Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward


Department of Pediatrics, Carver College of Medicine, University of Iowa, Iowa City, IA 52242, USA
*
Author to whom correspondence should be addressed.
Genes 2018, 9(11), 538; https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110538
Submission received: 27 September 2018
/
Revised: 30 October 2018
/
Accepted: 31 October 2018
/
Published: 7 November 2018
(This article belongs to the Special Issue Cystic Fibrosis: Therapy and Genetics)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that encodes a cAMP-regulated anion channel. Although CF is a multi-organ system disease, most people with CF die of progressive lung disease that begins early in childhood and is characterized by chronic bacterial infection and inflammation. Nearly 90% of people with CF have at least one copy of the ΔF508 mutation, but there are hundreds of CFTR mutations that result in a range of disease severities. A CFTR gene replacement approach would be efficacious regardless of the disease-causing mutation. After the discovery of the CFTR gene in 1989, the in vitro proof-of-concept for gene therapy for CF was quickly established in 1990. In 1993, the first of many gene therapy clinical trials attempted to rescue the CF defect in airway epithelia. Despite the initial enthusiasm, there is still no FDA-approved gene therapy for CF. Here we discuss the history of CF gene therapy, from the discovery of the CFTR gene to current state-of-the-art gene delivery vector designs. While implementation of CF gene therapy has proven more challenging than initially envisioned; thanks to continued innovation, it may yet become a reality.
1. Introduction: A Brief Summary of Cystic Fibrosis Today
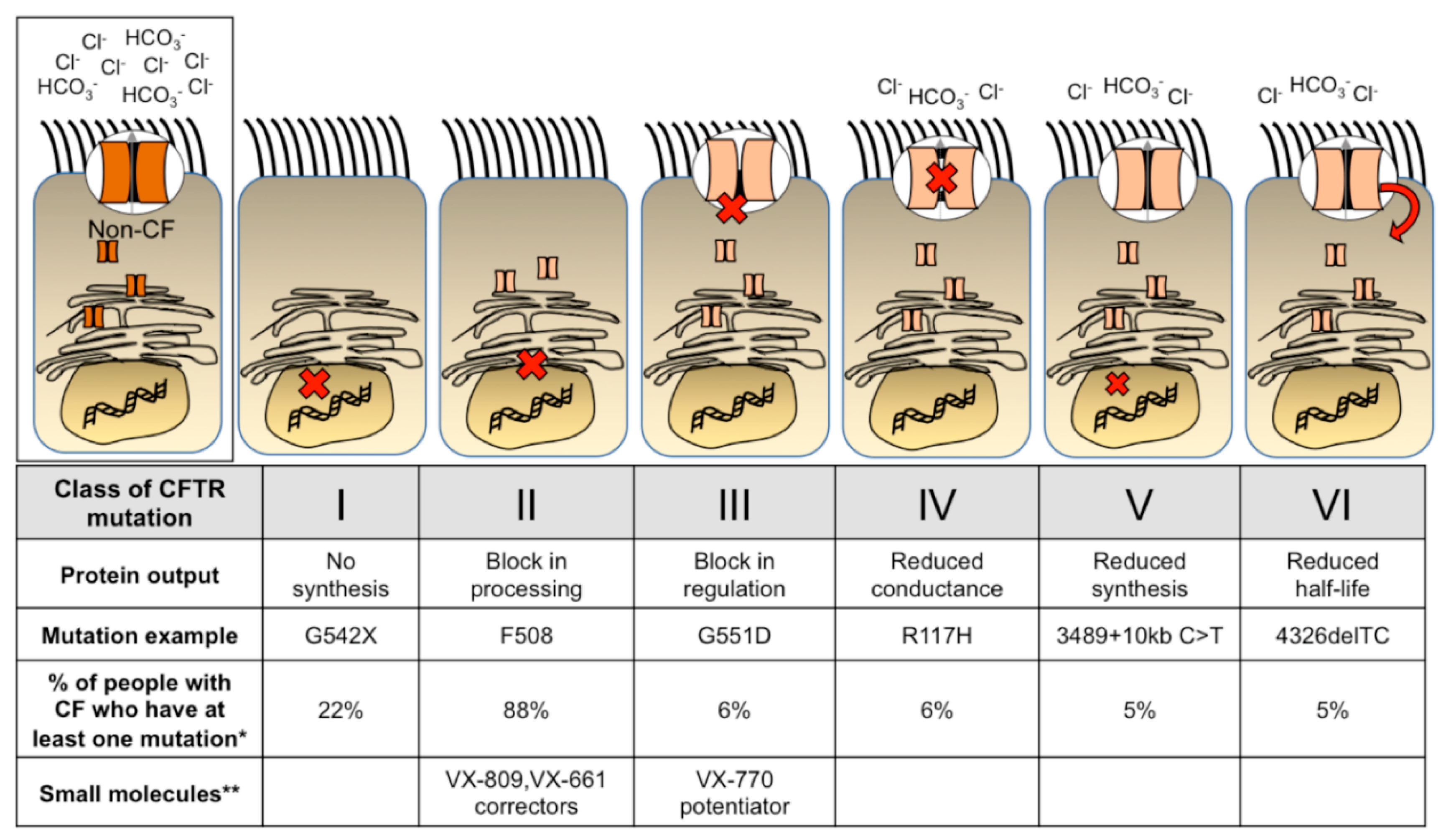
Cystic Fibrosis (CF) is a common autosomal recessive genetic disease that affects multiple organ systems. In 1938, Dr. Dorothy Andersen first described the disease as “cystic fibrosis of the pancreas”, which correlated with malnutrition. Since this discovery, aggressive early interventions have been established to improve the quality of life of people with CF, however progressive lung disease remains difficult to manage and is the leading cause of morbidity and mortality. Therapeutic small molecules [1,2,3] provide benefit to a growing percentage of people with CF. Although this is astounding progress, these cystic fibrosis transmembrane conductance regulator (CFTR) modulators are expensive and require lifetime treatment. A one-time treatment administered early in life for people with CF might prevent the onset of lung disease. Figure 1 outlines the six classes of CFTR mutations, examples of each mutation, and the prevalence within the U.S. population of people with CF. Small molecule therapeutics for each class are also labeled. Individuals may have different mutations on each allele and individual mutations may fit into more than one category. Furthermore, new mutation subclasses have been proposed based on the potential corrective therapy potential [4]. As small molecule treatments continue to improve and expand among different mutations, perhaps additional classes or subclasses will be added. Although the development of potentiator and corrector small molecule treatments provide relief for many people with CF, there remains an unmet need for those who have mutations that do not benefit from these treatments. Thus, gene therapy is a mutation agnostic approach and has the potential to repair the phenotypic defect for all people with CF.
CF gene therapy clinical trials pioneered the logistics of endpoint assays and for CF as well as other pulmonary diseases. For example, nasal potential difference (NPD) and forced expiratory volume in 1 s (FEV1) were used as clinical outcome measures in CF trials to quantify improvement of lung function (as previously reviewed [6]). The improved quality of life following small molecule treatment is now shedding light on other disease targets, such as inflammation. Lessons learned from these treatments will surely impact future gene therapy clinical trial designs.
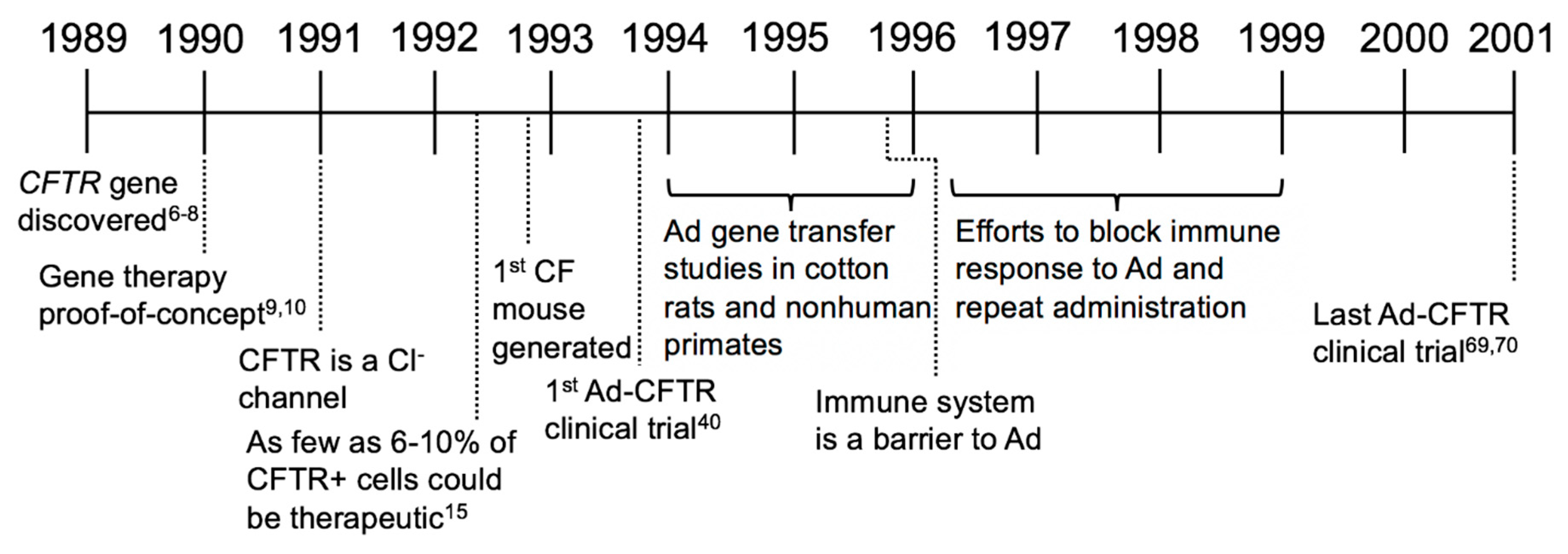
In this review, we will focus on the history of CF gene therapy, beginning with the discovery of the CFTR gene, continuing with the major milestones that have impacted the field, and looking to the future of CF gene therapy (Figure 2).
2. Establishing Benchmarks of Success and Adenovirus-Based Gene Therapy Trials (1989–2001)
In 1989, the gene responsible for CF was identified as CFTR [7,8,9]. Sequencing identified multiple CFTR mutations, most commonly a three-base deletion that results in the loss of phenylalanine at position 508 (ΔF508) [7,8]. Cloning CFTR was a major leap for studying CF and quickly launched the concept of gene-based therapeutics. Within a year of discovering CFTR, two groups independently demonstrated the proof-of-concept for gene therapy by expressing full length CFTR complementary DNA (cDNA) in CF cells and restoring anion channel activity. Restoring CFTR activity was proposed as a “cure all” for CF [10,11]. Soon after, several studies using viral and nonviral approaches to deliver CFTR and correct the CF defect were advanced, including an adeno-associated virus (AAV)-based vector [12], adenovirus (Ad)-based vector [13], plasmids formulated with cationic liposomes [14], and a retroviral vector [15]. At this time, complementing CFTR in CF patients was considered an achievable, near-term goal.
After ensuring that CFTR complementation restored Cl− current in CF cells, an important next question was to determine the percentage of corrected cells necessary to be therapeutically beneficial. Johnson et al. performed the first studies by mixing CF and MLV-CFTR transduced CF cells in varied ratios in vitro and found that as few as 6–10% of airway cells expressing CFTR achieved non-CF levels of Cl− transport [16]. These studies confirmed that CFTR gene delivery was a potential curative strategy and established a common benchmark of success for gene therapy (i.e., transduction of as few as 6% of airway cells). With relatively good agreement, this and other later studies suggest that expressing CFTR in 5–15% of cells restores Cl− to wild-type levels. Whether this benchmark truly translates into clinical efficacy is an open and complex question; however, as will be discussed, this important question may be addressable with improved gene delivery tools and animal models.
Also during this time, functional CFTR assays and new model systems were being rapidly developed. Experiments were performed on patient-derived immortalized cells such as CF pancreatic cells [17], human bronchial epithelial cells [18], CF bronchial epithelial cells [19], and IB3-1 cells [20]. Established metrics to quantify CFTR expression levels and activity included messenger RNA (mRNA) abundance, the ratio of CFTR protein band C to band B as measured by western blot, in situ hybridization, iodide efflux, patch clamp, and bioelectric properties measured in Ussing chambers [10]. Prior to 1992, no CF animal models existed to test functional gene transfer efficacy in vivo. However, within a short period of time, three groups independently generated CF mice by targeted knockout of endogenous Cftr [21,22,23,24,25]. These mice exhibited an increase in steady-state NPD compared to non-CF mice, altered Cl− transport, abnormal mucus accumulation, and disease-related changes in the lung and reproductive tract, but mice did not develop classic CF lung disease. Similar to humans, intestinal obstruction was also reported [26]. Consistent with in vitro experiments, in a CF mouse model, transduction of as few as 5% of cells with a CFTR expressing vector yielded 50% of the non-CF Cl− secretion [27].
At the time these studies were conducted, Ad-based vectors were widely available and production methods were established. Multiple in vivo experiments examined Ad-based lung gene transfer in various models. Repeat doses of Ad-LacZ to cotton rats or nonhuman primates showed that Ad transduced cells within the proximal bronchi and bronchioles including ciliated, secretory, undifferentiated, basal cells [28], and even submucosal glands [29]. The safety of repeated Ad administration was evaluated and studies concluded that Ad delivery was safe, exhibiting little to no immune response upon repeat administration [30,31,32,33]. These and other studies [31,34,35,36,37,38,39,40] suggested that Ad was a promising vector for CF gene therapy.
In 1993, Zabner et al. was the first to publish results from an early CF gene therapy clinical trial [41]. Three CF patients received an Ad vector (serotype 2) carrying CFTR [42]. The vector was applied to nasal turbinates with an applicator for 30 min and then removed by suction. One or three days later, one side was biopsied and the contralateral side was used to measure NPD. The authors reported a restored NPD response to a cAMP agonist in the nasal epithelia; however, CFTR mRNA and protein were undetectable. In retrospect, airway injury from the delivery method may have facilitated viral transduction. In parallel with the Zabner study, three additional Ad-based clinical trials ensued in efforts to restore CFTR function in CF patients. Crystal et al. performed a phase I dose-escalation study in 4 people with CF and concluded that up to 2 × 109 pfu of Ad2 led to CFTR DNA expression in the airway epithelium [43]. One subject that received 2 × 109 pfu in the right lower lobe experienced transient systemic inflammation in the first 24 h, but the symptoms quickly resolved and a 6-month follow up showed no long-term effects [43]. The NPD results in this study were reported as inconclusive but Ad delivery to the nose and lungs appeared to be safe [43]. Knowles et al. used an Ad serotype 5 vector in a larger study of 12 CF participants. Here, Ad-mediated delivery of CFTR did not correct the functional defects, perhaps due to inflammatory responses [44,45,46]. Hay et al. delivered Ad5-CFTR to nasal epithelia and reported that NPD decreased toward normal compared to the contralateral, untreated nostril [47,48]. In general, these studies suggested that Ad-based vectors could partially correct the Cl− transport defect in CF airway epithelia; however, the effects were transient and inflammatory responses were observed.
Although several studies showed partial CFTR correction in cell and animal models using Ad vectors [49,50,51], the results of clinical trials raised questions about safety and efficacy in humans. Both innate and cellular immune responses presented obstacles to achieving long-term and efficient gene transfer with Ad-based vectors. A nonhuman primate study reported increased alveolar inflammation with high doses of Ad vector [52,53]. Although Ad efficiently delivered genes to the lung, transient expression meant that repeat administration would be required [52,53]. Studies during this time elucidated one mechanism for transgene elimination as mediated by MHC-II presentation of viral antigens which led to activated CD8+ T cells in mice [54]. Immune responses also induced neutralizing antibody formation [55]. These serotype-specific neutralizing antibodies against Ad viral capsid antigens were found to prevent readministration of the same Ad serotype [56]. Efforts to facilitate repeat administration included: Blocking IgA antibodies [57], blocking neutralizing antibodies [58], and a nondepleting hCD4 antibody [59], none of which substantially improved repeat Ad administration.
To determine if Ad-vector readministration could achieve long-term CFTR expression in people with CF, clinical trials tested safety and effectiveness of repeat Ad administration. A trial in 1996 featured dose-escalation with 5 repeated doses up to 1010 pfu in the nasal epithelium of CF subjects. They concluded that the vector partially corrected the Cl− transport defect but observed significant variability between subjects. Of note, they found less CFTR-correction with each administration, likely due to the immune responses [60,61,62]. Another Ad5 clinical trial by Bellon et al. reported no toxic effects using doses of 105–108 pfu and detectable CFTR DNA and mRNA [63,64]. Other clinical trials occurring during this time using Ad5 with additional E3 or E4 deletions reported transient expression after a second administration [65] or low efficiency in the lower respiratory tract with short duration of expression [66]. Overall, repeated doses of Ad were not an effective therapeutic option.
In 1999, in a clinical trial for ornithine transcarbamylase (OTC) deficiency, an Ad5 vector carrying the OTC gene was delivered to an 18-year-old male subject named Jesse Gelsinger. He received a dose of 3.8 × 1013 viral particles via the femoral artery for delivery to the liver [67,68,69]. Four days after administration, he died from multiple organ failure and associated cytokine storm. Although this disease is unrelated to CF, it tragically demonstrated that high systemic doses of Ad can be fatal. In total, 9 CF clinical trials used Ad as the delivery vehicle, the last published in 2001 [70,71].
In the first decade of CF gene therapy the concept of CFTR complementation was firmly established; however, scientists and clinicians learned that delivery of a functional CFTR to the airways of humans was more challenging than originally anticipated. CF clinical trials using Ad-based vectors generally supported that gene therapy had potential therapeutic benefit, but improvements in vector design and delivery efficiency were necessary to correct CF lung disease (Figure 3).
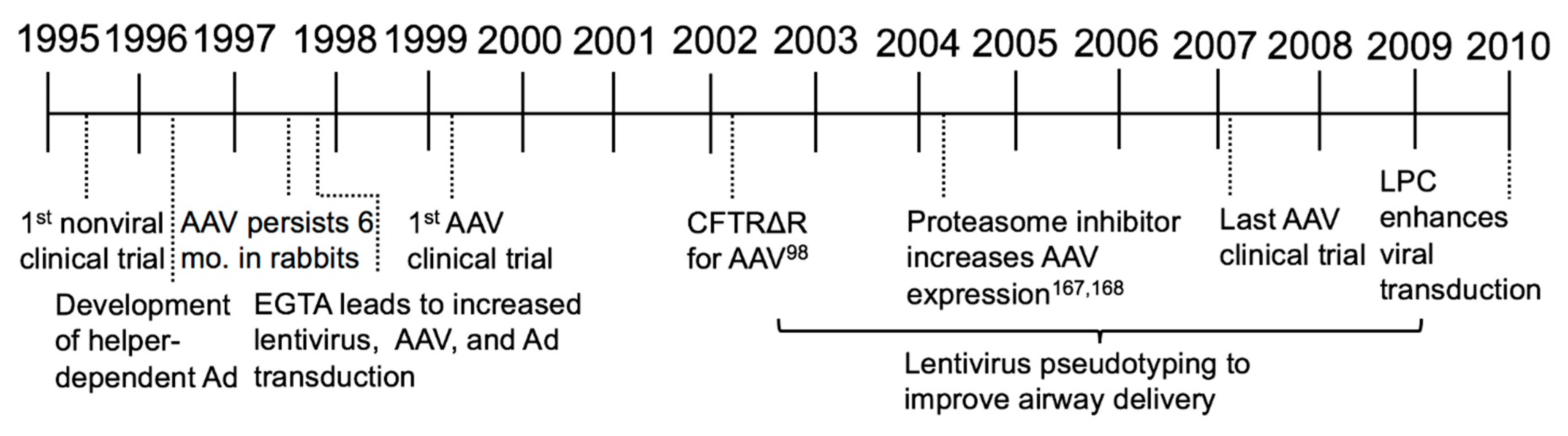
3. Alternatives to Ad (1995–2008)
The immunogenicity and transient expression of Ad-based vectors were recognized early on. In parallel to many of the aforementioned studies, development of other viral and non-viral vectors was underway. The delivery platforms that received the most attention were (1) AAV-based vectors, (2) non-viral (i.e., plasmid) vectors, and (3) retroviral- (or lentiviral)-based vectors. These vectors were evaluated for their ability to transduce airway cells in vivo and in vitro. Vector modifications and delivery agents were screened with the goal of improving titer, tropism, transduction efficiency, expression, and stability. The barriers to pulmonary gene transfer were examined in greater detail and better measurements for successful gene transfer were invented. In this era, research focused on improving gene delivery, measuring persistent expression, and understanding the host immune response to viral vectors and encoded transgenes.
3.1. Adeno-Associated Virus
Similar to Ad, recombinant AAV vectors can transduce terminally differentiated and non-dividing cells [72]. AAV persists episomally and does not integrate in the absence of exogenous Rep protein [73]. In preclinical studies, CFTR expression from an AAV serotype 2 vector persisted in rhesus macaques for 3 months [74] and a single delivery to the lung was shown to be safe [75]. In the newborn rabbit, AAV transduced alveolar epithelial cells, tracheobronchial and ciliated lung cells [76,77] and vector DNA was detectable for up to 6 months [78]. Functional CFTR activity was demonstrated in cultured cells by an iodide efflux assay [79]. Although early persistence results were encouraging, AAV expression decreased over time. Loss of expression was attributed to the dilution of transduced cells through lung growth and lack of stem cell transduction [80]. AAV could be readministered and persist for 8 months following transient immunosuppression with an anti-CD40 ligand antibody and a soluble CTLA4-immunoglobulin fusion protein [81]. Thus, a single dose of AAV transduced airways and conferred stable expression for many months.
During this time period, AAV vectors were the forefront of CF gene therapy. Many studies showed their safety and efficacy in repeat dosing to nonhuman primates [82,83] and mice, even in the presence of neutralizing antibodies [84]. Serotype studies in cells cultured from ferrets and pigs showed that AAV1, 2, and 5 transduction patterns closely mirrored human airway epithelia, but mice did not reflect this trend [85,86,87]. AAV6 studies in mice evaluated various promoters, which ultimately led to the generation of a hybrid β-actin promoter that included a β-actin splice donor and β-globin splice acceptor, termed CAG promoter [88]. This promoter substantially increased transgene expression in airway epithelia in vitro [88]. Delivering CFTR with AAV helped uncover features of CFTR biology. For example, AAV-mediated CFTR delivery to mouse airways exposed a correlation between gene transfer and mRNA levels which suggested a regulatory role of low levels of CFTR mRNA as an activator of other chloride channels [89]. Multiple in vitro and in vivo studies suggested AAV could efficiently restore CFTR expression in airway epithelial cells.
In 1999, the first AAV serotype 2 vector clinical trial tested single and two dose treatments, reporting safe dose-dependent gene transfer to the maxillary sinus with little to no cytopathic or host immune response [90]. In the midst of designing and testing new vectors, three additional single dose AAV clinical trials took place [91,92,93]. Results from the dose escalation AAV clinical trials for CF were published in 2004 and 2007 [94,95]. These trials used an AAV carrying the full length CFTR cDNA driven by the promoter activity from the AAV ITR. In total, the AAV clinical trials failed to meet their trial endpoints of detecting CFTR DNA from nasal brushings by PCR, a change in NPD, a change in specified metrics as measured by high resolution CT scan, and an improved FEV1. In general, AAV delivery was considered safe; however, the resultant levels of CFTR expression were typically below the limit of detection. Despite low CFTR expression levels, the results were encouraging because they demonstrated the safety of the vector.
A limitation of AAV for CF gene therapy is its small carrying capacity of ~4.6 kb. The AAV inverted terminal repeat (ITR) was shown to possess promoter activity and mediate stable, but low level CFTR expression in vitro [79]. The addition of an internal promoter and polyadenylation signal was necessary but exceeded the carrying capacity [96,97,98]. Deletion analysis showed that removing a portion of the R-domain shortened CFTR by nearly 300 base pairs and still retained anion channel function to a similar extent as wild-type CFTR while addressing AAV packaging constraints [99]. Additional studies to shorten other expression cassette components led to the generation of a shortened CMVie (Cytomegalovirus immediate early) promoter and polyA [100]. AAV vectors with these modifications delivered a functional CFTR and corrected the anion channel defect in vitro and the CFTR minigene complemented the intestinal defect in CFTR null mice [101].
3.2. Nonviral Vectors
As an alternative to viral vectors, new focus was directed toward a nonviral strategy for CFTR delivery. Nonviral vector delivery is an enticing approach because there are no size constraints; however, efficient delivery to cells is challenging. This challenge is most commonly addressed by formulating plasmid DNA with a cationic lipid. An important mechanistic study during this time showed that plasmids complexed with lipids traffic to vesicles for unpacking prior to reaching the nucleus [102]. Many cationic lipids tested were reported to be safe plasmid delivery vehicles for gene transfer [103,104,105]. In the first nonviral clinical trial by Caplen et al., a cationic lipid complexed with CFTR showed partial NPD correction of cAMP-mediated Cl− transport in the nasal epithelium of six people with CF, that approximated 20% of non-CF levels [106,107,108]. Hyde et al. performed a repeat administration trial with a nonviral complex delivered to the nose and reported no loss of efficacy with repeat administration [109]. Seven subsequent nonviral clinical trials all reported partial, transient expression lasting no longer than 4 weeks [110,111,112,113,114,115,116,117]. After a wave of nonviral clinical trials from 1995 to 2004, there was a lull in activity for nearly a decade to focus on improved delivery and gene expression. In perhaps the most ambitious nonviral clinical trial to date, in 2013 Alton and colleagues nebulized CFTR plasmid formulated with the cationic lipid pGM169/GL67A to the lungs of people with CF and reported a 3.7% increase in FEV1 [118,119]. However, despite these encouraging results, a nonviral approach to delivering CFTR was not enough to achieve clear phenotypic correction.
3.3. Retroviral and Lentiviral Vectors
In the late 1990s, viral vector delivery methods steadily improved and details of vector/host interactions were elucidated. For example, MuLV-based vectors only integrate into dividing cells. Keratinocyte growth factor (KGF) was found to stimulate proliferation of airway epithelial cells and improve MuLV gene transfer efficacy [120]. Additionally, ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) treatment or formulation enhanced transepithelial permeability and gene transfer with VSV-G pseudotyped MuLV [121], AAV2 [122] and Ad5 [123]. Other studies using retroviral vectors [124] reported enhanced gene transfer in mice after injury with sulfur dioxide [125]. Retrovirus studies in rabbit tracheal epithelial cells also reported persistent expression following transduction of airway epithelial cells; however, transduction only occurred in wounded trachea [126]. These were the first studies to shed light on how receptor access and cell polarity influenced the transduction efficiency.
Lentiviral vectors were welcomed as a promising therapeutic option because genomic integration could provide long-term expression [127]. Primate and non-primate lentiviral vectors quickly replaced retroviral vectors as they could effectively transduce nondividing airway cells [128]. Specifically, feline immunodeficiency virus (FIV) vectors transduced nondividing airway epithelial cells, persistently expressed transgenes of interest, and corrected the CFTR defect in vitro [129]. Further modifications to lentiviral vectors also improved gene transfer [130]. Although typically pseudotyped with vesicular stomatitis virus (VSVG), its basolateral entry preference hindered efficient airway gene transfer [129,131]. Envelope glycoproteins from respiratory syncytial virus (RSV) [131], Marburg and Ebola virus [132,133], influenza HA-M2 [134], severe acute respiratory syndrome (SARS) spike protein [135], Jaagsiekte sheep retrovirus (JSRV) [136,137], baculovirus GP64 [138], and Sendai F/HN [139,140] were evaluated for apical entry properties. Led by these studies, lentiviruses became a new interest in the field of CF gene therapy.
Many studies confirmed that primate and non-primate lentiviral vectors were promising gene therapy candidates. VSVG-pseudotyped human immunodeficiency virus (HIV)-CFTR with a lysophosphatidylcholine (LPC) pretreatment achieved persistent CFTR expression for at least 12 months in CF-null mice [141,142]. Additionally, the Sendai virus F/HN proteins were used to pseudotype a simian immunodeficiency virus (SIV)-derived lentiviral vector carrying CFTR and correct the CF defects in vitro [139,140]. A GP64 pseudotyped FIV was shown to support gene transfer in a pig model [143]. HIV-based lentiviruses also transduced marmoset lungs [144] and achieved long-term correction in CF mice [145].
3.4. Addressing Barriers to Gene Transfer
Although much progress was made in understanding viral and nonviral vector delivery methods, several barriers remained. For example, since wild-type adenovirus is a respiratory virus, it was assumed to enter airway epithelial cells from the apical surface. However, the transduction inefficiency in vivo suggested that Ad preferred basolateral entry, perhaps due to receptor-mediated entry [146]. This was supported by the finding that EGTA treatment enhanced Ad gene transfer in mouse airways [147] by disrupting tight junctions [148]. The discovery of the Coxsackievirus and Adenovirus receptor (CAR) in 1997 confirmed that the receptor for Ad2 and Ad5 was indeed localized to the basolateral surface [149]. Studies redirecting CAR to the apical surface of human airway epithelial cells using a glycosylphosphatidylinositol (GPI) linkage increased apical gene transfer [123]. This series of events exemplifies a use of vehicle formulations to overcome physical and immunological barriers to the vector.
Indeed, many vehicle formulations were evaluated for their ability to improve receptor access or reduce immunostimulation upon entry as a means to increase transduction efficiency. Viral vector formulation with polycations neutralized the negative charge of the vector surface glycoproteins, increasing transduction and transgene expression. Examples included polybrene, protamine, diethylaminoethyl (DEAE)-dextran, poly-L-lysine, and cationic lipids [150,151]. Viscoelastic gel formulations also improved apical gene transfer for more than one vector class [150,151]. Agents such as the natural airway surfactant LPC or the calcium chelator EGTA were employed to transiently disrupt epithelial tight junctions and improved VSVG-HIV entry in vivo [152]. Precipitation of Ad using calcium phosphate enhanced gene transfer by receptor-independent endocytosis [153,154,155,156]. Formulating Ad with dexamethasone to reduce inflammation [157], using polidocanol to disrupt tight junctions [158], and using a u7-peptide to improve apical binding [159] were all methods that enhanced gene transfer. Many of these vehicles and experiments have since become widely used in preclinical studies and played a major role in enhancing receptor access and gene transfer.
Additional efforts to improve transgene delivery and understand CFTR expression were explored through vector design. Selection of promoters and polyA signals with the appropriate strength might improve CFTR expression and increase anion channel expression, potentially achieving therapeutic levels of correction with few cells transduced. The PGK, EF1α, and CMV promoters were compared in context with bovine growth hormone (BgH) or SV40 poly A signals [160,161]. A novel hybrid CMV enhancer/ EF1α (termed hCEF) promoter developed for nonviral clinical trials proved efficacious [162]. Due to the size limits to the AAV packaging capacity, the development of a minimal synthetic promoter F5Tg83 also facilitated improved CFTR expression from an AAV vector [163]. The inclusion of inducible promoters regulated by transgene expression levels and modified CFTR expression cassettes were novel ways to regulate gene expression through vector design [164,165,166]. Selective transgene expression in epithelia was first documented by showing that a cytokeratin 18 promoter drives CFTR expression in airway epithelial cells [167].
Several advances in vector design helped AAV gene transfer steadily move forward. Co-delivery of AAV with proteasome inhibitors such as doxorubicin and LLnL increased expression in airway cells by preventing proteasomal degradation of the vector [168,169]. Directed evolution of AAV capsids on human airway epithelial cells led to the development of AAV2.5T capsid with improved apical transduction properties to human airway epithelia [170]. Novel AAV capsid variants derived from AAV1, 6, and 9 were generated by directed evolution to improve CFTR delivery [171]. In a dual reporter gene study in chimpanzee airways, AAV1 was shown to transduce 20-fold higher than AAV5 at 90 days post-delivery [172]. Other studies using AAV showed that incorporating peptide motifs into the AAV capsid improved gene transfer in human airway epithelial cells [173].
To circumvent the harmful immunostimulatory properties of Ad, a helper-dependent Ad (HDAd) (also known as delta-rAd or gutted Ad) was developed, expressing no viral proteins [174]. This was a strategy to reduce immune responses to viral antigens in efforts to provide a safer Ad vector option for CF gene therapy [175]. Studies comparing first generation Ad to HDAd focused on understanding the innate immune responses. For example, systemic IL-6 levels were elevated following multiple routes of vector instillation [176], however use of HDAd led to reduced levels of inflammation [177]. Interestingly, both Ad5-CFTR and HDAd5-CFTR decreased lung bacterial infections through restoration of CFTR to the airways in mice [178,179,180,181]. HDAd can be readministered to the mouse lung [182] and reduces the innate immune response [183]. These continued improvements in gene transfer vectors provide an expanded toolbox of delivery options (Figure 4).
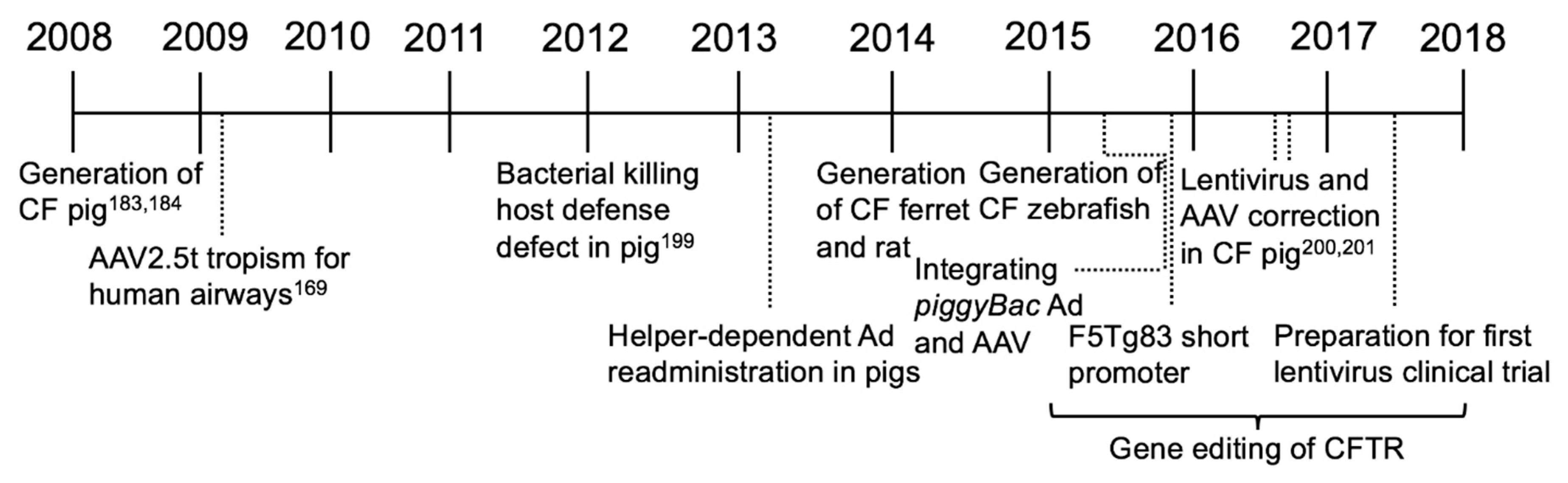
4. New Models for Preclinical Studies (2008–2018)
In this most recent era of CF gene therapy, it was evident that animal models that developed lung disease would help advance studies of disease pathogenesis and provide a preclinical model to study delivery and efficacy. Because the CF mouse does not develop spontaneous lung disease like people with CF, a model that more closely represents human lung disease would be an advancement. Studies from new models could lead to a better understanding at the basic level of the relationship between loss of CFTR and development of lung disease.
Several CF animal models are now available. In 2008, the development of a CF pig was reported [184,185]. The pig was chosen because it is a large animal model with many pulmonary, anatomical, physiological, and biochemical similarities to humans. Unlike CF mice, the CF pig recapitulates several features of human lung disease [185,186]. Development of this model has changed the way we think about CF gene therapy. One limitation of the CF pig was the 100% penetrance of meconium ileus that required surgical intervention for long term survival. Creating a “gut corrected” CF pig expressing CFTR in the gastrointestinal tract by the fatty acid binding protein (FABP) promoter alleviated meconium ileus, making longitudinal studies more feasible [187].
A CF ferret first reported in 2008 is another large animal model with a lung disease phenotype [188,189]. CFTR-knockout ferrets recapitulated many disease similarities to humans including defective airway Cl− transport, meconium ileus, and the spontaneous development of bacterial lung infections [190]. Studies in non-CF ferrets show that rAAV2/1 and lentiviral vectors facilitate gene transfer in newborn airways, and thus hold promise as vector platforms for CF studies. In addition, a novel parvovirus vector derived from human bocavirus transduces ferret airways [191].
Other newer CF animal models include the rat [192,193], zebrafish [194], and sheep [195]. The CFTR null rat was created in 2014 using zinc finger nucleases (ZFNs) for gene targeting and exhibits an NPD and bioelectric properties similar to humans. Additionally, histological abnormalities in the ileum parallel intestinal complications seen in other animal models. Efforts to characterize a CF rabbit model are ongoing [196]. Novel CF mouse models have been generated to study specific CFTR mutations such as the premature stop codon G542X [197] and gating effects caused by R117H [198]. In addition, a conditional null allele for Cftr in mice was generated [199]. Advancements in animal models have provided the essential insight into underlying causes of CF.
An important advance in understanding CF disease pathogenesis was elucidated using the CF pig by Pezzulo et al. in 2012. Here, they provided evidence that a reduced airway surface liquid pH decreased the bacterial killing ability in newborn CF pigs [200]. From these studies, relevant assays were devised to measure CFTR correction in vivo. These assays were subsequently used to show that delivery of CFTR by FIV [201], AAV [202], and Ad [203] vectors to CF pig airways partially restored anion channel activity, increased the airway surface liquid (ASL) pH, and improved bacterial killing ability. Further understanding of CF host defenses was applied from these studies to define relationships between CFTR expression and Cl− and HCO3− transport that have important implications for CF gene therapies [204].
Gene Editing and the Era of Precision Medicine
As a complementary approach to gene addition, the ability to enzymatically modify nucleic acids sequences has advanced a new field of study in the CF research community. Gene editing tools include ZFNs [205], TALENs [206], meganucleases [207], and CRISPR/Cas9 [208]. Zinc finger nucleases were early gene editing tools; however, the ease of CRISPR/Cas9 engineering and guide RNA production allowed broader access of genome modification tools. Gene editing holds the promise of repairing the endogenous CFTR gene; restoring CFTR function, for repair of individual mutation, insertion, or partial or full-length cDNA to “safe-site” targeted integration. However, in vivo gene repair faces obstacles that extend beyond those associated with “simple” gene delivery. For each CFTR mutation, the efficiency of introducing double stranded breaks with different guide RNAs and the efficiency of homologous recombination with different repair templates will need to be assessed. CFTR gene editing by both ZFNs [209,210], TALEN [211], and CRISPR/Cas9 [212] methods have been evaluated in vitro. Importantly, both gene addition and gene repair strategies rely on efficient in vivo gene delivery. Interests include both delivery to respiratory epithelia, and systemic delivery strategies that might correct CF defects in multiple affected organs.
Whether the goal is gene addition or gene repair, for gene therapy to be successful, the vector selected must either integrate into a basal cell or other progenitor cell types or be readministered. While AAV, Ad, and HDAd are generally considered to be non-integrating, development of a hybrid nonviral transposon/viral integrating vector system led to persistent expression in mice for at least a year. Previously, the DNA transposon piggyBac has been shown to promote persistent gene transfer in mice [213]. In this system, the piggyBac/AAV and piggyBac/Ad vector system confers persistent expression in mouse airways only in the presence of the transposase [214]. A recent study in the pig model documents whole lung distribution by a piggyBac/Ad vector and phenotypic correction of a CF pig model [203]. Promising work evaluating HDAd suggests that this vector efficiently transduces airway epithelia [215], including submucosal glands [216], and can be readministered with use of an immunosuppressant [217,218]. Perhaps an integrating piggyBac/HDAd could provide long-term, efficient correction and be readministered if necessary.
Advances in gene therapy from other genetic disease continue to inform the field. In December 2017, the FDA approved voretigene neparvovec-rzyl (Luxturna) as a gene therapy treatment for individuals with inherited retinal diseases such as retinitis pigmentosa and Leber’s congential amaurosis. For this treatment, an AAV2 vector carrying RPE65 is delivered by injection into the subretinal space [219]. Individuals who received this treatment exhibited increased full-field light sensitivity and improved fMRI results [220]. Exciting advancements in CAR-T therapy for cancer treatments include tisagenlecleucel (Kymriah) [221] and axicabtagene ciloleucel (Yescarta) [222]. For both, anti-CD19 chimeric antigen receptors are delivered by lentiviral vectors to autologous T cells, with some patients achieving complete remission. Gene therapies approved for use in Europe include Alofisel, Spherox, Zalmoxis, Stimvelis, Imlygic, and Holoclar (European Medicines Agency). The steadily growing list of clinically approved gene therapy therapeutics suggests that we may be entering a new era where gene therapy is a commonplace option.
Currently, there are exciting new developments in CF gene therapy. Nanoparticle delivery chemistries with enhanced tropism are emerging [223] and CF clinical trials using lentiviral vectors are currently being pursued [224]. The major breakthroughs in gene therapy for other genetic diseases is fueling new interest in CF. The field of gene therapy has learned valuable lessons from the many viral and nonviral clinical trials to date and has developed new vectors that can efficiently and persistently correct the CFTR anion channel defect. New animal models that recapitulate features of human lung disease allow preclinical studies to refine delivery protocols and measure novel metrics of phenotypic correction. The early surge in enthusiasm to develop a gene therapy treatment for CF fell to the realization that delivering CFTR to the lung was more complex than expected. Improvements in vector design and delivery methods, understanding lung biology and disease development, and the ability to use new animal models are all reenergizing the gene therapy field and adding to the momentum that we hope will lead to the generation of an effective therapeutic reagent for CF.
Author Contributions
Writing—Original Draft Preparation, A.L.C. Writing—Review & Editing, A.L.C., P.L.S. and P.B.M.J.
Funding
This work was supported by the National Institutes of Health [NIH P01 HL-51670, NIH P01 HL-091842, NIH R01 HL-133089, NIH R01 HL-105821]; the Center for Gene Therapy of Cystic Fibrosis [NIH P30 DK-054759]; and the Cystic Fibrosis Foundation [SINN15XX0]. Funding for open access charge: National Institutes of Health. Additional support by the Roy J. Carver Endowed Chair in Lung Research.
Acknowledgments
We thank Miguel Ortiz, Laura Marquez Loza, Cami Hippee, and Lynda Ostedgaard for their critical review of this manuscript.
Conflicts of Interest
P.B.M.J. is a founder of and holds equity in Talee Bio. The other authors have no conflicts of interests to declare.
References
- Condren, M.E.; Bradshaw, M.D. Ivacaftor: A novel gene-based therapeutic approach for cystic fibrosis. J. Pediatr. Pharmacol. Ther. 2013, 18, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala, M.A.; Jain, M. Tezacaftor for the treatment of cystic fibrosis. Expert Rev. Respir. Med. 2018, 12, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e37–e38. [Google Scholar] [CrossRef]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- VanDevanter, D.R.; Konstan, M.W. Outcome measures for clinical trials assessing treatment of cystic fibrosis lung disease. Clin. Investig. 2012, 2, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Drumm, M.L.; Pope, H.A.; Cliff, W.H.; Rommens, J.M.; Marvin, S.A.; Tsui, L.C.; Collins, F.S.; Frizzell, R.A.; Wilson, J.M. Correction of the cystic fibrosis defect in vitro by retrovirus-mediated gene transfer. Cell 1990, 62, 1227–1233. [Google Scholar] [CrossRef]
- Rich, D.P.; Anderson, M.P.; Gregory, R.J.; Cheng, S.H.; Paul, S.; Jefferson, D.M.; McCann, J.D.; Klinger, K.W.; Smith, A.E.; Welsh, M.J. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature 1990, 347, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Solow, R.; Owens, R.A.; Afione, S.; Zeitlin, P.L.; Carter, B.J. Gene expression from adeno-associated virus vectors in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 1992, 7, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.A.; Yoshimura, K.; Trapnell, B.C.; Yoneyama, K.; Rosenthal, E.R.; Dalemans, W.; Fukayama, M.; Bargon, J.; Stier, L.E.; Stratford-Perricaudet, L.; et al. In vivo transfer of the human cystic fibrosis transmembrane conductance regulator gene to the airway epithelium. Cell 1992, 68, 143–155. [Google Scholar] [CrossRef]
- Yoshimura, K.; Rosenfeld, M.A.; Nakamura, H.; Scherer, E.M.; Pavirani, A.; Lecocq, J.P.; Crystal, R.G. Expression of the human cystic fibrosis transmembrane conductance regulator gene in the mouse lung after in vivo intratracheal plasmid-mediated gene transfer. Nucleic Acids Res. 1992, 20, 3233–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, J.C.; Johnson, L.G.; Stutts, M.J.; Sarkadi, B.; Yankaskas, J.R.; Swanstrom, R.; Boucher, R.C. Correction of the apical membrane chloride permeability defect in polarized cystic fibrosis airway epithelia following retroviral-mediated gene transfer. Hum. Gene Ther. 1992, 3, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.G.; Olsen, J.C.; Sarkadi, B.; Moore, K.L.; Swanstrom, R.; Boucher, R.C. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat. Genet. 1992, 2, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Schoumacher, R.A.; Ram, J.; Iannuzzi, M.C.; Bradbury, N.A.; Wallace, R.W.; Hon, C.T.; Kelly, D.R.; Schmid, S.M.; Gelder, F.B.; Rado, T.A.; et al. A cystic fibrosis pancreatic adenocarcinoma cell line. Proc. Natl. Acad. Sci. USA 1990, 87, 4012–4016. [Google Scholar] [CrossRef] [PubMed]
- Cozens, A.L.; Yezzi, M.J.; Kunzelmann, K.; Ohrui, T.; Chin, L.; Eng, K.; Finkbeiner, W.E.; Widdicombe, J.H.; Gruenert, D.C. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1994, 10, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Schwiebert, E.M.; Zeitlin, P.L.; Kuo, W.L.; Stanton, B.A.; Gruenert, D.C. An immortalized cystic fibrosis tracheal epithelial cell line homozygous for the delta F508 CFTR mutation. Am. J. Respir. Cell Mol. Biol. 1993, 8, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, P.L.; Lu, L.; Rhim, J.; Cutting, G.; Stetten, G.; Kieffer, K.A.; Craig, R.; Guggino, W.B. A cystic fibrosis bronchial epithelial cell line: Immortalization by adeno-12-SV40 infection. Am. J. Respir. Cell Mol. Biol. 1991, 4, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Dorin, J.R.; Dickinson, P.; Emslie, E.; Clarke, A.R.; Dobbie, L.; Hooper, M.L.; Halford, S.; Wainwright, B.J.; Porteous, D.J. Successful targeting of the mouse cystic fibrosis transmembrane conductance regulator gene in embryonal stem cells. Transgenic Res. 1992, 1, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Dorin, J.R.; Dickinson, P.; Alton, E.W.; Smith, S.N.; Geddes, D.M.; Stevenson, B.J.; Kimber, W.L.; Fleming, S.; Clarke, A.R.; Hooper, M.L.; et al. Cystic fibrosis in the mouse by targeted insertional mutagenesis. Nature 1992, 359, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Dorin, J.R.; Porteous, D.J. Cystic fibrosis mice with disease-related changes in lung and reproductive tract. Lancet 1992, 340, 984. [Google Scholar] [CrossRef]
- Ratcliff, R.; Evans, M.J.; Doran, J.; Wainwright, B.J.; Williamson, R.; Colledge, W.H. Disruption of the cystic fibrosis transmembrane conductance regulator gene in embryonic stem cells by gene targeting. Transgenic Res. 1992, 1, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Snouwaert, J.N.; Brigman, K.K.; Latour, A.M.; Malouf, N.N.; Boucher, R.C.; Smithies, O.; Koller, B.H. An animal model for cystic fibrosis made by gene targeting. Science 1992, 257, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Colledge, W.H.; Ratcliff, R.; Foster, D.; Williamson, R.; Evans, M.J. Cystic fibrosis mouse with intestinal obstruction. Lancet 1992, 340, 680. [Google Scholar] [CrossRef]
- Dorin, J.R.; Farley, R.; Webb, S.; Smith, S.N.; Farini, E.; Delaney, S.J.; Wainwright, B.J.; Alton, E.W.; Porteous, D.J. A demonstration using mouse models that successful gene therapy for cystic fibrosis requires only partial gene correction. Gene Ther. 1996, 3, 797–801. [Google Scholar] [PubMed]
- Mastrangeli, A.; Danel, C.; Rosenfeld, M.A.; Stratford-Perricaudet, L.; Perricaudet, M.; Pavirani, A.; Lecocq, J.P.; Crystal, R.G. Diversity of airway epithelial cell targets for in vivo recombinant adenovirus-mediated gene transfer. J. Clin. Investig. 1993, 91, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Pilewski, J.M.; Engelhardt, J.F.; Bavaria, J.E.; Kaiser, L.R.; Wilson, J.M.; Albelda, S.M. Adenovirus-mediated gene transfer to human bronchial submucosal glands using xenografts. Am. J. Physiol. 1995, 268, L657–L665. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Petersen, D.M.; Puga, A.P.; Graham, S.M.; Couture, L.A.; Keyes, L.D.; Lukason, M.J.; St George, J.A.; Gregory, R.J.; Smith, A.E.; et al. Safety and efficacy of repetitive adenovirus-mediated transfer of CFTR cDNA to airway epithelia of primates and cotton rats. Nat. Genet. 1994, 6, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Yei, S.; Mittereder, N.; Tang, K.; O’Sullivan, C.; Trapnell, B.C. Adenovirus-mediated gene transfer for cystic fibrosis: Quantitative evaluation of repeated in vivo vector administration to the lung. Gene Ther. 1994, 1, 192–200. [Google Scholar] [PubMed]
- St George, J.A.; Pennington, S.E.; Kaplan, J.M.; Peterson, P.A.; Kleine, L.J.; Smith, A.E.; Wadsworth, S.C. Biological response of nonhuman primates to long-term repeated lung exposure to Ad2/CFTR-2. Gene Ther. 1996, 3, 103–116. [Google Scholar] [PubMed]
- Kaplan, J.M.; St George, J.A.; Pennington, S.E.; Keyes, L.D.; Johnson, R.P.; Wadsworth, S.C.; Smith, A.E. Humoral and cellular immune responses of nonhuman primates to long-term repeated lung exposure to Ad2/CFTR-2. Gene Ther. 1996, 3, 117–127. [Google Scholar] [PubMed]
- Mittereder, N.; Yei, S.; Bachurski, C.; Cuppoletti, J.; Whitsett, J.A.; Tolstoshev, P.; Trapnell, B.C. Evaluation of the efficacy and safety of in vitro, adenovirus-mediated transfer of the human cystic fibrosis transmembrane conductance regulator cDNA. Hum. Gene Ther. 1994, 5, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Yei, S.; Mittereder, N.; Wert, S.; Whitsett, J.A.; Wilmott, R.W.; Trapnell, B.C. In vivo evaluation of the safety of adenovirus-mediated transfer of the human cystic fibrosis transmembrane conductance regulator cDNA to the lung. Hum. Gene Ther. 1994, 5, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Nunes, F.A.; Berencsi, K.; Gonczol, E.; Engelhardt, J.F.; Wilson, J.M. Inactivation of E2a in recombinant adenoviruses improves the prospect for gene therapy in cystic fibrosis. Nat. Genet. 1994, 7, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.F.; Litzky, L.; Wilson, J.M. Prolonged transgene expression in cotton rat lung with recombinant adenoviruses defective in E2a. Hum. Gene Ther. 1994, 5, 1217–1229. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.F.; Ye, X.; Doranz, B.; Wilson, J.M. Ablation of E2A in recombinant adenoviruses improves transgene persistence and decreases inflammatory response in mouse liver. Proc. Natl. Acad. Sci. USA 1994, 91, 6196–6200. [Google Scholar] [CrossRef] [PubMed]
- Bout, A.; Imler, J.L.; Schultz, H.; Perricaudet, M.; Zurcher, C.; Herbrink, P.; Valerio, D.; Pavirani, A. In vivo adenovirus-mediated transfer of human CFTR cDNA to rhesus monkey airway epithelium: Efficacy, toxicity and safety. Gene Ther. 1994, 1, 385–394. [Google Scholar] [PubMed]
- McDonald, R.J.; Lukason, M.J.; Raabe, O.G.; Canfield, D.R.; Burr, E.A.; Kaplan, J.M.; Wadsworth, S.C.; St George, J.A. Safety of airway gene transfer with Ad2/CFTR2: Aerosol administration in the nonhuman primate. Hum. Gene Ther. 1997, 8, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Couture, L.A.; Gregory, R.J.; Graham, S.M.; Smith, A.E.; Welsh, M.J. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell 1993, 75, 207–216. [Google Scholar] [CrossRef]
- Rich, D.P.; Couture, L.A.; Cardoza, L.M.; Guiggio, V.M.; Armentano, D.; Espino, P.C.; Hehir, K.; Welsh, M.J.; Smith, A.E.; Gregory, R.J. Development and analysis of recombinant adenoviruses for gene therapy of cystic fibrosis. Hum. Gene Ther. 1993, 4, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G.; McElvaney, N.G.; Rosenfeld, M.A.; Chu, C.S.; Mastrangeli, A.; Hay, J.G.; Brody, S.L.; Jaffe, H.A.; Eissa, N.T.; Danel, C. Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat. Genet. 1994, 8, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Boucher, R.C.; Knowles, M.R.; Johnson, L.G.; Olsen, J.C.; Pickles, R.; Wilson, J.M.; Engelhardt, J.; Yang, Y.; Grossman, M. Gene Therapy for cystic fibrosis using E1-deleted adenovirus: A phase I trial in the nasal cavity. The University of North Carolina at Chapel Hill. Hum. Gene Ther. 1994, 5, 615–639. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Hohneker, K.W.; Zhou, Z.; Olsen, J.C.; Noah, T.L.; Hu, P.C.; Leigh, M.W.; Engelhardt, J.F.; Edwards, L.J.; Jones, K.R.; et al. A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N. Engl. J. Med. 1995, 333, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Engelhardt, J.F.; Grossman, M.; Simon, R.H.; Yang, Y. Gene Therapy of cystic fibrosis lung disease using E1 deleted adenoviruses: A phase I trial. Hum. Gene Ther. 1994, 5, 501–519. [Google Scholar] [CrossRef] [PubMed]
- Hay, J.G.; McElvaney, N.G.; Herena, J.; Crystal, R.G. Modification of nasal epithelial potential differences of individuals with cystic fibrosis consequent to local administration of a normal CFTR cDNA adenovirus gene transfer vector. Hum. Gene Ther. 1995, 6, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G.; Jaffe, A.; Brody, S.; Mastrangeli, A.; McElvaney, N.G.; Rosenfeld, M.; Chu, C.S.; Danel, C.; Hay, J.; Eissa, T. A phase 1 study, in cystic fibrosis patients, of the safety, toxicity, and biological efficacy of a single administration of a replication deficient, recombinant adenovirus carrying the cDNA of the normal cystic fibrosis transmembrane conductance regulator gene in the lung. Hum. Gene Ther. 1995, 6, 643–666. [Google Scholar] [PubMed]
- Zabner, J.; Couture, L.A.; Smith, A.E.; Welsh, M.J. Correction of cAMP-stimulated fluid secretion in cystic fibrosis airway epithelia: Efficiency of adenovirus-mediated gene transfer in vitro. Hum. Gene Ther. 1994, 5, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.A.; Chu, C.S.; Seth, P.; Danel, C.; Banks, T.; Yoneyama, K.; Yoshimura, K.; Crystal, R.G. Gene transfer to freshly isolated human respiratory epithelial cells in vitro using a replication-deficient adenovirus containing the human cystic fibrosis transmembrane conductance regulator cDNA. Hum. Gene Ther. 1994, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.G.; Boyles, S.E.; Wilson, J.; Boucher, R.C. Normalization of raised sodium absorption and raised calcium-mediated chloride secretion by adenovirus-mediated expression of cystic fibrosis transmembrane conductance regulator in primary human cystic fibrosis airway epithelial cells. J. Clin. Investig. 1995, 95, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.H.; Engelhardt, J.F.; Yang, Y.; Zepeda, M.; Weber-Pendleton, S.; Grossman, M.; Wilson, J.M. Adenovirus-mediated transfer of the CFTR gene to lung of nonhuman primates: Toxicity study. Hum. Gene Ther. 1993, 4, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.F.; Simon, R.H.; Yang, Y.; Zepeda, M.; Weber-Pendleton, S.; Doranz, B.; Grossman, M.; Wilson, J.M. Adenovirus-mediated transfer of the CFTR gene to lung of nonhuman primates: Biological efficacy study. Hum. Gene Ther. 1993, 4, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Su, Q.; Wilson, J.M. Role of viral antigens in destructive cellular immune responses to adenovirus vector-transduced cells in mouse lungs. J. Virol. 1996, 70, 7209–7212. [Google Scholar] [PubMed]
- Eissa, N.T.; Chu, C.S.; Danel, C.; Crystal, R.G. Evaluation of the respiratory epithelium of normals and individuals with cystic fibrosis for the presence of adenovirus E1a sequences relevant to the use of E1a-adenovirus vectors for gene therapy for the respiratory manifestations of cystic fibrosis. Hum. Gene Ther. 1994, 5, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xiang, Z.; Ertl, H.C.; Wilson, J.M. Upregulation of class I major histocompatibility complex antigens by interferon gamma is necessary for T-cell-mediated elimination of recombinant adenovirus-infected hepatocytes in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 7257–7261. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Trinchieri, G.; Wilson, J.M. Recombinant IL-12 prevents formation of blocking IgA antibodies to recombinant adenovirus and allows repeated gene therapy to mouse lung. Nat. Med. 1995, 1, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Scaria, A.; St George, J.A.; Gregory, R.J.; Noelle, R.J.; Wadsworth, S.C.; Smith, A.E.; Kaplan, J.M. Antibody to CD40 ligand inhibits both humoral and cellular immune responses to adenoviral vectors and facilitates repeated administration to mouse airway. Gene Ther. 1997, 4, 611–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirmule, N.; Truneh, A.; Haecker, S.E.; Tazelaar, J.; Gao, G.; Raper, S.E.; Hughes, J.V.; Wilson, J.M. Repeated administration of adenoviral vectors in lungs of human CD4 transgenic mice treated with a nondepleting CD4 antibody. J. Immunol. 1999, 163, 448–455. [Google Scholar] [PubMed]
- Welsh, M.J.; Smith, A.E.; Zabner, J.; Rich, D.P.; Graham, S.M.; Gregory, R.J.; Pratt, B.M.; Moscicki, R.A. Cystic fibrosis gene therapy using an adenovirus vector: In vivo safety and efficacy in nasal epithelium. Hum. Gene Ther. 1994, 5, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Welsh, M.J.; Zabner, J.; Graham, S.M.; Smith, A.E.; Moscicki, R.; Wadsworth, S. Adenovirus-mediated gene transfer for cystic fibrosis: Part A. Safety of dose and repeat administration in the nasal epithelium. Part B. Clinical efficacy in the maxillary sinus. Hum. Gene Ther. 1995, 6, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Ramsey, B.W.; Meeker, D.P.; Aitken, M.L.; Balfour, R.P.; Gibson, R.L.; Launspach, J.; Moscicki, R.A.; Richards, S.M.; Standaert, T.A.; et al. Repeat administration of an adenovirus vector encoding cystic fibrosis transmembrane conductance regulator to the nasal epithelium of patients with cystic fibrosis. J. Clin. Investig. 1996, 97, 1504–1511. [Google Scholar] [CrossRef] [PubMed]
- Bellon, G.; Calmard, L.; Thouvenot, D.; Levrey, H.; Jagneaux, V.; Poitevin, F.; Malcus, C.; Accart, N.; Sene, C.; Layani, M.P.; et al. Aerosol administration of a replication defective recombinant adenovirus expressing normal human cDNA-CFTR in the respiratory tractus in patients with cystic fibrosis. C R Seances Soc. Biol. Fil. 1996, 190, 109–142. [Google Scholar] [PubMed]
- Bellon, G.; Michel-Calemard, L.; Thouvenot, D.; Jagneaux, V.; Poitevin, F.; Malcus, C.; Accart, N.; Layani, M.P.; Aymard, M.; Bernon, H.; et al. Aerosol administration of a recombinant adenovirus expressing CFTR to cystic fibrosis patients: A phase I clinical trial. Hum. Gene Ther. 1997, 8, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.G.; Leopold, P.L.; Hackett, N.R.; Grasso, T.M.; Williams, P.M.; Tucker, A.L.; Kaner, R.J.; Ferris, B.; Gonda, I.; Sweeney, T.D.; et al. Airway epithelial CFTR mRNA expression in cystic fibrosis patients after repetitive administration of a recombinant adenovirus. J. Clin. Investig. 1999, 104, 1245–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuckerman, J.B.; Robinson, C.B.; McCoy, K.S.; Shell, R.; Sferra, T.J.; Chirmule, N.; Magosin, S.A.; Propert, K.J.; Brown-Parr, E.C.; Hughes, J.V.; et al. A phase I study of adenovirus-mediated transfer of the human cystic fibrosis transmembrane conductance regulator gene to a lung segment of individuals with cystic fibrosis. Hum. Gene Ther. 1999, 10, 2973–2985. [Google Scholar] [CrossRef] [PubMed]
- Lehrman, S. Virus treatment questioned after gene therapy death. Nature 1999, 401, 517–518. [Google Scholar] [CrossRef] [PubMed]
- Marshall, E. Gene therapy death prompts review of adenovirus vector. Science 1999, 286, 2244–2245. [Google Scholar] [CrossRef] [PubMed]
- Raper, S.E.; Wilson, J.M.; Yudkoff, M.; Robinson, M.B.; Ye, X.; Batshaw, M.L. Developing adenoviral-mediated in vivo gene therapy for ornithine transcarbamylase deficiency. J. Inherit. Metab. Dis. 1998, 21 (Suppl. 1), 119–137. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.M.; O’Sullivan, B.P.; Lapey, A.; Dorkin, H.; Oren, J.; Balfour, R.; Perricone, M.A.; Rosenberg, M.; Wadsworth, S.C.; Smith, A.E.; et al. Aerosol and lobar administration of a recombinant adenovirus to individuals with cystic fibrosis. I. Methods, safety, and clinical implications. Hum. Gene Ther. 2001, 12, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Perricone, M.A.; Morris, J.E.; Pavelka, K.; Plog, M.S.; O’Sullivan, B.P.; Joseph, P.M.; Dorkin, H.; Lapey, A.; Balfour, R.; Meeker, D.P.; et al. Aerosol and lobar administration of a recombinant adenovirus to individuals with cystic fibrosis. II. Transfection efficiency in airway epithelium. Hum. Gene Ther. 2001, 12, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Wendtner, C.M. Recombinant adeno-associated virus (rAAV) vectors for somatic gene therapy: Recent advances and potential clinical applications. Cytokines Mol. Ther. 1996, 2, 69–79. [Google Scholar] [PubMed]
- Kearns, W.G.; Afione, S.A.; Fulmer, S.B.; Pang, M.C.; Erikson, D.; Egan, M.; Landrum, M.J.; Flotte, T.R.; Cutting, G.R. Recombinant adeno-associated virus (AAV-CFTR) vectors do not integrate in a site-specific fashion in an immortalized epithelial cell line. Gene Ther. 1996, 3, 748–755. [Google Scholar] [PubMed]
- Afione, S.A.; Conrad, C.K.; Kearns, W.G.; Chunduru, S.; Adams, R.; Reynolds, T.C.; Guggino, W.B.; Cutting, G.R.; Carter, B.J.; Flotte, T.R. In vivo model of adeno-associated virus vector persistence and rescue. J. Virol. 1996, 70, 3235–3241. [Google Scholar] [PubMed]
- Conrad, C.K.; Allen, S.S.; Afione, S.A.; Reynolds, T.C.; Beck, S.E.; Fee-Maki, M.; Barrazza-Ortiz, X.; Adams, R.; Askin, F.B.; Carter, B.J.; et al. Safety of single-dose administration of an adeno-associated virus (AAV)-CFTR vector in the primate lung. Gene Ther. 1996, 3, 658–668. [Google Scholar] [PubMed]
- Zeitlin, P.L.; Chu, S.; Conrad, C.; McVeigh, U.; Ferguson, K.; Flotte, T.R.; Guggino, W.B. Alveolar stem cell transduction by an adeno-associated viral vector. Gene Ther. 1995, 2, 623–631. [Google Scholar] [PubMed]
- Halbert, C.L.; Standaert, T.A.; Aitken, M.L.; Alexander, I.E.; Russell, D.W.; Miller, A.D. Transduction by adeno-associated virus vectors in the rabbit airway: Efficiency, persistence, and readministration. J. Virol. 1997, 71, 5932–5941. [Google Scholar] [PubMed]
- Flotte, T.R.; Afione, S.A.; Conrad, C.; McGrath, S.A.; Solow, R.; Oka, H.; Zeitlin, P.L.; Guggino, W.B.; Carter, B.J. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc. Natl. Acad. Sci. USA 1993, 90, 10613–10617. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Afione, S.A.; Solow, R.; Drumm, M.L.; Markakis, D.; Guggino, W.B.; Zeitlin, P.L.; Carter, B.J. Expression of the cystic fibrosis transmembrane conductance regulator from a novel adeno-associated virus promoter. J. Biol. Chem. 1993, 268, 3781–3790. [Google Scholar] [PubMed]
- Rubenstein, R.C.; McVeigh, U.; Flotte, T.R.; Guggino, W.B.; Zeitlin, P.L. CFTR gene transduction in neonatal rabbits using an adeno-associated virus (AAV) vector. Gene Ther. 1997, 4, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Standaert, T.A.; Wilson, C.B.; Miller, A.D. Successful readministration of adeno-associated virus vectors to the mouse lung requires transient immunosuppression during the initial exposure. J. Virol. 1998, 72, 9795–9805. [Google Scholar] [PubMed]
- Fischer, A.C.; Beck, S.E.; Smith, C.I.; Laube, B.L.; Askin, F.B.; Guggino, S.E.; Adams, R.J.; Flotte, T.R.; Guggino, W.B. Successful transgene expression with serial doses of aerosolized rAAV2 vectors in rhesus macaques. Mol. Ther. 2003, 8, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.C.; Smith, C.I.; Cebotaru, L.; Zhang, X.; Askin, F.B.; Wright, J.; Guggino, S.E.; Adams, R.J.; Flotte, T.; Guggino, W.B. Expression of a truncated cystic fibrosis transmembrane conductance regulator with an AAV5-pseudotyped vector in primates. Mol. Ther. 2007, 15, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Limberis, M.P.; Wilson, J.M. Adeno-associated virus serotype 9 vectors transduce murine alveolar and nasal epithelia and can be readministered. Proc. Natl. Acad. Sci. USA 2006, 103, 12993–12998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Luo, M.; Guo, C.; Yan, Z.; Wang, Y.; Engelhardt, J.F. Comparative biology of rAAV transduction in ferret, pig and human airway epithelia. Gene Ther. 2007, 14, 1543–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Luo, M.; Zhang, L.N.; Yan, Z.; Zak, R.; Ding, W.; Mansfield, S.G.; Mitchell, L.G.; Engelhardt, J.F. Spliceosome-mediated RNA trans-splicing with recombinant adeno-associated virus partially restores cystic fibrosis transmembrane conductance regulator function to polarized human cystic fibrosis airway epithelial cells. Hum. Gene Ther. 2005, 16, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yan, Z.; Luo, M.; Engelhardt, J.F. Species-specific differences in mouse and human airway epithelial biology of recombinant adeno-associated virus transduction. Am. J. Respir. Cell Mol. Biol. 2006, 34, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Lam, S.L.; Miller, A.D. High-efficiency promoter-dependent transduction by adeno-associated virus type 6 vectors in mouse lung. Hum. Gene Ther. 2007, 18, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Schwiebert, E.M.; Zeitlin, P.L.; Carter, B.J.; Guggino, W.B. Correlation between DNA transfer and cystic fibrosis airway epithelial cell correction after recombinant adeno-associated virus serotype 2 gene therapy. Hum. Gene Ther. 2005, 16, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Messner, A.H.; Moran, M.L.; Daifuku, R.; Kouyama, K.; Desch, J.K.; Manley, S.; Norbash, A.M.; Conrad, C.K.; Friborg, S.; et al. Safety and biological efficacy of an adeno-associated virus vector-cystic fibrosis transmembrane regulator (AAV-CFTR) in the cystic fibrosis maxillary sinus. Laryngoscope 1999, 109, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Nepomuceno, I.B.; Messner, A.H.; Moran, M.L.; Batson, E.P.; Dimiceli, S.; Brown, B.W.; Desch, J.K.; Norbash, A.M.; Conrad, C.K.; et al. A phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Hum. Gene Ther. 2002, 13, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Zeitlin, P.L.; Reynolds, T.C.; Heald, A.E.; Pedersen, P.; Beck, S.; Conrad, C.K.; Brass-Ernst, L.; Humphries, M.; Sullivan, K.; et al. Phase I trial of intranasal and endobronchial administration of a recombinant adeno-associated virus serotype 2 (rAAV2)-CFTR vector in adult cystic fibrosis patients: A two-part clinical study. Hum. Gene Ther. 2003, 14, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Aitken, M.L.; Moss, R.B.; Waltz, D.A.; Dovey, M.E.; Tonelli, M.R.; McNamara, S.C.; Gibson, R.L.; Ramsey, B.W.; Carter, B.J.; Reynolds, T.C. A phase I study of aerosolized administration of tgAAVCF to cystic fibrosis subjects with mild lung disease. Hum. Gene Ther. 2001, 12, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B.; Milla, C.; Colombo, J.; Accurso, F.; Zeitlin, P.L.; Clancy, J.P.; Spencer, L.T.; Pilewski, J.; Waltz, D.A.; Dorkin, H.L.; et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: A randomized placebo-controlled phase 2B trial. Hum. Gene Ther. 2007, 18, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B.; Rodman, D.; Spencer, L.T.; Aitken, M.L.; Zeitlin, P.L.; Waltz, D.; Milla, C.; Brody, A.S.; Clancy, J.P.; Ramsey, B.; et al. Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regulator gene transfer to the lungs of patients with cystic fibrosis: A multicenter, double-blind, placebo-controlled trial. Chest 2004, 125, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.Y.; Fan, P.D.; Frizzell, R.A. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 1996, 7, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, D.; Fischer, H.; Fan, P.D.; Widdicombe, J.H.; Kan, Y.W.; Dong, J.Y. Efficient expression of CFTR function with adeno-associated virus vectors that carry shortened CFTR genes. Proc. Natl. Acad. Sci. USA 1998, 95, 10158–10163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Fischer, H.; Zhang, L.; Fan, P.; Ding, R.X.; Dong, J. Efficient CFTR expression from AAV vectors packaged with promoters—The second generation. Gene Ther. 1999, 6, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Ostedgaard, L.S.; Zabner, J.; Vermeer, D.W.; Rokhlina, T.; Karp, P.H.; Stecenko, A.A.; Randak, C.; Welsh, M.J. CFTR with a partially deleted R domain corrects the cystic fibrosis chloride transport defect in human airway epithelia in vitro and in mouse nasal mucosa in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 3093–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostedgaard, L.S.; Rokhlina, T.; Karp, P.H.; Lashmit, P.; Afione, S.; Schmidt, M.; Zabner, J.; Stinski, M.F.; Chiorini, J.A.; Welsh, M.J. A shortened adeno-associated virus expression cassette for CFTR gene transfer to cystic fibrosis airway epithelia. Proc. Natl. Acad. Sci. USA 2005, 102, 2952–2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostedgaard, L.S.; Meyerholz, D.K.; Vermeer, D.W.; Karp, P.H.; Schneider, L.; Sigmund, C.D.; Welsh, M.J. Cystic fibrosis transmembrane conductance regulator with a shortened R domain rescues the intestinal phenotype of CFTR−/− mice. Proc. Natl. Acad. Sci. USA 2011, 108, 2921–2926. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Fasbender, A.J.; Moninger, T.; Poellinger, K.A.; Welsh, M.J. Cellular and molecular barriers to gene transfer by a cationic lipid. J. Biol. Chem. 1995, 270, 18997–19007. [Google Scholar] [CrossRef] [PubMed]
- Kollen, W.J.; Midoux, P.; Erbacher, P.; Yip, A.; Roche, A.C.; Monsigny, M.; Glick, M.C.; Scanlin, T.F. Gluconoylated and glycosylated polylysines as vectors for gene transfer into cystic fibrosis airway epithelial cells. Hum. Gene Ther. 1996, 7, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- McDonald, R.J.; Liggitt, H.D.; Roche, L.; Nguyen, H.T.; Pearlman, R.; Raabe, O.G.; Bussey, L.B.; Gorman, C.M. Aerosol delivery of lipid:DNA complexes to lungs of rhesus monkeys. Pharm. Res. 1998, 15, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Fasbender, A.; Marshall, J.; Moninger, T.O.; Grunst, T.; Cheng, S.; Welsh, M.J. Effect of co-lipids in enhancing cationic lipid-mediated gene transfer in vitro and in vivo. Gene Ther. 1997, 4, 716–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, P.G.; Geddes, D.M.; Alton, E.W. Protocols for in vivo measurement of the ion transport defects in cystic fibrosis nasal epithelium. Eur. Respir. J. 1994, 7, 2050–2056. [Google Scholar] [PubMed]
- Middleton, P.G.; Caplen, N.J.; Gao, X.; Huang, L.; Gaya, H.; Geddes, D.M.; Alton, E.W. Nasal application of the cationic liposome DC-Chol:DOPE does not alter ion transport, lung function or bacterial growth. Eur. Respir. J. 1994, 7, 442–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplen, N.J.; Alton, E.W.; Middleton, P.G.; Dorin, J.R.; Stevenson, B.J.; Gao, X.; Durham, S.R.; Jeffery, P.K.; Hodson, M.E.; Coutelle, C.; et al. Liposome-mediated CFTR gene transfer to the nasal epithelium of patients with cystic fibrosis. Nat. Med. 1995, 1, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Hyde, S.C.; Southern, K.W.; Gileadi, U.; Fitzjohn, E.M.; Mofford, K.A.; Waddell, B.E.; Gooi, H.C.; Goddard, C.A.; Hannavy, K.; Smyth, S.E.; et al. Repeat administration of DNA/liposomes to the nasal epithelium of patients with cystic fibrosis. Gene Ther. 2000, 7, 1156–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noone, P.G.; Hohneker, K.W.; Zhou, Z.; Johnson, L.G.; Foy, C.; Gipson, C.; Jones, K.; Noah, T.L.; Leigh, M.W.; Schwartzbach, C.; et al. Safety and biological efficacy of a lipid-CFTR complex for gene transfer in the nasal epithelium of adult patients with cystic fibrosis. Mol. Ther. 2000, 1, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.R.; Southern, K.W.; Mofford, K.A.; Seddon, T.; Huang, L.; Sorgi, F.; Thomson, A.; MacVinish, L.J.; Ratcliff, R.; Bilton, D.; et al. A placebo-controlled study of liposome-mediated gene transfer to the nasal epithelium of patients with cystic fibrosis. Gene Ther. 1997, 4, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porteous, D.J.; Dorin, J.R.; McLachlan, G.; Davidson-Smith, H.; Davidson, H.; Stevenson, B.J.; Carothers, A.D.; Wallace, W.A.; Moralee, S.; Hoenes, C.; et al. Evidence for safety and efficacy of DOTAP cationic liposome mediated CFTR gene transfer to the nasal epithelium of patients with cystic fibrosis. Gene Ther. 1997, 4, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLachlan, G.; Ho, L.P.; Davidson-Smith, H.; Samways, J.; Davidson, H.; Stevenson, B.J.; Carothers, A.D.; Alton, E.W.; Middleton, P.G.; Smith, S.N.; et al. Laboratory and clinical studies in support of cystic fibrosis gene therapy using pCMV-CFTR-DOTAP. Gene Ther. 1996, 3, 1113–1123. [Google Scholar] [PubMed]
- Zabner, J.; Cheng, S.H.; Meeker, D.; Launspach, J.; Balfour, R.; Perricone, M.A.; Morris, J.E.; Marshall, J.; Fasbender, A.; Smith, A.E.; et al. Comparison of DNA-lipid complexes and DNA alone for gene transfer to cystic fibrosis airway epithelia in vivo. J. Clin. Investig. 1997, 100, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Alton, E.W.; Stern, M.; Farley, R.; Jaffe, A.; Chadwick, S.L.; Phillips, J.; Davies, J.; Smith, S.N.; Browning, J.; Davies, M.G.; et al. Cationic lipid-mediated CFTR gene transfer to the lungs and nose of patients with cystic fibrosis: A double-blind placebo-controlled trial. Lancet 1999, 353, 947–954. [Google Scholar] [CrossRef]
- Ruiz, F.E.; Clancy, J.P.; Perricone, M.A.; Bebok, Z.; Hong, J.S.; Cheng, S.H.; Meeker, D.P.; Young, K.R.; Schoumacher, R.A.; Weatherly, M.R.; et al. A clinical inflammatory syndrome attributable to aerosolized lipid-DNA administration in cystic fibrosis. Hum. Gene Ther. 2001, 12, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; Davis, P.B.; Wagener, J.S.; Hilliard, K.A.; Stern, R.C.; Milgram, L.J.; Kowalczyk, T.H.; Hyatt, S.L.; Fink, T.L.; Gedeon, C.R.; et al. Compacted DNA nanoparticles administered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate partial to complete cystic fibrosis transmembrane regulator reconstitution. Hum. Gene Ther. 2004, 15, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Alton, E.W.; Boyd, A.C.; Cheng, S.H.; Cunningham, S.; Davies, J.C.; Gill, D.R.; Griesenbach, U.; Higgins, T.; Hyde, S.C.; Innes, J.A.; et al. A randomised, double-blind, placebo-controlled phase IIB clinical trial of repeated application of gene therapy in patients with cystic fibrosis. Thorax 2013, 68, 1075–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alton, E.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. A Randomised, Double-Blind, Placebo-Controlled Trial of Repeated Nebulisation of Non-Viral Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Gene Therapy in Patients with Cystic Fibrosis; NIHR Journals Library: Southampton, UK, 2016. [Google Scholar] [CrossRef]
- Wang, G.; Slepushkin, V.A.; Bodner, M.; Zabner, J.; van Es, H.H.; Thomas, P.; Jolly, D.J.; Davidson, B.L.; McCray, P.B., Jr. Keratinocyte growth factor induced epithelial proliferation facilitates retroviral-mediated gene transfer to distal lung epithelia in vivo. J. Gene Med. 1999, 1, 22–30. [Google Scholar] [CrossRef]
- Wang, G.; Davidson, B.L.; Melchert, P.; Slepushkin, V.A.; van Es, H.H.; Bodner, M.; Jolly, D.J.; McCray, P.B., Jr. Influence of cell polarity on retrovirus-mediated gene transfer to differentiated human airway epithelia. J. Virol. 1998, 72, 9818–9826. [Google Scholar] [PubMed]
- Duan, D.; Yue, Y.; Yan, Z.; McCray, P.B., Jr.; Engelhardt, J.F. Polarity influences the efficiency of recombinant adenoassociated virus infection in differentiated airway epithelia. Hum. Gene Ther. 1998, 9, 2761–2776. [Google Scholar] [CrossRef] [PubMed]
- Walters, R.W.; van’t Hof, W.; Yi, S.M.; Schroth, M.K.; Zabner, J.; Crystal, R.G.; Welsh, M.J. Apical localization of the coxsackie-adenovirus receptor by glycosyl-phosphatidylinositol modification is sufficient for adenovirus-mediated gene transfer through the apical surface of human airway epithelia. J. Virol. 2001, 75, 7703–7711. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Sinn, P.L.; McCray, P.B., Jr. Development of retroviral vectors for gene transfer to airway epithelia. Curr. Opin. Mol. Ther. 2000, 2, 497–506. [Google Scholar] [PubMed]
- Johnson, L.G.; Olsen, J.C.; Naldini, L.; Boucher, R.C. Pseudotyped human lentiviral vector-mediated gene transfer to airway epithelia in vivo. Gene Ther. 2000, 7, 568–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbert, C.L.; Aitken, M.L.; Miller, A.D. Retroviral vectors efficiently transduce basal and secretory airway epithelial cells in vitro resulting in persistent gene expression in organotypic culture. Hum. Gene Ther. 1996, 7, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Lee, P.S.; Yang, J.S.; Wilson, J.M. Lentiviral vectors for gene therapy of cystic fibrosis. Hum. Gene Ther. 1997, 8, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Slepushkin, V.; Zabner, J.; Keshavjee, S.; Johnston, J.C.; Sauter, S.L.; Jolly, D.J.; Dubensky, T.W., Jr.; Davidson, B.L.; McCray, P.B., Jr. Feline immunodeficiency virus vectors persistently transduce nondividing airway epithelia and correct the cystic fibrosis defect. J. Clin. Investig. 1999, 104, R55–R62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinn, P.L.; Goreham-Voss, J.D.; Arias, A.C.; Hickey, M.A.; Maury, W.; Chikkanna-Gowda, C.P.; McCray, P.B., Jr. Enhanced gene expression conferred by stepwise modification of a nonprimate lentiviral vector. Hum. Gene Ther. 2007, 18, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Peeples, M.E.; Boucher, R.C.; Collins, P.L.; Pickles, R.J. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J. Virol. 2002, 76, 5654–5666. [Google Scholar] [CrossRef] [PubMed]
- Sinn, P.L.; Hickey, M.A.; Staber, P.D.; Dylla, D.E.; Jeffers, S.A.; Davidson, B.L.; Sanders, D.A.; McCray, P.B., Jr. Lentivirus vectors pseudotyped with filoviral envelope glycoproteins transduce airway epithelia from the apical surface independently of folate receptor α. J. Virol. 2003, 77, 5902–5910. [Google Scholar] [CrossRef] [PubMed]
- Kobinger, G.P.; Weiner, D.J.; Yu, Q.C.; Wilson, J.M. Filovirus-pseudotyped lentiviral vector can efficiently and stably transduce airway epithelia in vivo. Nat. Biotechnol. 2001, 19, 225–230. [Google Scholar] [CrossRef] [PubMed]
- McKay, T.; Patel, M.; Pickles, R.J.; Johnson, L.G.; Olsen, J.C. Influenza M2 envelope protein augments avian influenza hemagglutinin pseudotyping of lentiviral vectors. Gene Ther. 2006, 13, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobinger, G.P.; Limberis, M.P.; Somanathan, S.; Schumer, G.; Bell, P.; Wilson, J.M. Human immunodeficiency viral vector pseudotyped with the spike envelope of severe acute respiratory syndrome coronavirus transduces human airway epithelial cells and dendritic cells. Hum. Gene Ther. 2007, 18, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Sinn, P.L.; Penisten, A.K.; Burnight, E.R.; Hickey, M.A.; Williams, G.; McCoy, D.M.; Mallampalli, R.K.; McCray, P.B. Gene transfer to respiratory epithelia with lentivirus pseudotyped with Jaagsiekte sheep retrovirus envelope glycoprotein. Hum. Gene Ther. 2005, 16, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.K.; DeMartini, J.C.; Miller, A.D. Retrovirus vectors bearing jaagsiekte sheep retrovirus Env transduce human cells by using a new receptor localized to chromosome 3p21.3. J. Virol. 2000, 74, 4698–4704. [Google Scholar] [CrossRef] [PubMed]
- Kremer, K.L.; Dunning, K.R.; Parsons, D.W.; Anson, D.S. Gene delivery to airway epithelial cells in vivo: A direct comparison of apical and basolateral transduction strategies using pseudotyped lentivirus vectors. J. Gene Med. 2007, 9, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Mitomo, K.; Griesenbach, U.; Inoue, M.; Somerton, L.; Meng, C.; Akiba, E.; Tabata, T.; Ueda, Y.; Frankel, G.M.; Farley, R.; et al. Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with Sendai virus envelopes. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Griesenbach, U.; Inoue, M.; Meng, C.; Farley, R.; Chan, M.; Newman, N.K.; Brum, A.; You, J.; Kerton, A.; Shoemark, A.; et al. Assessment of F/HN-pseudotyped lentivirus as a clinically relevant vector for lung gene therapy. Am. J. Respir. Crit. Care Med. 2012, 186, 846–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, A.G.; Kremer, K.L.; Koldej, R.; Miller, D.S.; Anson, D.S.; Parsons, D.W. Single-dose lentiviral gene transfer for lifetime airway gene expression. J. Gene Med. 2009, 11, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Cmielewski, P.; Anson, D.S.; Parsons, D.W. Lysophosphatidylcholine as an adjuvant for lentiviral vector mediated gene transfer to airway epithelium: Effect of acyl chain length. Respir. Res. 2010, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Sinn, P.L.; Cooney, A.L.; Oakland, M.; Dylla, D.E.; Wallen, T.J.; Pezzulo, A.A.; Chang, E.H.; McCray, P.B., Jr. Lentiviral vector gene transfer to porcine airways. Mol. Ther. Nucleic Acids 2012, 1, e56. [Google Scholar] [CrossRef] [PubMed]
- Farrow, N.; Miller, D.; Cmielewski, P.; Donnelley, M.; Bright, R.; Parsons, D.W. Airway gene transfer in a non-human primate: Lentiviral gene expression in marmoset lungs. Sci. Rep. 2013, 3, 1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cmielewski, P.; Donnelley, M.; Parsons, D.W. Long-term therapeutic and reporter gene expression in lentiviral vector treated cystic fibrosis mice. J. Gene Med. 2014, 16, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Walters, R.W.; Grunst, T.; Bergelson, J.M.; Finberg, R.W.; Welsh, M.J.; Zabner, J. Basolateral localization of fiber receptors limits adenovirus infection from the apical surface of airway epithelia. J. Biol. Chem. 1999, 274, 10219–10226. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; St George, J.A.; Lukason, M.; Cheng, S.H.; Scheule, R.K.; Eastman, S.J. EGTA enhancement of adenovirus-mediated gene transfer to mouse tracheal epithelium in vivo. Hum. Gene Ther. 2001, 12, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zabner, J.; Deering, C.; Launspach, J.; Shao, J.; Bodner, M.; Jolly, D.J.; Davidson, B.L.; McCray, P.B., Jr. Increasing epithelial junction permeability enhances gene transfer to airway epithelia in vivo. Am. J. Respir. Cell Mol. Biol. 2000, 22, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.P.; Luner, P.; Moninger, T.O.; Karp, P.H.; Keshavjee, S.; Zabner, J. Thixotropic solutions enhance viral-mediated gene transfer to airway epithelia. Am. J. Respir. Cell Mol. Biol. 2002, 27, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Sinn, P.L.; Shah, A.J.; Donovan, M.D.; McCray, P.B., Jr. Viscoelastic gel formulations enhance airway epithelial gene transfer with viral vectors. Am. J. Respir. Cell Mol. Biol. 2005, 32, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, J. Detergent-lentiviral combination gives gene therapy hope for cystic fibrosis. Lancet 2002, 360, 1306. [Google Scholar] [CrossRef]
- Fasbender, A.; Lee, J.H.; Walters, R.W.; Moninger, T.O.; Zabner, J.; Welsh, M.J. Incorporation of adenovirus in calcium phosphate precipitates enhances gene transfer to airway epithelia in vitro and in vivo. J. Clin. Investig. 1998, 102, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Zabner, J.; Welsh, M.J. Delivery of an adenovirus vector in a calcium phosphate coprecipitate enhances the therapeutic index of gene transfer to airway epithelia. Hum. Gene Ther. 1999, 10, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Walters, R.; Welsh, M. Mechanism by which calcium phosphate coprecipitation enhances adenovirus-mediated gene transfer. Gene Ther. 1999, 6, 1845–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Welsh, M.J. Enhancement of calcium phosphate-mediated transfection by inclusion of adenovirus in coprecipitates. Gene Ther. 1999, 6, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otake, K.; Ennist, D.L.; Harrod, K.; Trapnell, B.C. Nonspecific inflammation inhibits adenovirus-mediated pulmonary gene transfer and expression independent of specific acquired immune responses. Hum. Gene Ther. 1998, 9, 2207–2222. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Grubb, B.R.; Johnson, L.G.; Boucher, R.C. Enhanced in vivo airway gene transfer via transient modification of host barrier properties with a surface-active agent. Hum. Gene Ther. 1998, 9, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Drapkin, P.T.; O’Riordan, C.R.; Yi, S.M.; Chiorini, J.A.; Cardella, J.; Zabner, J.; Welsh, M.J. Targeting the urokinase plasminogen activator receptor enhances gene transfer to human airway epithelia. J. Clin. Investig. 2000, 105, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabner, J.; Wadsworth, S.C.; Smith, A.E.; Welsh, M.J. Adenovirus-mediated generation of cAMP-stimulated Cl-transport in cystic fibrosis airway epithelia in vitro: Effect of promoter and administration method. Gene Ther. 1996, 3, 458–465. [Google Scholar] [PubMed]
- Zabner, J.; Zeiher, B.G.; Friedman, E.; Welsh, M.J. Adenovirus-mediated gene transfer to ciliated airway epithelia requires prolonged incubation time. J. Virol. 1996, 70, 6994–7003. [Google Scholar] [PubMed]
- Alton, E.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef]
- Yan, Z.; Sun, X.; Feng, Z.; Li, G.; Fisher, J.T.; Stewart, Z.A.; Engelhardt, J.F. Optimization of Recombinant Adeno-Associated Virus-Mediated Expression for Large Transgenes, Using a Synthetic Promoter and Tandem Array Enhancers. Hum. Gene Ther. 2015, 26, 334–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Singh, R.N.; Crystal, R.G. Regulatable promoters for use in gene therapy applications: Modification of the 5’-flanking region of the CFTR gene with multiple cAMP response elements to support basal, low-level gene expression that can be upregulated by exogenous agents that raise intracellular levels of cAMP. Hum. Gene Ther. 1996, 7, 1883–1893. [Google Scholar] [PubMed]
- Suzuki, M.; Singh, R.N.; Crystal, R.G. Ability of a chimeric cAMP-responsive promoter to confer pharmacologic control of CFTR cDNA expression and cAMP-mediated Cl− secretion. Gene Ther. 1997, 4, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; O’Connor, S.P.; Armentano, D.; Berthelette, P.B.; Schiavi, S.C.; Jefferson, D.M.; Smith, A.E.; Wadsworth, S.C.; Cheng, S.H. Ability of adenovirus vectors containing different CFTR transcriptional cassettes to correct ion transport defects in CF cells. Am. J. Physiol. 1996, 271, L527–L537. [Google Scholar] [CrossRef] [PubMed]
- Chow, Y.H.; O’Brodovich, H.; Plumb, J.; Wen, Y.; Sohn, K.J.; Lu, Z.; Zhang, F.; Lukacs, G.L.; Tanswell, A.K.; Hui, C.C.; et al. Development of an epithelium-specific expression cassette with human DNA regulatory elements for transgene expression in lung airways. Proc. Natl. Acad. Sci. USA 1997, 94, 14695–14700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Zak, R.; Zhang, Y.; Ding, W.; Godwin, S.; Munson, K.; Peluso, R.; Engelhardt, J.F. Distinct classes of proteasome-modulating agents cooperatively augment recombinant adeno-associated virus type 2 and type 5-mediated transduction from the apical surfaces of human airway epithelia. J. Virol. 2004, 78, 2863–2874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Karp, P.; Gerard, C.J.; Pastor, E.; Laux, D.; Munson, K.; Yan, Z.; Liu, X.; Godwin, S.; Thomas, C.P.; et al. Dual therapeutic utility of proteasome modulating agents for pharmaco-gene therapy of the cystic fibrosis airway. Mol. Ther. 2004, 10, 990–1002. [Google Scholar] [CrossRef] [PubMed]
- Excoffon, K.J.; Koerber, J.T.; Dickey, D.D.; Murtha, M.; Keshavjee, S.; Kaspar, B.K.; Zabner, J.; Schaffer, D.V. Directed evolution of adeno-associated virus to an infectious respiratory virus. Proc. Natl. Acad. Sci. USA 2009, 106, 3865–3870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, L.; Johnson, J.S.; Zhijian, W.; Grieger, J.C.; Ping-Jie, X.; Drouin, L.M.; Agbandje-McKenna, M.; Pickles, R.J.; Samulski, R.J. Generation of novel AAV variants by directed evolution for improved CFTR delivery to human ciliated airway epithelium. Mol. Ther. 2009, 17, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Fischer, A.C.; Goetzmann, J.; Mueller, C.; Cebotaru, L.; Yan, Z.; Wang, L.; Wilson, J.M.; Guggino, W.B.; Engelhardt, J.F. Dual reporter comparative indexing of rAAV pseudotyped vectors in chimpanzee airway. Mol. Ther. 2010, 18, 594–600. [Google Scholar] [CrossRef] [PubMed]
- White, A.F.; Mazur, M.; Sorscher, E.J.; Zinn, K.R.; Ponnazhagan, S. Genetic modification of adeno-associated viral vector type 2 capsid enhances gene transfer efficiency in polarized human airway epithelial cells. Hum. Gene Ther. 2008, 19, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Parks, R.J.; Chen, L.; Anton, M.; Sankar, U.; Rudnicki, M.A.; Graham, F.L. A helper-dependent adenovirus vector system: Removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc. Natl. Acad. Sci. USA 1996, 93, 13565–13570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, K.J.; Choi, H.; Burda, J.; Chen, S.J.; Wilson, J.M. Recombinant adenovirus deleted of all viral genes for gene therapy of cystic fibrosis. Virology 1996, 217, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ben-Gary, H.; McKinney, R.L.; Rosengart, T.; Lesser, M.L.; Crystal, R.G. Systemic interleukin-6 responses following administration of adenovirus gene transfer vectors to humans by different routes. Mol. Ther. 2002, 6, 287–297. [Google Scholar] [PubMed]
- Toietta, G.; Koehler, D.R.; Finegold, M.J.; Lee, B.; Hu, J.; Beaudet, A.L. Reduced inflammation and improved airway expression using helper-dependent adenoviral vectors with a K18 promoter. Mol. Ther. 2003, 7, 649–658. [Google Scholar] [CrossRef]
- Van Heeckeren, A.M.; Scaria, A.; Schluchter, M.D.; Ferkol, T.W.; Wadsworth, S.; Davis, P.B. Delivery of CFTR by adenoviral vector to cystic fibrosis mouse lung in a model of chronic Pseudomonas aeruginosa lung infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L717–L726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Heeckeren, A.M.; Schluchter, M.D.; Drumm, M.L.; Davis, P.B. Role of Cftr genotype in the response to chronic Pseudomonas aeruginosa lung infection in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L944–L952. [Google Scholar] [CrossRef] [PubMed]
- Koehler, D.R.; Sajjan, U.; Chow, Y.H.; Martin, B.; Kent, G.; Tanswell, A.K.; McKerlie, C.; Forstner, J.F.; Hu, J. Protection of Cftr knockout mice from acute lung infection by a helper-dependent adenoviral vector expressing Cftr in airway epithelia. Proc. Natl. Acad. Sci. USA 2003, 100, 15364–15369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oceandy, D.; McMorran, B.J.; Smith, S.N.; Schreiber, R.; Kunzelmann, K.; Alton, E.W.; Hume, D.A.; Wainwright, B.J. Gene complementation of airway epithelium in the cystic fibrosis mouse is necessary and sufficient to correct the pathogen clearance and inflammatory abnormalities. Hum. Mol. Genet. 2002, 11, 1059–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehler, D.R.; Martin, B.; Corey, M.; Palmer, D.; Ng, P.; Tanswell, A.K.; Hu, J. Readministration of helper-dependent adenovirus to mouse lung. Gene Ther. 2006, 13, 773–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushwah, R.; Cao, H.; Hu, J. Potential of helper-dependent adenoviral vectors in modulating airway innate immunity. Cell. Mol. Immunol. 2007, 4, 81–89. [Google Scholar] [PubMed]
- Rogers, C.S.; Abraham, W.M.; Brogden, K.A.; Engelhardt, J.F.; Fisher, J.T.; McCray, P.B., Jr.; McLennan, G.; Meyerholz, D.K.; Namati, E.; Ostedgaard, L.S.; et al. The porcine lung as a potential model for cystic fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L240–L263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008, 321, 1837–1841. [Google Scholar] [CrossRef] [PubMed]
- Klymiuk, N.; Mundhenk, L.; Kraehe, K.; Wuensch, A.; Plog, S.; Emrich, D.; Langenmayer, M.C.; Stehr, M.; Holzinger, A.; Kroner, C.; et al. Sequential targeting of CFTR by BAC vectors generates a novel pig model of cystic fibrosis. J. Mol. Med. 2012, 90, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, D.A.; Rokhlina, T.; Ernst, S.E.; Pezzulo, A.A.; Ostedgaard, L.S.; Karp, P.H.; Samuel, M.S.; Reznikov, L.R.; Rector, M.V.; Gansemer, N.D.; et al. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J. Clin. Investig. 2013, 123, 2685–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Sui, H.; Fisher, J.T.; Yan, Z.; Liu, X.; Cho, H.J.; Joo, N.S.; Zhang, Y.; Zhou, W.; Yi, Y.; et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J. Clin. Investig. 2010, 120, 3149–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Yan, Z.; Yi, Y.; Li, Z.; Lei, D.; Rogers, C.S.; Chen, J.; Zhang, Y.; Welsh, M.J.; Leno, G.H.; et al. Adeno-associated virus-targeted disruption of the CFTR gene in cloned ferrets. J. Clin. Investig. 2008, 118, 1578–1583. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Olivier, A.K.; Liang, B.; Yi, Y.; Sui, H.; Evans, T.I.; Zhang, Y.; Zhou, W.; Tyler, S.R.; Fisher, J.T.; et al. Lung phenotype of juvenile and adult cystic fibrosis transmembrane conductance regulator-knockout ferrets. Am. J. Respir. Cell Mol. Biol. 2014, 50, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Feng, Z.; Sun, X.; Zhang, Y.; Zou, W.; Wang, Z.; Jensen-Cody, C.; Liang, B.; Park, S.Y.; Qiu, J.; et al. Human Bocavirus Type-1 Capsid Facilitates the Transduction of Ferret Airways by Adeno-Associated Virus Genomes. Hum. Gene Ther. 2017, 28, 612–625. [Google Scholar] [CrossRef] [PubMed]
- Tuggle, K.L.; Birket, S.E.; Cui, X.; Hong, J.; Warren, J.; Reid, L.; Chambers, A.; Ji, D.; Gamber, K.; Chu, K.K.; et al. Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats. PLoS ONE 2014, 9, e91253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birket, S.E.; Davis, J.M.; Fernandez, C.M.; Tuggle, K.L.; Oden, A.M.; Chu, K.K.; Tearney, G.J.; Fanucchi, M.V.; Sorscher, E.J.; Rowe, S.M. Development of an airway mucus defect in the cystic fibrosis rat. JCI Insight 2018, 3, 97199. [Google Scholar] [CrossRef] [PubMed]
- Navis, A.; Bagnat, M. Loss of cftr function leads to pancreatic destruction in larval zebrafish. Dev. Biol. 2015, 399, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Perisse, I.V.; Cotton, C.U.; Regouski, M.; Meng, Q.; Domb, C.; Van Wettere, A.J.; Wang, Z.; Harris, A.; White, K.L.; et al. A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene. JCI Insight 2018, 3, 123529. [Google Scholar] [CrossRef] [PubMed]
- Kamaruzaman, N.A.; Kardia, E.; Kamaldin, N.; Latahir, A.Z.; Yahaya, B.H. The rabbit as a model for studying lung disease and stem cell therapy. BioMed Res. Int. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.R.; Steele, M.S.; Valerio, D.M.; Miron, A.; Mann, R.J.; LePage, D.F.; Conlon, R.A.; Cotton, C.U.; Drumm, M.L.; Hodges, C.A. A G542X cystic fibrosis mouse model for examining nonsense mutation directed therapies. PLoS ONE 2018, 13, e0199573. [Google Scholar] [CrossRef] [PubMed]
- Darrah, R.J.; Mitchell, A.L.; Campanaro, C.K.; Barbato, E.S.; Litman, P.; Sattar, A.; Hodges, C.A.; Drumm, M.L.; Jacono, F.J. Early pulmonary disease manifestations in cystic fibrosis mice. J. Cyst. Fibros. 2016, 15, 736–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, C.A.; Cotton, C.U.; Palmert, M.R.; Drumm, M.L. Generation of a conditional null allele for Cftr in mice. Genesis 2008, 46, 546–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, A.L.; Abou Alaiwa, M.H.; Shah, V.S.; Bouzek, D.C.; Stroik, M.R.; Powers, L.S.; Gansemer, N.D.; Meyerholz, D.K.; Welsh, M.J.; Stoltz, D.A.; et al. Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight 2016, 1, e88730. [Google Scholar] [CrossRef] [PubMed]
- Steines, B.; Dickey, D.D.; Bergen, J.; Excoffon, K.J.; Weinstein, J.R.; Li, X.; Yan, Z.; Abou Alaiwa, M.H.; Shah, V.S.; Bouzek, D.C.; et al. CFTR gene transfer with AAV improves early cystic fibrosis pig phenotypes. JCI Insight 2016, 1, e88728. [Google Scholar] [CrossRef] [PubMed]
- Cooney, A.L.; Singh, B.K.; Loza, L.M.; Thornell, I.M.; Hippee, C.E.; Powers, L.S.; Ostedgaard, L.S.; Meyerholz, D.K.; Wohlford-Lenane, C.; Stoltz, D.A.; et al. Widespread airway distribution and short-term phenotypic correction of cystic fibrosis pigs following aerosol delivery of piggyBac/adenovirus. Nucleic Acids Res. 2018, 46, 9561–9600. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.S.; Ernst, S.; Tang, X.X.; Karp, P.H.; Parker, C.P.; Ostedgaard, L.S.; Welsh, M.J. Relationships among CFTR expression, HCO3− secretion, and host defense may inform gene- and cell-based cystic fibrosis therapies. Proc. Natl. Acad. Sci. USA 2016, 113, 5382–5387. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H.; Carroll, D. Gene targeting using zinc finger nucleases. Nat. Biotechnol. 2005, 23, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Epinat, J.C.; Arnould, S.; Chames, P.; Rochaix, P.; Desfontaines, D.; Puzin, C.; Patin, A.; Zanghellini, A.; Paques, F.; Lacroix, E. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res. 2003, 31, 2952–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Flynn, R.; Hollywood, J.A.; Scallan, M.F.; Harrison, P.T. Correction of the DeltaF508 Mutation in the Cystic Fibrosis Transmembrane Conductance Regulator Gene by Zinc-Finger Nuclease Homology-Directed Repair. Biores. Open Access 2012, 1, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Thibodeau-Beganny, S.; Osiak, A.; Wright, D.A.; Anthony, R.M.; Eichtinger, M.; Jiang, T.; Foley, J.E.; Winfrey, R.J.; Townsend, J.A.; et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol. Cell 2008, 31, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.; Annaluru, N.; Kandavelou, K.; Chandrasegaran, S. TALEN-mediated generation and genetic correction of disease-specific human induced pluripotent stem cells. Curr. Gene Ther. 2014, 14, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Hollywood, J.A.; Lee, C.M.; Scallan, M.F.; Harrison, P.T. Analysis of gene repair tracts from Cas9/gRNA double-stranded breaks in the human CFTR gene. Sci. Rep. 2016, 6, 32230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnight, E.R.; Staber, J.M.; Korsakov, P.; Li, X.; Brett, B.T.; Scheetz, T.E.; Craig, N.L.; McCray, P.B., Jr. A Hyperactive Transposase Promotes Persistent Gene Transfer of a piggyBac DNA Transposon. Mol. Ther. Nucleic Acids 2012, 1, e50. [Google Scholar] [CrossRef] [PubMed]
- Cooney, A.L.; Singh, B.K.; Sinn, P.L. Hybrid nonviral/viral vector systems for improved piggyBac DNA transposon in vivo delivery. Mol. Ther. 2015, 23, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Ouyang, H.; Ip, W.; Du, K.; Duan, W.; Avolio, J.; Wu, J.; Duan, C.; Yeger, H.; Bear, C.E.; et al. Testing gene therapy vectors in human primary nasal epithelial cultures. Mol. Ther. Methods Clin. Dev. 2015, 2, 15034. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Machuca, T.N.; Yeung, J.C.; Wu, J.; Du, K.; Duan, C.; Hashimoto, K.; Linacre, V.; Coates, A.L.; Leung, K.; et al. Efficient gene delivery to pig airway epithelia and submucosal glands using helper-dependent adenoviral vectors. Mol. Ther. Nucleic Acids 2013, 2, e127. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Yang, T.; Li, X.F.; Wu, J.; Duan, C.; Coates, A.L.; Hu, J. Readministration of helper-dependent adenoviral vectors to mouse airway mediated via transient immunosuppression. Gene Ther. 2011, 18, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wu, J.; Duan, C.; Du, K.; Lee, C.M.; Yeger, H.; Hu, J. Long-Term Expression of the Human CFTR Gene in Mouse Airway via Helper-Dependent Adenoviral Vector Delivery and Transient Immunosuppression. Hum. Gene Ther. 2016, 27, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Ashtari, M.; Nikonova, E.S.; Marshall, K.A.; Young, G.J.; Aravand, P.; Pan, W.; Ying, G.S.; Willett, A.E.; Mahmoudian, M.; Maguire, A.M.; et al. The Role of the Human Visual Cortex in Assessment of the Long-Term Durability of Retinal Gene Therapy in Follow-on RPE65 Clinical Trial Patients. Ophthalmology 2017, 124, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.D.; Bachmeier, C.A.; Phuoc, V.H.; Chavez, J.C. Axicabtagene ciloleucel (KTE-C19), an anti-CD19 CAR T therapy for the treatment of relapsed/refractory aggressive B-cell non-Hodgkin’s lymphoma. Ther. Clin. Risk Manag. 2018, 14, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Kolte, A.; Patil, S.; Lesimple, P.; Hanrahan, J.W.; Misra, A. PEGylated composite nanoparticles of PLGA and polyethylenimine for safe and efficient delivery of pDNA to lungs. Int. J. Pharm. 2017, 524, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Alton, E.W.; Beekman, J.M.; Boyd, A.C.; Brand, J.; Carlon, M.S.; Connolly, M.M.; Chan, M.; Conlon, S.; Davidson, H.E.; Davies, J.C.; et al. Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 2017, 72, 137–147. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Classes of cystic fibrosis transmembrane conductance regulator (CFTR) mutations. Cystic fibrosis transmembrane conductance regulator mutations are categorized into 6 classes based on the mutation function or protein output [5]. A red “x” or arrow indicates where each CFTR mutant protein is affected. A common mutation example is listed for each class. * People with CF can have more than one mutation; thus, the percentage is representative of the entire population and does not add up to 100. Percentages acquired from the Cystic Fibrosis Foundation (U.S., 2017). ** Potentiators and correctors provide relief to some people with CF in these classes. Additional mutations have been approved for use of CFTR modulators.
Figure 1.
Classes of cystic fibrosis transmembrane conductance regulator (CFTR) mutations. Cystic fibrosis transmembrane conductance regulator mutations are categorized into 6 classes based on the mutation function or protein output [5]. A red “x” or arrow indicates where each CFTR mutant protein is affected. A common mutation example is listed for each class. * People with CF can have more than one mutation; thus, the percentage is representative of the entire population and does not add up to 100. Percentages acquired from the Cystic Fibrosis Foundation (U.S., 2017). ** Potentiators and correctors provide relief to some people with CF in these classes. Additional mutations have been approved for use of CFTR modulators.

Figure 2.
Timelines of CF gene therapy eras: Important milestones impacting the CF field are represented in timelines at the beginning of each era. The timelines are intended to orient the reader to new developments relative to other events and are not comprehensive of all contributions to the field (1989–2001).
Figure 2.
Timelines of CF gene therapy eras: Important milestones impacting the CF field are represented in timelines at the beginning of each era. The timelines are intended to orient the reader to new developments relative to other events and are not comprehensive of all contributions to the field (1989–2001).

Figure 3.
Important milestones for the second era of CF gene therapy (1995–2010).

Figure 4.
Important milestones for the third era of CF gene therapy (2008–2018).

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cooney, A.L.; McCray, P.B., Jr.; Sinn, P.L. Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward. Genes 2018, 9, 538. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110538
AMA Style
Cooney AL, McCray PB Jr., Sinn PL. Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward. Genes. 2018; 9(11):538. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110538
Chicago/Turabian StyleCooney, Ashley L., Paul B. McCray, Jr., and Patrick L. Sinn. 2018. "Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward" Genes 9, no. 11: 538. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110538
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.