in autoantibodies stratified type 1 diabetes patients further confirmed IL2 association in pediatric patients and individuals with late onset of T1D [30]. Additional studies are required to elucidate the culprit genes and their mechanism at the 15q24 and 22q13 loci.
Meta-analysis by T1DGC [31] provided evidence of T1D association for 41 distinct genomic locations (p < 10−6) by using datasets from WTCCC [19], the GoKind study [28], and controls and family sets from Type 1 Diabetes Genetics Consortium (T1DGC). The study confirmed a number of previously reported associations [32,33,34] and discovered 22 novel, of which 18 regions were replicated (p < 5 × 10−8) and four additional regions provided nominal evidence of replication (p < 0.05). The meta-analysis observed association to 1q32.1 (which harbors the immunoregulatory interleukin genes IL10, IL19 and IL20), 9p24.2 contains only Glis family zinc finger protein 3 (GLIS3), which was first suggested by us in [35], 12p13.31 which harbors a number of immunoregulatory genes including CD69 and 16p11.2 harboring IL27. Our in silico replication efforts [36] further confirmed the associations previously reported by the T1DGC [31]. The entire Barrett et al. study was later replicated in 2012 by T1DGC to exclude the possibility that any of the 18 loci were false-positives due to population stratification. Seventeen of the 18 susceptibility loci reached nominal levels of significance (p < 0.05) in the expanded family collection, with 14q24.1 just falling short (p = 0.055) [37]. All susceptibility loci had consistent direction of effects with the original study.
To identify additional genetic loci for T1D susceptibility, we examined associations in the largest meta-analysis to date between the disease and ~2.54 million SNPs in a combined cohort of 9,934 T1D cases and 16,956 controls [38]. Targeted follow-up of 53 SNPs in 1,120 affected trios uncovered three new loci associated with T1D that reached genome wide significance. The most significantly associated SNP (rs539514, p = 5.66 × 10−11) resided in an intronic region of the LMO7 (LIM domain only 7) gene on 13q22. The second most significantly associated SNP (rs478222, p = 3.50 × 10−9) resided in an intronic region of the EFR3B (protein EFR3 homolog B) gene on 2p23; however the region of linkage disequilibrium is approximately 800 kb and harbors additional multiple genes, including NCOA1, C2orf79, CENPO, ADCY3, DNAJC27, POMC, and DNMT3A. The third most significantly associated SNP (rs924043, p = 8.06 × 10−9) was in an intergenic region on 6q27, where the region of association is approximately 900 kb and harbors additional genes including WDR27, C6orf120, PHF10, TCTE3, C6orf208, LOC154449, DLL1, FAM120B, PSMB1, TBP, and PCD2. These latest associations add to the growing repertoire of gene networks predisposing to T1D. Table 1 summarizes all T1D associated loci reported to date.

{kind=link}
{kind=link}
{kind=link}
| Reference | Study Type | Main Findings | Sample Size | Replication Sample Size | Ethnic Group | |
|---|---|---|---|---|---|---|
| Hakonarson et al., 2007 [18] | GWAS | HLA-DRB1, HLA-DQA2, CLEC16A, INS, PTPN22 | 467 trios, 561 cases, 1,143 controls | 2,350 individuals in 549 families; 390 trios | European ancestry | |
| WTCCC 2007 [19] | GWAS | HLA-DRB1, INS, CTLA4, PTPN22, IL2RA, IFIH1, PPARG, KCNJ11, TCF7L2 | 1,963 cases, 2,938 controls | see Todd et al., 2007 | European, British | |
| Todd et al., 2007 [20] | GWAS | PHTF1-PTPN22, ERBB3, CLEC16A, C12orf30 | see WTCCC 2007 | 2,997 trios, 4,000 cases, 5,000 controls | European, British | |
| Hakonarson et al., 2008 [21] | GWAS | SUOX-IKZF4 | 467 trios, 561 cases, 1,143 controls | 549 families, 364 trios | European ancestry | |
| Concannon et al., 2008 [22] | GWAS | INS, IFIH1, CLEC16A, UBASH3A | 2,496 families | 2,214 trios, 7,721 cases, 9,679 controls | European ancestry | |
| Cooper et al., 2008 [29] | GWAS meta-analysis | PTPN22, CTLA4, HLA, IL2RA, ERRB3, C12orf30, CLEC16A, PTPN2 | 3,561 cases, 4,646 controls | 6,225 cases, 6,946 controls, 3,064 trios | European ancestry | |
| Grant et al., 2009 [35] | GWAS | EDG7, BACH2, GLIS3, UBASH3A, RASGRP1 | 563 cases, 1,146 controls, 483 case-parents trios | 636 families, 3,303 cases, 4,673 controls | European ancestry | |
| Awata et al., 2009 [39] | TaqMan genotyping | ERBB3, CLEC16A | 735 cases, 621 controls | − | Japanese | |
| Zoledziewska et al., 2009 [440] | TaqMan genotyping | CLEC16A | 1037 cases, 1706 controls | − | European, Sardinian | |
| Fung et al., 2009 [33] | TaqMan genotyping | STAT4, STAT3, ERAP1, TNFAIP3, KIF5A/PIP4K2C | 8010 cases, 9733 controls | − | European, British | |
| Wu et al., 2009 [41] | TaqMan genotyping | CLEC16A | 205 cases, 422 controls | − | Han Chinese | |
| Barrett et al., 2009 [31] | GWAS meta-analysis | MHC, PTPN22, INS, C10orf59, SH2B3, ERBB3, CLEC16A, CTLA4, PTPN2, IL2RA, IL27, C6orf173, IL2, ORMDL3, GLIS3, CD69, IL10, IFIH1, UBASH3A, COBL, BACH2, CTSH, PRKCQ, C1QTNF6, PGM1 | 7,514 cases, 9,045 controls | 4,267 cases, 4,670 controls, 4,342 trios | European | |
| Wallace et al., 2010 [42] | GWAS meta-analysis | DLK1, TYK2 | 7,514 cases, 9,045 controls | 4,840 cases, 2,670 controls, 4,152 trios | European ancestry | |
| Wang et al., 2010 [43] | GWAS | PTPN22, IL10, IFIH1, KIAA0746, BACH2, C6orf173, TAGAP, GLIS3, L2R, INS, ERBB3, C14orf181, IL27, PRKD2, HERC2, CLEC16A, IFNG, IL26 | 989 cases, 6,197 controls | − | European ancestry | |
| Reddy et al., 2011 [44] | TaqMan genotyping | PTPN22, INS, IFIH1, SH2B3, ERBB3, CTLA4, C14orf181, CTSH, CLEC16A, CD69, ITPR3, CENPW, SKAP2, PRKCQ, RNLS, IL27, SIRPG, CTRB2 | 1,434 cases, 1,864 controls | − | European ancestry, southeast USA | |
| Bradfield et al., 2011 [38] | GWAS meta-analysis | LMO7, EFR3B, 6q27, TNFRSF11B, LOC100128081, FOSL2 | 9,934 cases, 16,956 controls | 1,120 trios | European ancestry | |
| Asad et al., 2012 [45] | Genotyping andsequencing | HTR1A, RFN180 | 424 families, 3,078 cases, 1,363 controls | − | European, Scandinavians | |
| Huang et al., 2012 [46] | Genomes-based imputation | CUX2, IL2RA | 16,179 individuals | − | European ancestry | |
2.4. Immune Components in T1D
The immune system is well organized and well regulated with a basic function of protecting the host against pathogens. This places the immune system in a vital position between healthy and diseased states of the host. Its protective task is regulated by a complex regulatory mechanism involving a diverse army of cells and molecules of humoral and cellular factors working in concert to protect the body against invaders. Our immune system has two components: innate and adaptive. Innate immunity is comprised of physical, chemical, and microbiological barriers to the entry of antigen, and the elements of immune system (DC, macrophages, mast cells, NK cells, neutrophils, monocytes, complements, cytokines, and acute phase proteins), which provide immediate host defense. Adaptive immunity is the hallmark of the immune system of higher animals with T and B cells as the key cellular players that provide more specific life-long immunity [47].
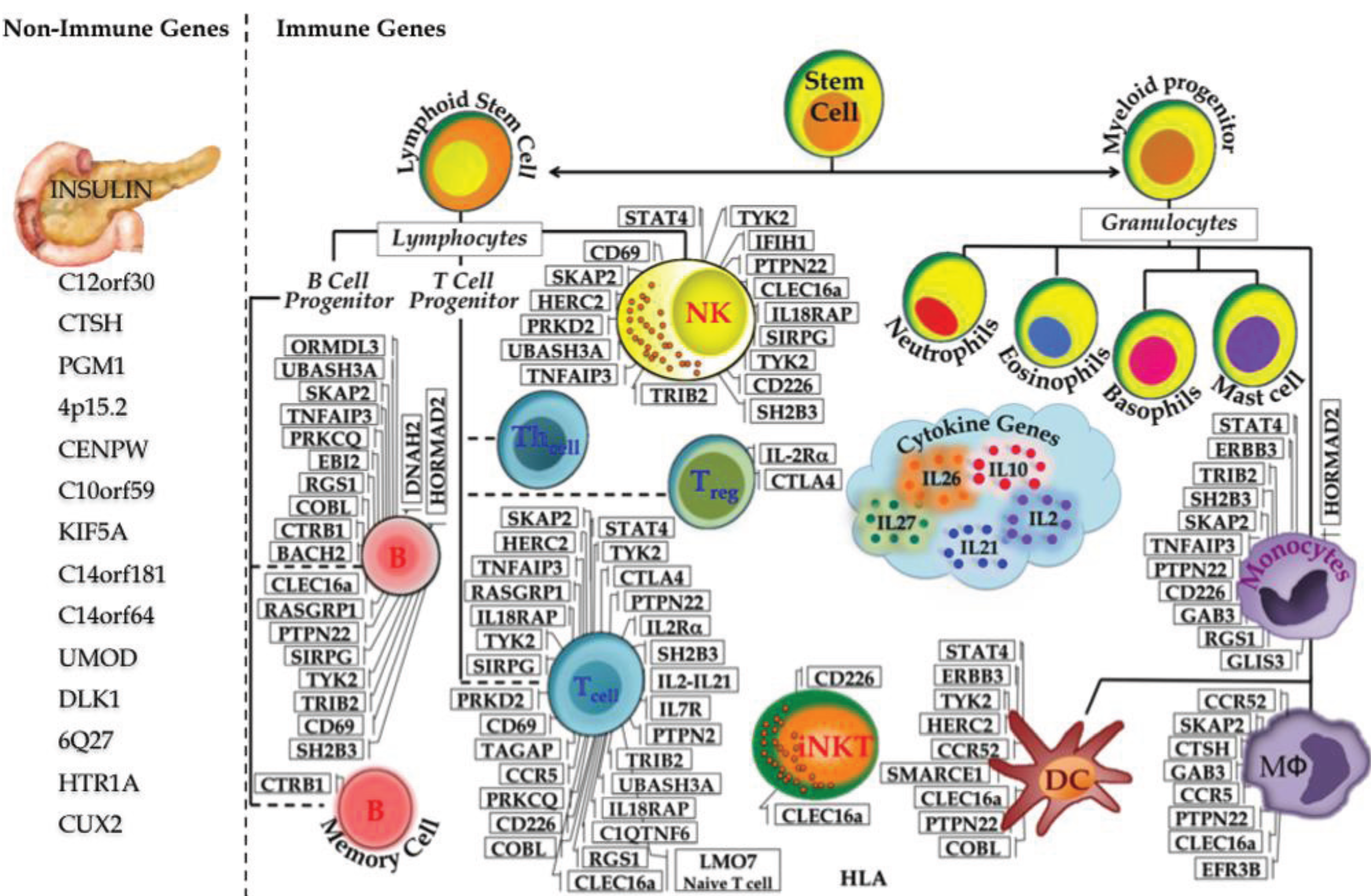
In T1D this system breaks down: insulin-producing β-cells are subjected to specific attack by the host immune system. To better understand the etiology of T1D for prevention and cure, a plethora of research has been done to link the systematic destruction of β-cells and the role of the immune system, however the exact mechanism of T1D pathogenesis is not completely elucidated. Linkage studies in the 1970s revealed MHC as the first key contributor to T1D susceptibility [8,9,10]. Further linkage analysis and candidate gene association studies uncovered additional T1D loci. Starting in 2007, GWAS has increased the number of loci associated with T1D to almost 60 [38]. As T1D is an immune-mediated disorder the majority of candidate genes exert their functions in immune cells. In Figure 2, we have made an attempt to classify all 59 T1D susceptibility loci/genes in keeping with their predominant function of either non-immune (14) vs. immune (45). However, recent studies indicate that many T1D candidate genes are also expressed in human islets suggesting that functions are not restricted to immune cells, but also play roles in the islets and β cells [48]. The functional aspects of some of the most interesting genes or biological pathways are discussed below.
Figure 2.
Immune and Non-immune T1D genes. The discovery of T1D susceptibility genes started as early as 1974, with six T1D genes identified by 2006. The advent of GWAS led to flurry of novel genes associated with T1D reaching the excess of 40 by 2009 and almost 60 by 2012.
Figure 2.
Immune and Non-immune T1D genes. The discovery of T1D susceptibility genes started as early as 1974, with six T1D genes identified by 2006. The advent of GWAS led to flurry of novel genes associated with T1D reaching the excess of 40 by 2009 and almost 60 by 2012.

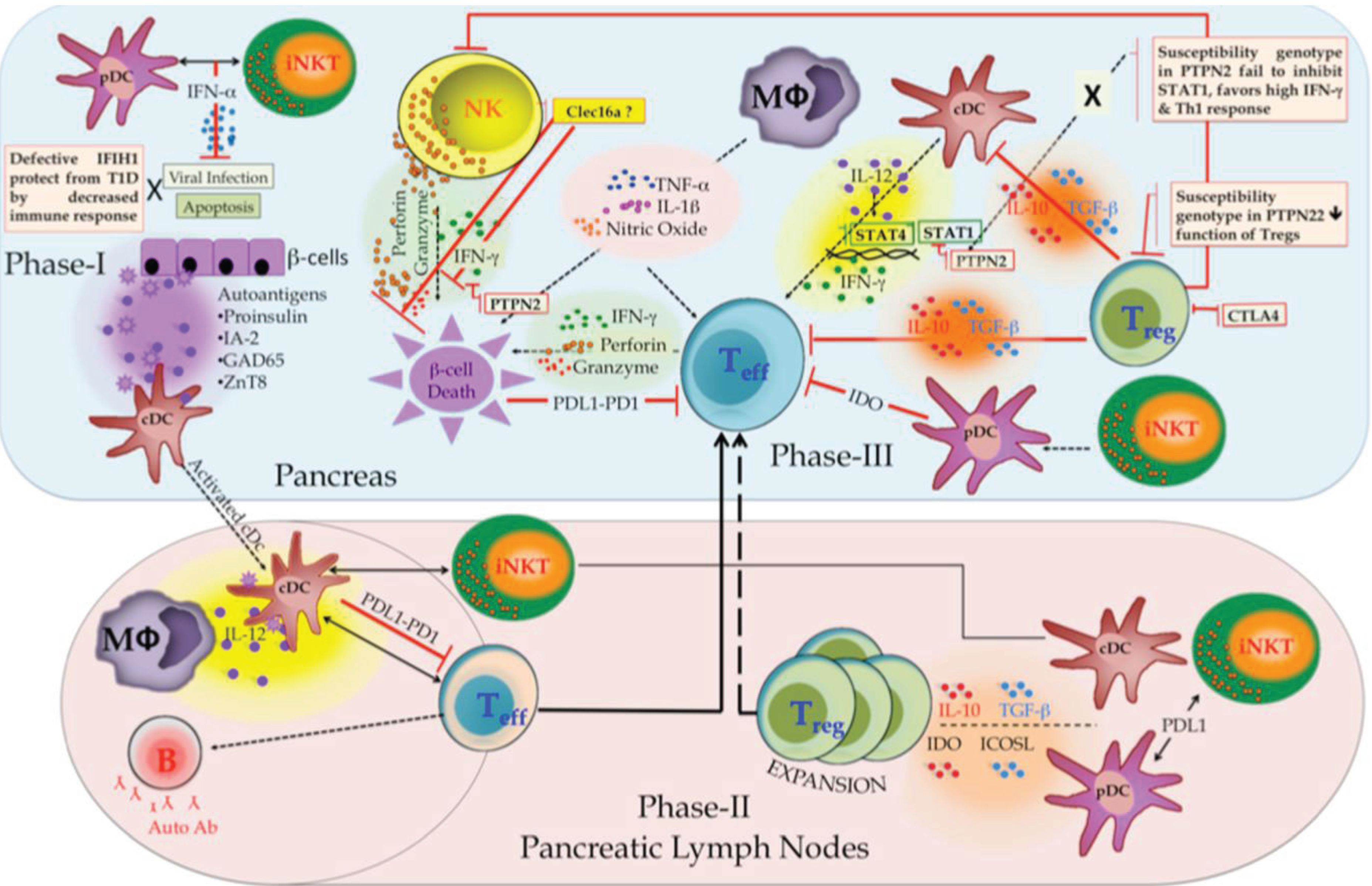
The complex crosstalk between innate and adaptive immune cells is broadly categorized in three phases, which results in the development or the prevention of T1D and is illustrated in Figure 3 as a hypothetical model.
Phase I (the initiation phase of T1D) involves β-cell death and APC activation. It takes place in the pancreas where conventional dendritic cells (cDCs) capture and process β-cell antigens. Natural cell death (apoptosis) or viral infection can lead to β-cell death. Antiviral responses are mediated by invariant natural killer T (iNKT) cells; crossplay between iNKT, and plasmacytoid DCs (pDCs) controls viral replication thus prevents subsequent inflammation, tissue damage, and downregulation of T1D pathogenesis [49].
Phase II (the expansion phase) involves expansion of self-antigens and specific T cells. Migration of activated cDCs to the draining lymph node primes pathogenic islet antigen-specific T cells. This activation is promoted by macrophages through IL12 secretion. B cells present β-cell antigen to diabetogenic T cells and secrete autoantibodies in response. The activation of islet antigen-specific T cells can be inhibited by cDCs through engagement of programmed cell death ligand 1 (PDL1). iNKT cells can further promote the recruitment of tolerogenic cDCs and pDCs. These DCs promote expansion of regulatory T (TReg) cells through the production of indoleamine 2,3-dioxygenase (IDO), IL10, transforming growth factor-β (TGFβ) and inducible T cell co-stimulator ligand (ICOSL) [50].
Figure 3.
Pathogenesis model of T1D involves complex interactions between innate and adaptive immune cell types.
Figure 3.
Pathogenesis model of T1D involves complex interactions between innate and adaptive immune cell types.

Phase III (immune cell crosstalk) occurs in the pancreas where β-cell can be killed by diabetogenic T cells (adaptive component) and NK cells (innate component) through the release of interferon-γ (IFNγ), granzymes, and perforin, as well as by macrophages through the production of tumor necrosis factor (TNF), IL-1β and nitric oxide (NO). IL12 produced by cDCs sustains the effector functions of activated diabetogenic T cells and NK cells. TReg cells that inhibit diabetogenic T cells and innate immune cells through IL10 and TGFβ can prevent β-cell damage. Tolerogenic pDCs stimulated by iNKT cells could also control diabetogenic T cells through IDO production. Lastly, β-cells can inhibit diabetogenic T cells by expressing PDL1 and escape the cell death [50,51].
There is increasing evidence that innate cells play critical roles in T1D onset. In our 2007 GWAS we identified CLEC16A as a novel T1D susceptibility gene [18]. CLEC16A is almost exclusively expressed in immune cells. As CLEC16A SNPs were associated with T1D protection and some of the highest expression of CLEC16A was identified in NK cells, we hypothesize that CLEC16A may function in NK cells to restrain secretory functions including cytokine release and cytotoxicity after activation (Figure 3).
2.5. Insights from T1D Specific Loci
Four decades of intensive studies have discovered nearly 60 T1D susceptibility loci; however the exact mechanisms by which associated loci confer T1D susceptibility remain elusive and require in depth characterization. Several novel T1D susceptibility genes are discussed below.
2.5.1. CLEC16A (16p13)
Our 2007 GWAS in a large pediatric cohort of European descent identified CLEC16A as a novel T1D susceptibility gene within a 233-kb linkage disequilibrium block on chromosome 16p13. Three common non-coding variants of the CLEC16A gene (rs2903692, rs725613, and rs17673553) reached genome-wide significance for association with T1D [18]. Importantly, the allele of CLEC16A linked to protection from T1D was also associated with higher levels of CLEC16A expression in NK cells [18]. The C-type lectin domain family 16, member A (CLEC16A) gene encodes protein with C-type lectin domain structure, which makes it potentially related to the immune response [52]. It is established that C-type lectins function both as adhesion and pathogen recognition receptors (PPRs) [53]. In addition, CLEC16A is almost exclusively expressed in immune cells including DCs, B lymphocytes, and NK cells.
The 2007 WTCCC study independently discovered CLEC16A (formally known as KIAA0350) as a T1D susceptibility locus associated with the non-coding variant rs12708716. This finding was confirmed immediately for T1D in populations of European descent [20,29]. To date, several SNPs (rs2903692, rs17673553, rs725613, rs12708716, rs12921922, rs12931878) within the CLEC16A gene have been reported to be associated with T1D in several populations: Sardinian [40], Spanish [54], south-east USA [44], Chinese [41,55], and Japanese [56]. Recently CLEC16A was also associated with adult-onset of autoimmune diabetes [57].
Several GWAS in different autoimmune diseases such as multiple sclerosis [40,58,59], primary adrenal insufficiency [60], systemic lupus erythematosus [61,62], Celiac disease [63], Crohn’s disease [64], selective immunoglobulin A deficiency [65], alopecia areata [65], juvenile idiopathic arthritis [66], rheumatoid arthritis [54,66], and primary biliary cirrhosis [67,68] also demonstrated association of the 16p13 loci with disease risk, implying that the 16p13 region contains a key regulator of the self-reactive immune response.
Recently, Davison et al. reported intron 19 of the CLEC16A gene behaves as a regulatory sequence, which affects the expression of a neighboring gene dexamethasone-induced (DEXI) [69]. While it is clear that intron 19 of CLEC16A is highly enriched for transcription-factor-binding events, more functional studies are needed to advance from GWAS to candidate causal genes and their biological functions.
Little is yet proven about CLEC16A functions. Kim et al., 2010 characterized an endosomal membrane protein “ema” to be required for endosomal trafficking and promotes endosomal maturation in fruit flies [70]. Expression of human orthologue of ema “CLEC16A” rescued the Drosophila mutant demonstrating conserved function of the protein. A more recent study by the same group also reported its requirement for the growth of autophagosomes and proposed that the Golgi is a membrane source for autophagosomal growth, and that ema facilitates this process [71]. Expression of CLEC16A rescued the autophagosome size defect in the ema mutant, suggesting that regulation of autophagosome morphogenesis may be one of the fundamental functions of CLEC16A. Another study elucidated the dynamic expression changes and localization of CLEC16A in lipopolysaccharide (LPS) induced neuroinflammatory processes in adult rats. CLEC16A expression was strongly induced in active astrocytes in inflamed cerebral cortex. In vitro studies indicated that the up-regulation of CLEC16A may be involved in astrocyte activation following LPS challenge [72].
CLEC16A is well-established T1D susceptibility gene, which probably contributes to the disease by modulating immunity and thus the encoded protein, is of high interest for further functional studies.
2.5.2. Latest Novel T1D Susceptibility Loci (2011–2013)
In our latest effort to identify additional genetic loci for T1D, we examined associations in the largest meta-analysis to date between T1D and ~2.54 million SNPs in a combined cohort of 9,934 cases and 16,956 controls. Targeted follow-up of 53 SNPs in 1,120 affected trios uncovered three novel loci associated with T1D that reached genome-wide significance [38].
2.5.2.1. Region 13q22
The most significantly associated SNP (rs539514, p = 5.66 × 10−11) resides in an intronic region of the LMO7 (LIM domain only 7) gene on 13q22 [38]. LMO7 is a multi-domain mammalian protein with a calponin homology (CH) domain, a discs-large homologous regions (DHR) domain, and a LIM domain. Proteins of this family are involved in protein-protein interactions, regulation of cell adhesion and signaling [73,74]. The expression of LMO7 is cell type specific and is essential for the development of muscle and heart tissues [75,76,77]. Mice with homozygous deletions of LMO7 display retinal, muscular, and growth retardation [78]. LMO7 is known to be upregulated in multiple cancers, especially at the metastatic stage [79]. In cultured rat ascites hepatoma cells, the upregulation of LMO7 correlates with the ability of transforming growth factor β (TGFβ) to enhance the invasiveness of these cells [80]. Recent GWAS meta-analysis from our group identified LMO7 association with T1D [38]. Although the function of LMO7 does not clearly relate to the etiology of T1D, LMO7 is expressed in pancreatic islets and thus is a plausible biological candidate at this locus [81].
2.5.2.2. Region 2q23
The second most significantly associated SNP (rs478222, p = 3.50 × 10−9) resides in an intronic region of the EFR3B (protein EFR3 homolog B) gene on 2p23; however, the region of linkage disequilibrium is approximately 800 kb and harbors additional multiple genes, including NCOA1, C2orf79, CENPO, ADCY3, DNAJC27, POMC, and DNMT3A. Protein EFR3B is an 817 amino acid and exists as three alternatively spliced isoforms. The gene encoding EFR3B maps to human chromosome 2p23.3. A number of genetic diseases have been linked to genes on chromosome 2 including Harlequin icthyosis [82], lipid metabolic disorder sitosterolemia [83], and Alstrom syndrome [84]. Our recent study showed novel association of 2q23 locus with T1D risk [38]. Though the 2q23 region harbors additional multiple genes, including NCOA1, C2orf79, CENPO, ADCY3, DNAJC27, POMC, and DNMT3A, location of SNP rs478222 in the intronic region of EFR3B makes it a good candidate gene.
Nuclear receptor coactivator 1 protein (NCOA1) is a member of the p160/steroid receptor co-activator (SRC) family. The product of this gene binds to a variety of nuclear hormone receptors in a ligand-dependent manner suggesting that NCOA1 may play a role as a bridging molecule between nuclear hormone receptors and general transcription factors [85,86].
C2orf79 is peptidyl-tRNA hydrolase domain containing 1 (PTRHD1) predicted protein with unknown function.
Centromere protein O gene (CENPO) encodes a component of the interphase centromere complex. The protein is localized to the centromere throughout cell division and is required for bipolar spindle assembly, chromosome segregation and checkpoint signaling during mitosis [87].
Adenylate cyclase 3 gene (ADCY3) encodes a membrane-associated enzyme. This protein catalyzes the formation of the secondary messenger cyclic adenosine monophosphate (cAMP) and is highly expressed in human placenta, testis, ovary, and colon [88]. Expression of adenylyl cyclase 2, 3, and 4 has been reported in olfactory cilia; ADCY3 mutants failed olfaction-based behavioral tests indicating that ADCY3 and cAMP signaling are critical for olfactory-dependent behavior [89].
DnaJ/Hsp40 homolog, subfamily C, member 27 gene (DNAJC27) encodes 273 amino acid protein with RAB-like GTPase and DNAJ domains. EST database reports high expression in nervous and reproductive systems [90].
Pro-opiomelanocortin gene (POMC) encodes a polypeptide hormone precursor protein synthesized mainly in corticotroph cells of the anterior pituitary. POMC is essential for normal steroidogenesis and maintenance of adrenal weight. Mutations in this gene have been associated with early onset of obesity, adrenal insufficiency, and red hair pigmentation [91,92]. The recent study in UK population suggested that POMC SNP haplotype GGCGAG may have a protective effect against T1D [93].
DNA (cytosine-5)-methyltransferase 3 alpha gene (DNMT3A) encodes a protein that functions as a de novo methyltransferase that can methylate unmethylated and hemimethylated DNA with equal efficiencies [94].
Additional fine gene mapping and functional studies are needed for above-mentioned genes to determine causal variants for 2q23 region and their role in T1D.
2.5.2.3. Intergenic Region 6q27
Intergenic region on 6q27 contained the third most significantly associated SNP (rs924043, p = 8.06 × 10−9) in our recent study [38]. The region of association is approximately 900 kb and harbors multiple genes including PHF10, TCTE3, DLL1, FAM120B, PSMB1, TBP, and PDCD2. The 6q27 region also includes several genes of unknown function: C6orf208/LINC00574 (long intergenic non-protein coding RNA 574), T-complex-associated-testis-expressed 3 (TCTE3), LOC154449, WD repeat domain 27 (WDR27), and chromosome 6 open reading frame 120 (C6orf120).
Plant Homeo Domain (PHD) finger protein 10 gene (PHF10) encodes a subunit of an ATP-dependent chromatin-remodeling complex that functions in neural precursor cells [95].
Delta-like 1-Drosophila gene (DLL1) is a human homolog of the Notch Delta ligand and a member of the delta/serrate/jagged family. It plays a role in mediating cell fate decisions during hematopoiesis and cell communication [96,97]. The protein is expressed in heart, pancreas and brain. Pancreatic regeneration in chronic pancreatitis requires activation of the notch signaling pathway [98].
The family with sequence similarity 120B gene (FAM120) encodes protein belonging to the constitutive coactivator of peroxisome proliferator-activated receptor gamma (PPARG) family. FAM120B functions in adipogenesis through PPARG activation in a ligand-independent manner [99].
Proteasome (prosome, macropain) subunit, beta type, 1 gene (PSMB1) encodes a member of the proteasome B-type family, also known as the T1B family, that is a 20S core beta subunit [100]. This gene encodes TBP, the TATA-binding protein, transcription factor that functions at the core of the DNA-binding multiprotein transcription factor IID (TFIID). Binding of TFIID to TBP is the initial transcriptional step of the pre-initiation complex (PIC) and plays a role in the activation of eukaryotic genes transcribed by RNA polymerase II [101].
Programmed cell death 2 gene (PDCD2) encodes a nuclear protein highly expressed in placenta, heart, pancreas, lung, and liver, and lowly expressed in spleen, lymph nodes, and thymus. Expression of this gene is shown to be repressed by B-cell CLL/lymphoma 6 (BCL6), a transcriptional repressor [102].
In addition, despite not reaching the genome wide significance, our study observed evidence for association at three additional loci containing the candidate genes LOC100128081, TNFRSF11B, and FOSL2 [38]. Of these, it is notable that the tumor necrosis factor receptor superfamily, member 11B (TNFRSF11B) is a strongly associated locus with bone mineral density, also discovered in GWAS, and the locus harboring LOC100128081 has also been reported in the context of a GWAS of SLE. FOS-like antigen 2 (FOSL2) gene encodes a leucine zipper protein that dimerizes with the JUN family proteins and forms the transcription factor complex activator protein 1 (AP-1). The FOS proteins have been implicated as regulators of cell proliferation, differentiation, and transformation [103].
2.5.2.4. Region 12q24
CUX2 (12q24): Huang et al., 2012 re-analyzed the original 2007 WTCCC study by using the 1,000 Genomes imputation and reported refined variant rs1265564 in Cut-like homeobox 2 (CUX2) region for association with T1D [46]. CUX2 is expressed exclusively in neural tissues. The protein belongs to the CUT homeobox family and contains three CUT domains and a homeodomain, both domains are DNA-binding motifs [104]. CUX2 gene has been shown to directly regulate the expression of NeuroD [105]. NeuroD/BETA2, a transcription factor of the insulin gene, is reported to be associated with T1D in Asian descent [106,107]. Thus, CUX2 is a plausible candidate for exploration in T1D pathogenesis.
2.5.2.5. Region 5p13-q13
HTR1A (5p13-q13): Asad et al. confirmed [45] the previously suggested association between the chromosome 5p13-q13 region and T1D in Scandinavian families [108]. None of the previous GWAS have reported any association of 5p13-q13 with T1D. This recent study identified the 5-hydroxytryptamine receptor 1A (HTR1A), and the ring finger protein 180 (RFN180) genes, to be associated with T1D in multiplex (Swedish and Danish) families. However, the conditional analysis indicated HTR1A has as a primary association with T1D. Both quantitative PCR and immunohistochemical analysis confirmed the presence of the HTR1A in human pancreas [45]. The study suggests that HTR1A may affect T1D susceptibility by modulating the initial autoimmune attack or either islet regeneration, insulin release, or both. The HTR1A gene is known to encode for a G-protein coupled receptor specific for serotonin, which mediates cellular signaling via the amine serotonin [109]. The HTR1A receptor is mainly known to mediate signal transduction in neurons in the central nervous system [110]. However, serotonin is also produced in pancreatic islets of several different species [111]. Studies in rodent islets show inhibition of insulin secretion by serotonin [112]. Sumatriptan (serotonin agonist) has an inhibitory effect on insulin secretion in humans [113]. Previously a decrease in expression of HTR1A with increased insulin release during pancreatic regeneration has been reported [114]. HTR1A also plays a role in the immune system by downregulating adenylate cyclase, which in turn regulates T-cell cytokine production and cytotoxicity [115]. Hence, polymorphisms in the HTR1A gene may affect insulin release and T-cell activity thereby increases the risk of developing T1D.
3. Conclusions
This review provides a summary of recent advances in the identification of risk variants associated with T1D. Genome wide association studies have revolutionized the discovery approach to autoimmune mediated disorders. In T1D only six genetic factors were well known before GWAS. GWAS has contributed greatly by expanding the number of established genetic variants to 59 loci. Most of these genes are novel and were not in any investigator’s favorite list. For the first time there is real consensus on the role of specific genetic factors underpinning T1D pathogenesis.
The discoveries of genetic factors involved in T1D through GWAS present the first step in a long process leading to cure. Genes uncovered using this approach are indeed fundamental to disease biology and will define the key molecular pathways leading to cure of T1D. However, such genome wide scans can lack coverage in certain regions where it is difficult to genotype, thus, it is possible that other loci with reasonable effect sizes remain to be uncovered through whole genome sequencing approaches.
To date most of T1D associated variants have been discovered utilizing cohorts of European ancestry because the SNP arrays were designed to optimally capture the haplotype diversity in this ethnicity. Novel SNP arrays are needed with the same degree of capture in diverse populations to elucidate the full role of each locus in a worldwide context.
In addition to identifying genes influencing disease susceptibility GWAS can be utilized to facilitate implementation of personalized medicine based on genetic make-up of the individuals. Our pilot study showed a proof-of-principle that use of whole-genome data, rather than a few ‘‘validated’’ susceptibility loci, could improve predictive accuracy [116]. This approach will have a greater impact on health care in the future; for example, by applying personalized intervention strategies on newborns who are at risk of developing T1D, we may reduce their risk of developing the disease or be better prepared to treat the disease.
The next challenge is to resolve the specific causal variants and determine how they affect the expression and function of these gene products. The Next-Generation Sequencing (NGS) technology has opened new avenues to elucidate the role of coding and noncoding RNAs in health and disease and is speeding up the identification of causative gene variants in T1D.
No doubt, the in vitro and in vivo biology of these genes will be fascinating areas of exploration for many scientists. Only after scientists have fully uncovered the functional context of T1D associated genes, is the promise of new therapies and preventive strategies likely to materialize.
Acknowledgments
This research was financially supported by grant from National Institute of Health (DP3 DK085708-01) and an Institute Development Award to the Center for Applied Genomics from the Children’s Hospital of Philadelphia.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Steyn, N.P.; Lambert, E.V.; Tabana, H. Conference on “multidisciplinary approaches to nutritional problems”. Symposium on “diabetes and health”. Nutrition interventions for the prevention of type 2 diabetes. Proc. Nutr. Soc. 2009, 68, 55–70. [Google Scholar] [CrossRef]
- International Diabetes Federation. Available online: http://www.idf.org/diabetesatlas/ (accessed on 11 July 2013).
- Eurodiab ACE study group. Variation and trends in incidence of childhood diabetes in europe. Lancet 2000, 355, 873–876. [CrossRef]
- Onkamo, P.; Vaananen, S.; Karvonen, M.; Tuomilehto, J. Worldwide increase in incidence of type Ι diabetes—The analysis of the data on published incidence trends. Diabetologia 1999, 42, 1395–1403. [Google Scholar] [CrossRef]
- Redondo, M.J.; Yu, L.; Hawa, M.; Mackenzie, T.; Pyke, D.A.; Eisenbarth, G.S.; Leslie, R.D. Heterogeneity of type Ι diabetes: Analysis of monozygotic twins in great britain and the united states. Diabetologia 2001, 44, 354–362. [Google Scholar] [CrossRef]
- Clayton, D.G. Prediction and interaction in complex disease genetics: Experience in type 1 diabetes. PLoS Genet. 2009, 5, e1000540. [Google Scholar] [CrossRef]
- Rich, S.S. Mapping genes in diabetes. Genetic epidemiological perspective. Diabetes 1990, 39, 1315–1319. [Google Scholar]
- Cudworth, A.G.; Woodrow, J.C. Evidence for hl-a-linked genes in “juvenile” diabetes mellitus. Br. Med. J. 1975, 3, 133–135. [Google Scholar] [CrossRef]
- Nerup, J.; Platz, P.; Andersen, O.O.; Christy, M.; Lyngsoe, J.; Poulsen, J.E.; Ryder, L.P.; Nielsen, L.S.; Thomsen, M.; Svejgaard, A. Hl-a antigens and diabetes mellitus. Lancet 1974, 2, 864–866. [Google Scholar]
- Singal, D.P.; Blajchman, M.A. Histocompatibility (hl-a) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes 1973, 22, 429–432. [Google Scholar]
- Bell, G.I.; Horita, S.; Karam, J.H. A polymorphic locus near the human insulin gene is associated with insulin-dependent diabetes mellitus. Diabetes 1984, 33, 176–183. [Google Scholar]
- Nistico, L.; Buzzetti, R.; Pritchard, L.E.; van der Auwera, B.; Giovannini, C.; Bosi, E.; Larrad, M.T.; Rios, M.S.; Chow, C.C.; Cockram, C.S.; et al. The ctla-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian diabetes registry. Hum. Mol. Genet. 1996, 5, 1075–1080. [Google Scholar] [CrossRef]
- Bottini, N.; Musumeci, L.; Alonso, A.; Rahmouni, S.; Nika, K.; Rostamkhani, M.; MacMurray, J.; Meloni, G.F.; Lucarelli, P.; Pellecchia, M.; et al. A functional variant of lymphoid tyrosine phosphatase is associated with type Ι diabetes. Nat. Genet. 2004, 36, 337–338. [Google Scholar] [CrossRef]
- Vella, A.; Cooper, J.D.; Lowe, C.E.; Walker, N.; Nutland, S.; Widmer, B.; Jones, R.; Ring, S.M.; McArdle, W.; Pembrey, M.E.; et al. Localization of a type 1 diabetes locus in the il2ra/cd25 region by use of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 2005, 76, 773–779. [Google Scholar] [CrossRef]
- Smyth, D.J.; Cooper, J.D.; Bailey, R.; Field, S.; Burren, O.; Smink, L.J.; Guja, C.; Ionescu-Tirgoviste, C.; Widmer, B.; Dunger, D.B.; et al. A genome-wide association study of nonsynonymous snps identifies a type 1 diabetes locus in the interferon-induced helicase (ifih1) region. Nat. Genet. 2006, 38, 617–619. [Google Scholar] [CrossRef]
- International HapMap Consortium. The international hapmap project. Nature 2003, 426, 789–796. [CrossRef]
- International HapMap Consortium. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [CrossRef]
- Hakonarson, H.; Grant, S.F.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Casalunovo, T.; Taback, S.P.; Frackelton, E.C.; et al. A genome-wide association study identifies kiaa0350 as a type 1 diabetes gene. Nature 2007, 448, 591–594. [Google Scholar] [CrossRef]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007, 447, 661–678. [CrossRef]
- Todd, J.A.; Walker, N.M.; Cooper, J.D.; Smyth, D.J.; Downes, K.; Plagnol, V.; Bailey, R.; Nejentsev, S.; Field, S.F.; Payne, F.; et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007, 39, 857–864. [Google Scholar] [CrossRef]
- Hakonarson, H.; Qu, H.Q.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Casalunovo, T.; Taback, S.P.; Frackelton, E.C.; et al. A novel susceptibility locus for type 1 diabetes on chr12q13 identified by a genome-wide association study. Diabetes 2008, 57, 1143–1146. [Google Scholar] [CrossRef]
- Concannon, P.; Onengut-Gumuscu, S.; Todd, J.A.; Smyth, D.J.; Pociot, F.; Bergholdt, R.; Akolkar, B.; Erlich, H.A.; Hilner, J.E.; Julier, C.; et al. A human type 1 diabetes susceptibility locus maps to chromosome 21q22.3. Diabetes 2008, 57, 2858–2861. [Google Scholar] [CrossRef]
- Tsygankov, A.Y. Multidomain sts/tula proteins are novel cellular regulators. IUBMB Life 2008, 60, 224–231. [Google Scholar] [CrossRef]
- Cohen, S.; Dadi, H.; Shaoul, E.; Sharfe, N.; Roifman, C.M. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, lyp. Blood 1999, 93, 2013–2024. [Google Scholar]
- Cooper, J.D.; Walker, N.M.; Smyth, D.J.; Downes, K.; Healy, B.C.; Todd, J.A. Follow-up of 1715 snps from the wellcome trust case control consortium genome-wide association study in type Ι diabetes families. Genes Immun. 2009, 10, S85–S94. [Google Scholar] [CrossRef]
- Johnson, K.; Wong, R.; Barriga, K.J.; Klingensmith, G.; Ziegler, A.G.; Rewers, M.J.; Steck, A.K. Rs11203203 is associated with type 1 diabetes risk in population pre-screened for high-risk hla-dr,dq genotypes. Pediatr. Diabetes 2012, 13, 611–615. [Google Scholar] [CrossRef]
- Manolio, T.A.; Rodriguez, L.L.; Brooks, L.; Abecasis, G.; Ballinger, D.; Daly, M.; Donnelly, P.; Faraone, S.V.; Frazer, K.; Gabriel, S.; et al. New models of collaboration in genome-wide association studies: The genetic association information network. Nat. Genet. 2007, 39, 1045–1051. [Google Scholar] [CrossRef]
- Mueller, P.W.; Rogus, J.J.; Cleary, P.A.; Zhao, Y.; Smiles, A.M.; Steffes, M.W.; Bucksa, J.; Gibson, T.B.; Cordovado, S.K.; Krolewski, A.S.; et al. Genetics of kidneys in diabetes (gokind) study: A genetics collection available for identifying genetic susceptibility factors for diabetic nephropathy in type 1 diabetes. J. Am. Soc. Nephrol. JASN 2006, 17, 1782–1790. [Google Scholar] [CrossRef]
- Cooper, J.D.; Smyth, D.J.; Smiles, A.M.; Plagnol, V.; Walker, N.M.; Allen, J.E.; Downes, K.; Barrett, J.C.; Healy, B.C.; Mychaleckyj, J.C.; et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat. Genet. 2008, 40, 1399–1401. [Google Scholar] [CrossRef]
- Espino-Paisan, L.; de La Calle, H.; Fernandez-Arquero, M.; Figueredo, M.A.; de La Concha, E.G.; Urcelay, E.; Santiago, J.L. Study of polymorphisms in 4q27, 10p15, and 22q13 regions in autoantibodies stratified type 1 diabetes patients. Autoimmunity 2011, 44, 624–630. [Google Scholar] [CrossRef]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.D.; Walker, N.M.; Healy, B.C.; Smyth, D.J.; Downes, K.; Todd, J.A. Analysis of 55 autoimmune disease and type ii diabetes loci: Further confirmation of chromosomes 4q27, 12q13.2 and 12q24.13 as type i diabetes loci, and support for a new locus, 12q13.3-q14.1. Genes Immun. 2009, 10, S95–S120. [Google Scholar] [CrossRef]
- Fung, E.Y.; Smyth, D.J.; Howson, J.M.; Cooper, J.D.; Walker, N.M.; Stevens, H.; Wicker, L.S.; Todd, J.A. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/tnfaip3 as a susceptibility locus. Genes Immun. 2009, 10, 188–191. [Google Scholar] [CrossRef]
- Smyth, D.J.; Plagnol, V.; Walker, N.M.; Cooper, J.D.; Downes, K.; Yang, J.H.; Howson, J.M.; Stevens, H.; McManus, R.; Wijmenga, C.; et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N. Eng. J. Med. 2008, 359, 2767–2777. [Google Scholar] [CrossRef]
- Grant, S.F.; Qu, H.Q.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Taback, S.P.; Frackelton, E.C.; Eckert, A.W.; et al. Follow-up analysis of genome-wide association data identifies novel loci for type 1 diabetes. Diabetes 2009, 58, 290–295. [Google Scholar] [CrossRef]
- Qu, H.Q.; Bradfield, J.P.; Li, Q.; Kim, C.; Frackelton, E.; Grant, S.F.; Hakonarson, H.; Polychronakos, C. In silico replication of the genome-wide association results of the type 1 diabetes genetics consortium. Hum. Mol. Genet. 2010, 19, 2534–2538. [Google Scholar] [CrossRef]
- Cooper, J.D.; Howson, J.M.; Smyth, D.; Walker, N.M.; Stevens, H.; Yang, J.H.; She, J.X.; Eisenbarth, G.S.; Rewers, M.; Todd, J.A.; et al. Confirmation of novel type 1 diabetes risk loci in families. Diabetologia 2012, 55, 996–1000. [Google Scholar] [CrossRef]
- Bradfield, J.P.; Qu, H.Q.; Wang, K.; Zhang, H.; Sleiman, P.M.; Kim, C.E.; Mentch, F.D.; Qiu, H.; Glessner, J.T.; Thomas, K.A.; et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011, 7, e1002293. [Google Scholar] [CrossRef]
- Awata, T.; Kawasaki, E.; Tanaka, S.; Ikegami, H.; Maruyama, T.; Shimada, A.; Nakanishi, K.; Kobayashi, T.; Iizuka, H.; Uga, M.; et al. Association of type 1 diabetes with two loci on 12q13 and 16p13 and the influence coexisting thyroid autoimmunity in japanese. J. Clin. Endocrinol. Metab. 2009, 94, 231–235. [Google Scholar]
- Zoledziewska, M.; Costa, G.; Pitzalis, M.; Cocco, E.; Melis, C.; Moi, L.; Zavattari, P.; Murru, R.; Lampis, R.; Morelli, L.; et al. Variation within the clec16a gene shows consistent disease association with both multiple sclerosis and type 1 diabetes in sardinia. Genes Immun. 2009, 10, 15–17. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, X.; Wang, X.; Ma, J.; Zhu, S.; Li, J.; Liu, Y. Intron polymorphism in the kiaa0350 gene is reproducibly associated with susceptibility to type 1 diabetes (t1d) in the han chinese population. Clin. Endocrinol. 2009, 71, 46–49. [Google Scholar] [CrossRef]
- Wallace, C.; Smyth, D.J.; Maisuria-Armer, M.; Walker, N.M.; Todd, J.A.; Clayton, D.G. The imprinted dlk1-meg3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat. Genet. 2010, 42, 68–71. [Google Scholar] [CrossRef]
- Wang, K.; Baldassano, R.; Zhang, H.; Qu, H.Q.; Imielinski, M.; Kugathasan, S.; Annese, V.; Dubinsky, M.; Rotter, J.I.; Russell, R.K.; et al. Comparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effects. Hum. Mol. Genet. 2010, 19, 2059–2067. [Google Scholar] [CrossRef]
- Reddy, M.V.; Wang, H.; Liu, S.; Bode, B.; Reed, J.C.; Steed, R.D.; Anderson, S.W.; Steed, L.; Hopkins, D.; She, J.X. Association between type 1 diabetes and gwas snps in the southeast us caucasian population. Genes Immun. 2011, 12, 208–212. [Google Scholar] [CrossRef]
- Asad, S.; Nikamo, P.; Gyllenberg, A.; Bennet, H.; Hansson, O.; Wierup, N.; Carlsson, A.; Forsander, G.; Ivarsson, S.A.; Larsson, H.; et al. Htr1a a novel type 1 diabetes susceptibility gene on chromosome 5p13-q13. PLoS One 2012, 7, e35439. [Google Scholar]
- Huang, J.; Ellinghaus, D.; Franke, A.; Howie, B.; Li, Y. 1,000 genomes-based imputation identifies novel and refined associations for the wellcome trust case control consortium phase 1 data. Eur. J. Hum. Genet. 2012, 20, 801–805. [Google Scholar] [CrossRef]
- Litman, G.W.; Cannon, J.P.; Dishaw, L.J. Reconstructing immune phylogeny: New perspectives. Nat. Rev. Immunol. 2005, 5, 866–879. [Google Scholar] [CrossRef]
- Storling, J.; Brorsson, C.A. Candidate genes expressed in human islets and their role in the pathogenesis of type 1 diabetes. Curr. Diab. Rep. 2013, in press. [Google Scholar]
- Van Belle, T.L.; Coppieters, K.T.; von Herrath, M.G. Type 1 diabetes: Etiology, immunology, and therapeutic strategies. Physiol. Rev. 2011, 91, 79–118. [Google Scholar] [CrossRef]
- Peakman, M. Immunological pathways to β-cell damage in type 1 diabetes. Diabet. Med. 2013, 30, 147–154. [Google Scholar] [CrossRef]
- Diana, J.; Gahzarian, L.; Simoni, Y.; Lehuen, A. Innate immunity in type 1 diabetes. Discov. Med. 2011, 11, 513–520. [Google Scholar]
- Robinson, M.J.; Sancho, D.; Slack, E.C.; LeibundGut-Landmann, S.; Reis e Sousa, C. Myeloid c-type lectins in innate immunity. Nat. Immunol. 2006, 7, 1258–1265. [Google Scholar] [CrossRef]
- Cambi, A.; Figdor, C.G. Dual function of c-type lectin-like receptors in the immune system. Curr. Opin. Cell Biol. 2003, 15, 539–546. [Google Scholar] [CrossRef]
- Martinez, A.; Perdigones, N.; Cenit, M.C.; Espino, L.; Varade, J.; Lamas, J.R.; Santiago, J.L.; Fernandez-Arquero, M.; de la Calle, H.; Arroyo, R.; et al. Chromosomal region 16p13: Further evidence of increased predisposition to immune diseases. Ann. Rheum. Dis. 2010, 69, 309–311. [Google Scholar] [CrossRef]
- Sang, Y.; Zong, W.; Yan, J.; Liu, M. The correlation between the clec16a gene and genetic susceptibility to type 1 diabetes in chinese children. Int. J. Endocrinol. 2012, 2012, Article ID 245384. [Google Scholar]
- Yamashita, H.; Awata, T.; Kawasaki, E.; Ikegami, H.; Tanaka, S.; Maruyama, T.; Shimada, A.; Nakanishi, K.; Takahashi, K.; Kobayashi, T.; et al. Analysis of the hla and non-hla susceptibility loci in japanese type 1 diabetes. Diabet. Metab. Res. Rev. 2011, 27, 844–848. [Google Scholar]
- Howson, J.M.; Rosinger, S.; Smyth, D.J.; Boehm, B.O.; Todd, J.A. Genetic analysis of adult-onset autoimmune diabetes. Diabetes 2011, 60, 2645–2653. [Google Scholar] [CrossRef]
- Nischwitz, S.; Cepok, S.; Kroner, A.; Wolf, C.; Knop, M.; Muller-Sarnowski, F.; Pfister, H.; Rieckmann, P.; Hemmer, B.; Ising, M.; et al. More clec16a gene variants associated with multiple sclerosis. Acta Neurol. Scand. 2011, 123, 400–406. [Google Scholar] [CrossRef]
- Zuvich, R.L.; Bush, W.S.; McCauley, J.L.; Beecham, A.H.; de Jager, P.L.; Ivinson, A.J.; Compston, A.; Hafler, D.A.; Hauser, S.L.; Sawcer, S.J.; et al. Interrogating the complex role of chromosome 16p13.13 in multiple sclerosis susceptibility: Independent genetic signals in the ciita-clec16a-socs1 gene complex. Hum. Mol. Genet. 2011, 20, 3517–3524. [Google Scholar] [CrossRef]
- Skinningsrud, B.; Husebye, E.S.; Pearce, S.H.; McDonald, D.O.; Brandal, K.; Wolff, A.B.; Lovas, K.; Egeland, T.; Undlien, D.E. Polymorphisms in clec16a and ciita at 16p13 are associated with primary adrenal insufficiency. J. Clin. Endocrinol. Metab. 2008, 93, 3310–3317. [Google Scholar]
- Gateva, V.; Sandling, J.K.; Hom, G.; Taylor, K.E.; Chung, S.A.; Sun, X.; Ortmann, W.; Kosoy, R.; Ferreira, R.C.; Nordmark, G.; et al. A large-scale replication study identifies tnip1, prdm1, jazf1, uhrf1bp1 and il10 as risk loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1228–1233. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, Y.; Zhou, X.; Li, Y.; Gao, J.; Han, J.; Quan, C.; He, S.; Lv, Y.; Hu, D.; et al. Polymorphisms at 16p13 are associated with systemic lupus erythematosus in the chinese population. J. Med. Genet. 2011, 48, 69–72. [Google Scholar] [CrossRef]
- Dubois, P.C.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.; Adany, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef]
- Marquez, A.; Varade, J.; Robledo, G.; Martinez, A.; Mendoza, J.L.; Taxonera, C.; Fernandez-Arquero, M.; Diaz-Rubio, M.; Gomez-Garcia, M.; Lopez-Nevot, M.A.; et al. Specific association of a clec16a/kiaa0350 polymorphism with nod2/card15(-) crohn’s disease patients. Eur. J. Hum. Genet. 2009, 17, 1304–1308. [Google Scholar] [CrossRef]
- Jagielska, D.; Redler, S.; Brockschmidt, F.F.; Herold, C.; Pasternack, S.M.; Garcia Bartels, N.; Hanneken, S.; Eigelshoven, S.; Refke, M.; Barth, S.; et al. Follow-up study of the first genome-wide association scan in alopecia areata: Il13 and kiaa0350 as susceptibility loci supported with genome-wide significance. J. Invest. Dermatol. 2012, 132, 2192–2197. [Google Scholar] [CrossRef]
- Skinningsrud, B.; Lie, B.A.; Husebye, E.S.; Kvien, T.K.; Forre, O.; Flato, B.; Stormyr, A.; Joner, G.; Njolstad, P.R.; Egeland, T.; et al. A clec16a variant confers risk for juvenile idiopathic arthritis and anti-cyclic citrullinated peptide antibody negative rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1471–1474. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Xie, G.; Lu, E.; Sun, Y.; Juran, B.D.; Chellappa, V.; Coltescu, C.; Mason, A.L.; Milkiewicz, P.; Myers, R.P; et al. Association of primary biliary cirrhosis with variants in the clec16a, socs1, spib and siae immunomodulatory genes. Genes Immun. 2012, 13, 328–335. [Google Scholar] [CrossRef]
- Mells, G.F.; Floyd, J.A.; Morley, K.I.; Cordell, H.J.; Franklin, C.S.; Shin, S.Y.; Heneghan, M.A.; Neuberger, J.M.; Donaldson, P.T.; Day, D.B.; et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 2011, 43, 329–332. [Google Scholar] [CrossRef]
- Davison, L.J.; Wallace, C.; Cooper, J.D.; Cope, N.F.; Wilson, N.K.; Smyth, D.J.; Howson, J.M.; Saleh, N.; Al-Jeffery, A.; Angus, K.L.; et al. Long-range DNA looping and gene expression analyses identify dexi as an autoimmune disease candidate gene. Hum. Mol. Genet. 2012, 21, 322–333. [Google Scholar] [CrossRef]
- Kim, S.; Wairkar, Y.P.; Daniels, R.W.; DiAntonio, A. The novel endosomal membrane protein ema interacts with the class c vps-hops complex to promote endosomal maturation. J. Cell Biol. 2010, 188, 717–734. [Google Scholar] [CrossRef]
- Kim, S.; Naylor, S.A.; DiAntonio, A. Drosophila golgi membrane protein ema promotes autophagosomal growth and function. Proc. Natl. Acad. Sci. USA 2012, 109, E1072–E1081. [Google Scholar] [CrossRef]
- Wu, X.; Li, J.; Chen, C.; Yan, Y.; Jiang, S.; Shao, B.; Xu, J.; Kang, L.; Huang, Y.; Zhu, L.; et al. Involvement of clec16a in activation of astrocytes after lps treated. Neurochem. Res. 2012, 37, 5–14. [Google Scholar] [CrossRef]
- Ooshio, T.; Irie, K.; Morimoto, K.; Fukuhara, A.; Imai, T.; Takai, Y. Involvement of lmo7 in the association of two cell-cell adhesion molecules, nectin and e-cadherin, through afadin and alpha-actinin in epithelial cells. J. Biol. Chem. 2004, 279, 31365–31373. [Google Scholar]
- Yamada, A.; Irie, K.; Fukuhara, A.; Ooshio, T.; Takai, Y. Requirement of the actin cytoskeleton for the association of nectins with other cell adhesion molecules at adherens and tight junctions in mdck cells. Genes Cells 2004, 9, 843–855. [Google Scholar] [CrossRef]
- Furuya, M.; Tsuji, N.; Endoh, T.; Moriai, R.; Kobayashi, D.; Yagihashi, A.; Watanabe, N. A novel gene containing pdz and lim domains, pcd1, is overexpressed in human colorectal cancer. Anticancer Res. 2002, 22, 4183–4186. [Google Scholar]
- Kang, S.; Xu, H.; Duan, X.; Liu, J.J.; He, Z.; Yu, F.; Zhou, S.; Meng, X.Q.; Cao, M.; Kennedy, G.C. Pcd1, a novel gene containing pdz and lim domains, is overexpressed in several human cancers. Cancer Res. 2000, 60, 5296–5302. [Google Scholar]
- Lindvall, J.M.; Blomberg, K.E.; Wennborg, A.; Smith, C.I. Differential expression and molecular characterisation of lmo7, myo1e, sash1, and mcoln2 genes in btk-defective b-cells. Cell. Immunol. 2005, 235, 46–55. [Google Scholar] [CrossRef]
- Semenova, E.; Wang, X.; Jablonski, M.M.; Levorse, J.; Tilghman, S.M. An engineered 800 kilobase deletion of uchl3 and lmo7 on mouse chromosome 14 causes defects in viability, postnatal growth and degeneration of muscle and retina. Hum. Mol. Genet. 2003, 12, 1301–1312. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Nakamura, H.; Mukai, M.; Komatsu, K.; Tanaka-Okamoto, M.; Itoh, Y.; Ishizaki, H.; Tatsuta, M.; Inoue, M.; Miyoshi, J. Transforming growth factor-beta1 induces lmo7 while enhancing the invasiveness of rat ascites hepatoma cells. Cancer Lett. 2005, 220, 95–99. [Google Scholar] [CrossRef]
- Kutlu, B.; Burdick, D.; Baxter, D.; Rasschaert, J.; Flamez, D.; Eizirik, D.L.; Welsh, N.; Goodman, N.; Hood, L. Detailed transcriptome atlas of the pancreatic beta cell. BMC Med. Genomics 2009, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.B.; Salen, G.; Hidaka, H.; Kwiterovich, P.O.; Stalenhoef, A.F.; Miettinen, T.A.; Grundy, S.M.; Lee, M.H.; Rubenstein, J.S.; Polymeropoulos, M.H.; et al. Mapping a gene involved in regulating dietary cholesterol absorption. The sitosterolemia locus is found at chromosome 2p21. J. Clin. Invest. 1998, 102, 1041–1044. [Google Scholar] [CrossRef]
- Shulenin, S.; Schriml, L.M.; Remaley, A.T.; Fojo, S.; Brewer, B.; Allikmets, R.; Dean, M. An atp-binding cassette gene (abcg5) from the abcg (white) gene subfamily maps to human chromosome 2p21 in the region of the sitosterolemia locus. Cytogenet. Cell Genet. 2001, 92, 204–208. [Google Scholar] [CrossRef]
- Zumsteg, U.; Muller, P.Y.; Miserez, A.R. Alstrom syndrome: Confirmation of linkage to chromosome 2p12-13 and phenotypic heterogeneity in three affected sibs. J. Med. Genet. 2000, 37, E8. [Google Scholar] [CrossRef]
- Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 1995, 270, 1354–1357. [Google Scholar]
- Torchia, J.; Rose, D.W.; Inostroza, J.; Kamei, Y.; Westin, S.; Glass, C.K.; Rosenfeld, M.G. The transcriptional co-activator p/cip binds cbp and mediates nuclear-receptor function. Nature 1997, 387, 677–684. [Google Scholar] [CrossRef]
- Okada, M.; Cheeseman, I.M.; Hori, T.; Okawa, K.; McLeod, I.X.; Yates, J.R., 3rd; Desai, A.; Fukagawa, T. The cenp-h-i complex is required for the efficient incorporation of newly synthesized cenp-a into centromeres. Nat. Cell Biol. 2006, 8, 446–457. [Google Scholar] [CrossRef]
- Ludwig, M.G.; Seuwen, K. Characterization of the human adenylyl cyclase gene family: Cdna, gene structure, and tissue distribution of the nine isoforms. J. Recept. Signal Transduct. Res. 2002, 22, 79–110. [Google Scholar] [CrossRef]
- Wong, S.T.; Trinh, K.; Hacker, B.; Chan, G.C.; Lowe, G.; Gaggar, A.; Xia, Z.; Gold, G.H.; Storm, D.R. Disruption of the type iii adenylyl cyclase gene leads to peripheral and behavioral anosmia in transgenic mice. Neuron 2000, 27, 487–497. [Google Scholar] [CrossRef]
- Nepomuceno-Silva, J.L.; de Melo, L.D.; Mendonca, S.M.; Paixao, J.C.; Lopes, U.G. Rjls: A new family of ras-related gtp-binding proteins. Gene 2004, 327, 221–232. [Google Scholar] [CrossRef]
- Hung, C.N.; Poon, W.T.; Lee, C.Y.; Law, C.Y.; Chan, A.Y. A case of early-onset obesity, hypocortisolism, and skin pigmentation problem due to a novel homozygous mutation in the proopiomelanocortin (pomc) gene in an indian boy. J. Pediatr. Endocrinol. Metab. 2012, 25, 175–179. [Google Scholar]
- Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Gruters, A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by pomc mutations in humans. Nat. Genet. 1998, 19, 155–157. [Google Scholar] [CrossRef]
- Martin, R.J.; Savage, D.A.; Carson, D.J.; McKnight, A.J.; Maxwell, A.P.; Patterson, C.C. Association analysis of proopiomelanocortin (pomc) haplotypes in type 1 diabetes in a uk population. Diabetes Metab. 2011, 37, 298–304. [Google Scholar] [CrossRef]
- Yanagisawa, Y.; Ito, E.; Yuasa, Y.; Maruyama, K. The human DNA methyltransferases dnmt3a and dnmt3b have two types of promoters with different cpg contents. Biochim. Biophys. Acta 2002, 1577, 457–465. [Google Scholar] [CrossRef]
- Yoo, A.S.; Staahl, B.T.; Chen, L.; Crabtree, G.R. Microrna-mediated switching of chromatin-remodelling complexes in neural development. Nature 2009, 460, 642–646. [Google Scholar]
- Dontje, W.; Schotte, R.; Cupedo, T.; Nagasawa, M.; Scheeren, F.; Gimeno, R.; Spits, H.; Blom, B. Delta-like1-induced notch1 signaling regulates the human plasmacytoid dendritic cell versus t-cell lineage decision through control of gata-3 and spi-b. Blood 2006, 107, 2446–2452. [Google Scholar] [CrossRef]
- Santos, M.A.; Sarmento, L.M.; Rebelo, M.; Doce, A.A.; Maillard, I.; Dumortier, A.; Neves, H.; Radtke, F.; Pear, W.S.; Parreira, L.; et al. Notch1 engagement by delta-like-1 promotes differentiation of b lymphocytes to antibody-secreting cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15454–15459. [Google Scholar] [CrossRef]
- Su, Y.; Buchler, P.; Gazdhar, A.; Giese, N.; Reber, H.A.; Hines, O.J.; Giese, T.; Buchler, M.W.; Friess, H. Pancreatic regeneration in chronic pancreatitis requires activation of the notch signaling pathway. J. Gastrointest. Surg. 2006, 10, 1230–1241. [Google Scholar] [CrossRef]
- Li, D.; Kang, Q.; Wang, D.M. Constitutive coactivator of peroxisome proliferator-activated receptor (ppargamma), a novel coactivator of ppargamma that promotes adipogenesis. Mol. Endocrinol. 2007, 21, 2320–2333. [Google Scholar] [CrossRef]
- Trachtulec, Z.; Hamvas, R.M.; Forejt, J.; Lehrach, H.R.; Vincek, V.; Klein, J. Linkage of tata-binding protein and proteasome subunit c5 genes in mice and humans reveals synteny conserved between mammals and invertebrates. Genomics 1997, 44, 1–7. [Google Scholar] [CrossRef]
- Keutgens, A.; Zhang, X.; Shostak, K.; Robert, I.; Olivier, S.; Vanderplasschen, A.; Chapelle, J.P.; Viatour, P.; Merville, M.P.; Bex, F.; et al. Bcl-3 degradation involves its polyubiquitination through a fbw7-independent pathway and its binding to the proteasome subunit psmb1. J. Biol. Chem. 2010, 285, 25831–25840. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the pd-1 antigen on the surface of stimulated mouse t and b lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Cohen, D.R.; Ferreira, P.C.; Gentz, R.; Franza, B.R., Jr.; Curran, T. The product of a fos-related gene, fra-1, binds cooperatively to the ap-1 site with jun: Transcription factor ap-1 is comprised of multiple protein complexs. Genes Dev. 1989, 3, 173–184. [Google Scholar] [CrossRef]
- Gingras, H.; Cases, O.; Krasilnikova, M.; Berube, G.; Nepveu, A. Biochemical characterization of the mammalian cux2 protein. Gene 2005, 344, 273–285. [Google Scholar] [CrossRef]
- Iulianella, A.; Sharma, M.; Durnin, M.; Vanden Heuvel, G.B.; Trainor, P.A. Cux2 (cutl2) integrates neural progenitor development with cell-cycle progression during spinal cord neurogenesis. Development 2008, 135, 729–741. [Google Scholar] [CrossRef]
- Iwata, I.; Nagafuchi, S.; Nakashima, H.; Kondo, S.; Koga, T.; Yokogawa, Y.; Akashi, T.; Shibuya, T.; Umeno, Y.; Okeda, T.; et al. Association of polymorphism in the neurod/beta2 gene with type 1 diabetes in the japanese. Diabetes 1999, 48, 416–419. [Google Scholar] [CrossRef]
- Kavvoura, F.K.; Ioannidis, J.P. Ala45thr polymorphism of the neurod1 gene and diabetes susceptibility: A meta-analysis. Hum. Genet. 2005, 116, 192–199. [Google Scholar] [CrossRef]
- Nerup, J.; Pociot, F. A genomewide scan for type 1-diabetes susceptibility in scandinavian families: Identification of new loci with evidence of interactions. Am. J. Hum. Genet. 2001, 69, 1301–1313. [Google Scholar] [CrossRef]
- Barnes, N.M.; Sharp, T. A review of central 5-ht receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef]
- Lesurtel, M.; Soll, C.; Graf, R.; Clavien, P.A. Role of serotonin in the hepato-gastrointestinal tract: An old molecule for new perspectives. Cell. Mol. life Sci. 2008, 65, 940–952. [Google Scholar] [CrossRef]
- Sundler, F.; Hakanson, R.; Loren, I.; Lundquist, I. Amine storage and function in peptide hormone-producing cells. Invest. Cell Pathol. 1980, 3, 87–103. [Google Scholar]
- Zawalich, W.S.; Tesz, G.J.; Zawalich, K.C. Effects of prior 5-hydroxytryptamine exposure on rat islet insulin secretory and phospholipase c responses. Endocrine 2004, 23, 11–16. [Google Scholar] [CrossRef]
- Coulie, B.; Tack, J.; Bouillon, R.; Peeters, T.; Janssens, J. 5-hydroxytryptamine-1 receptor activation inhibits endocrine pancreatic secretion in humans. Am. J. Physiol. 1998, 274, E317–E320. [Google Scholar]
- Mohanan, V.V.; Khan, R.; Paulose, C.S. Hypothalamic 5-ht functional regulation through 5-ht1a and 5-ht2c receptors during pancreatic regeneration. Life Sci. 2006, 78, 1603–1609. [Google Scholar] [CrossRef]
- Aune, T.M.; McGrath, K.M.; Sarr, T.; Bombara, M.P.; Kelley, K.A. Expression of 5ht1a receptors on activated human t cells. Regulation of cyclic amp levels and t cell proliferation by 5-hydroxytryptamine. J. Immunol. 1993, 151, 1175–1183. [Google Scholar]
- Wei, Z.; Wang, K.; Qu, H.Q.; Zhang, H.; Bradfield, J.; Kim, C.; Frackleton, E.; Hou, C.; Glessner, J.T.; Chiavacci, R.; et al. From disease association to risk assessment: An optimistic view from genome-wide association studies on type 1 diabetes. PLoS Genet. 2009, 5, e1000678. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Bakay, M.; Pandey, R.; Hakonarson, H. Genes Involved in Type 1 Diabetes: An Update. Genes 2013, 4, 499-521. https://0-doi-org.brum.beds.ac.uk/10.3390/genes4030499
AMA Style
Bakay M, Pandey R, Hakonarson H. Genes Involved in Type 1 Diabetes: An Update. Genes. 2013; 4(3):499-521. https://0-doi-org.brum.beds.ac.uk/10.3390/genes4030499
Chicago/Turabian StyleBakay, Marina, Rahul Pandey, and Hakon Hakonarson. 2013. "Genes Involved in Type 1 Diabetes: An Update" Genes 4, no. 3: 499-521. https://0-doi-org.brum.beds.ac.uk/10.3390/genes4030499