
Effect of Humidity on the Reactive Uptake of Ammonia and Dimethylamine by Nitrogen-Containing Secondary Organic Aerosol



Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
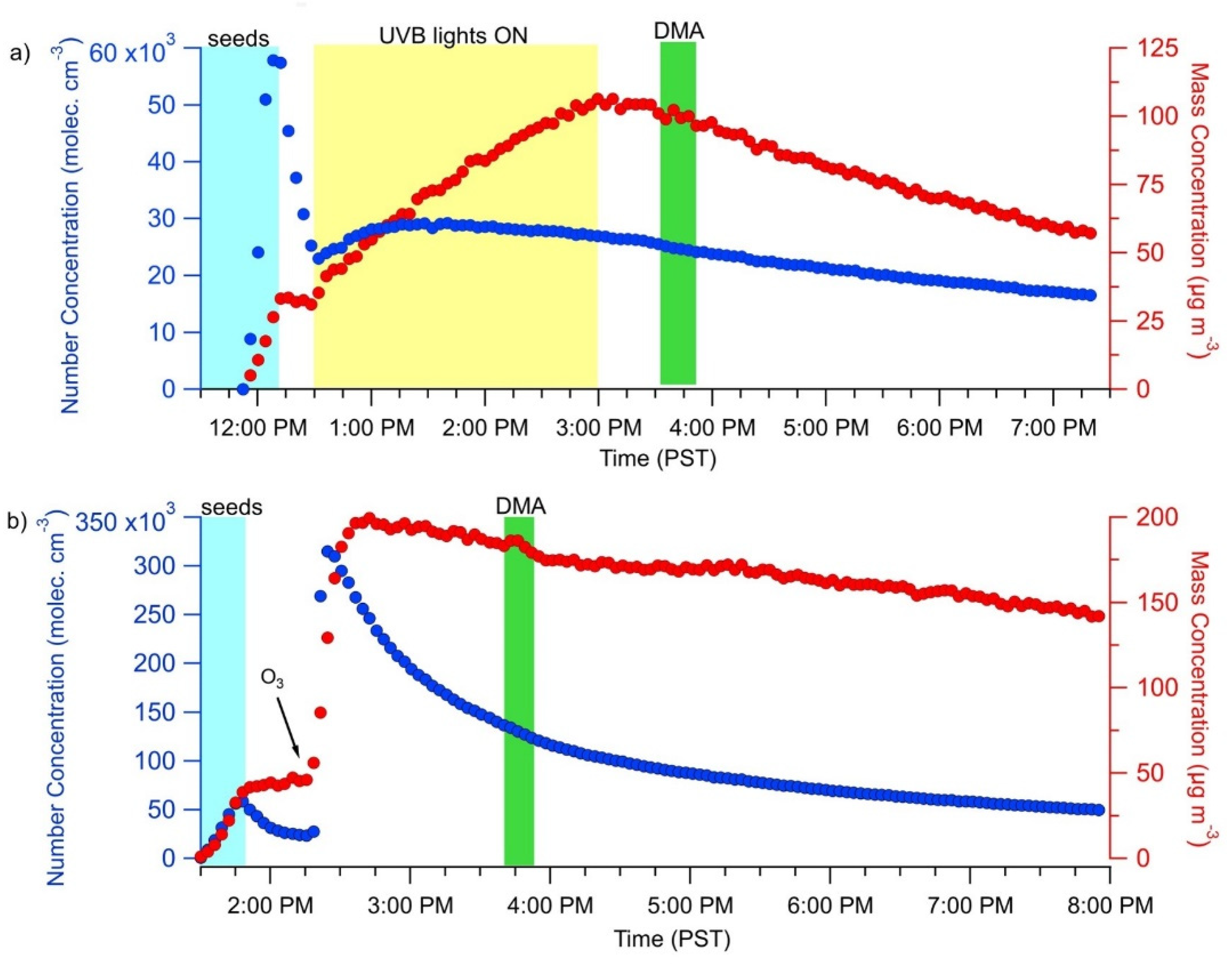
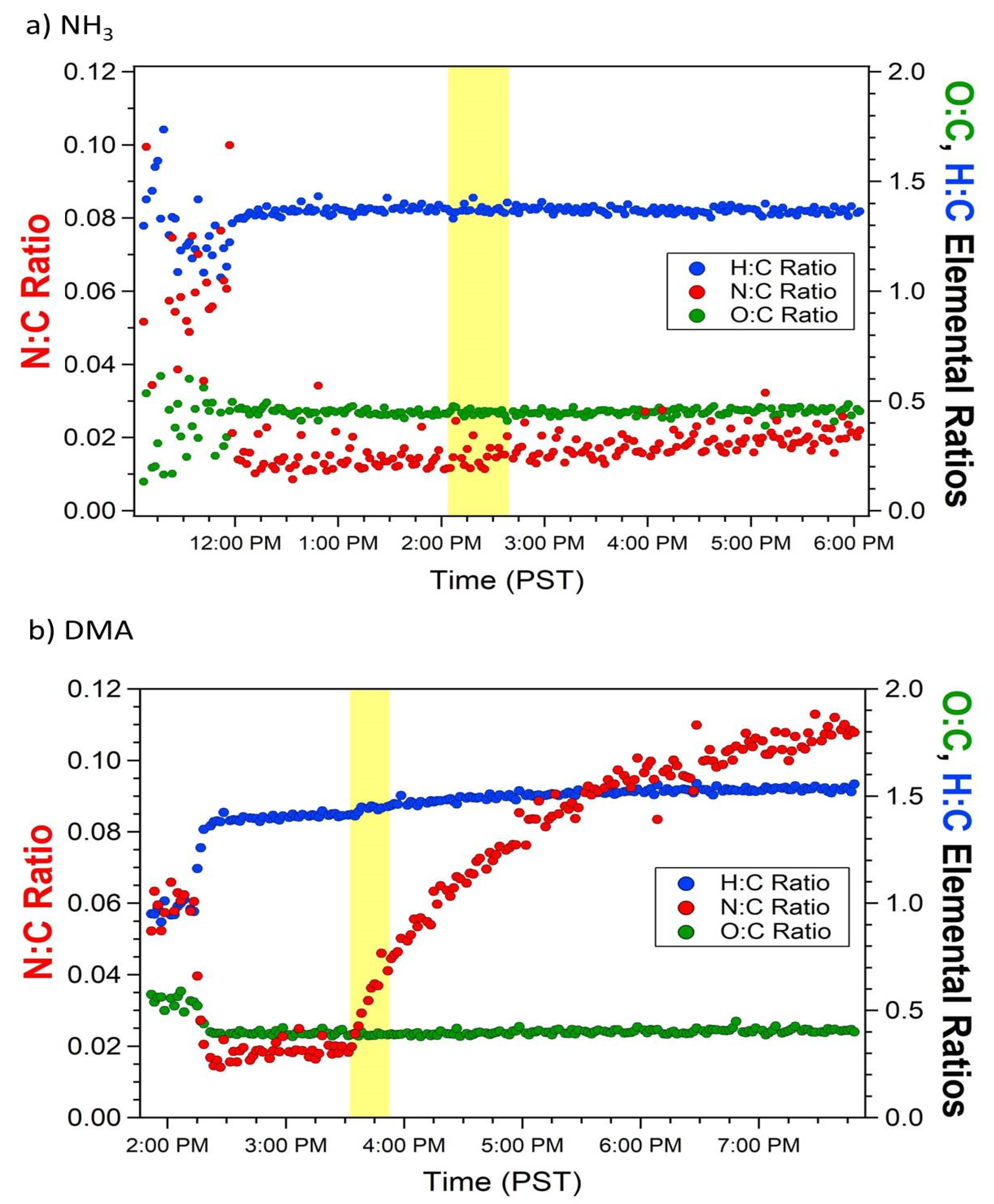
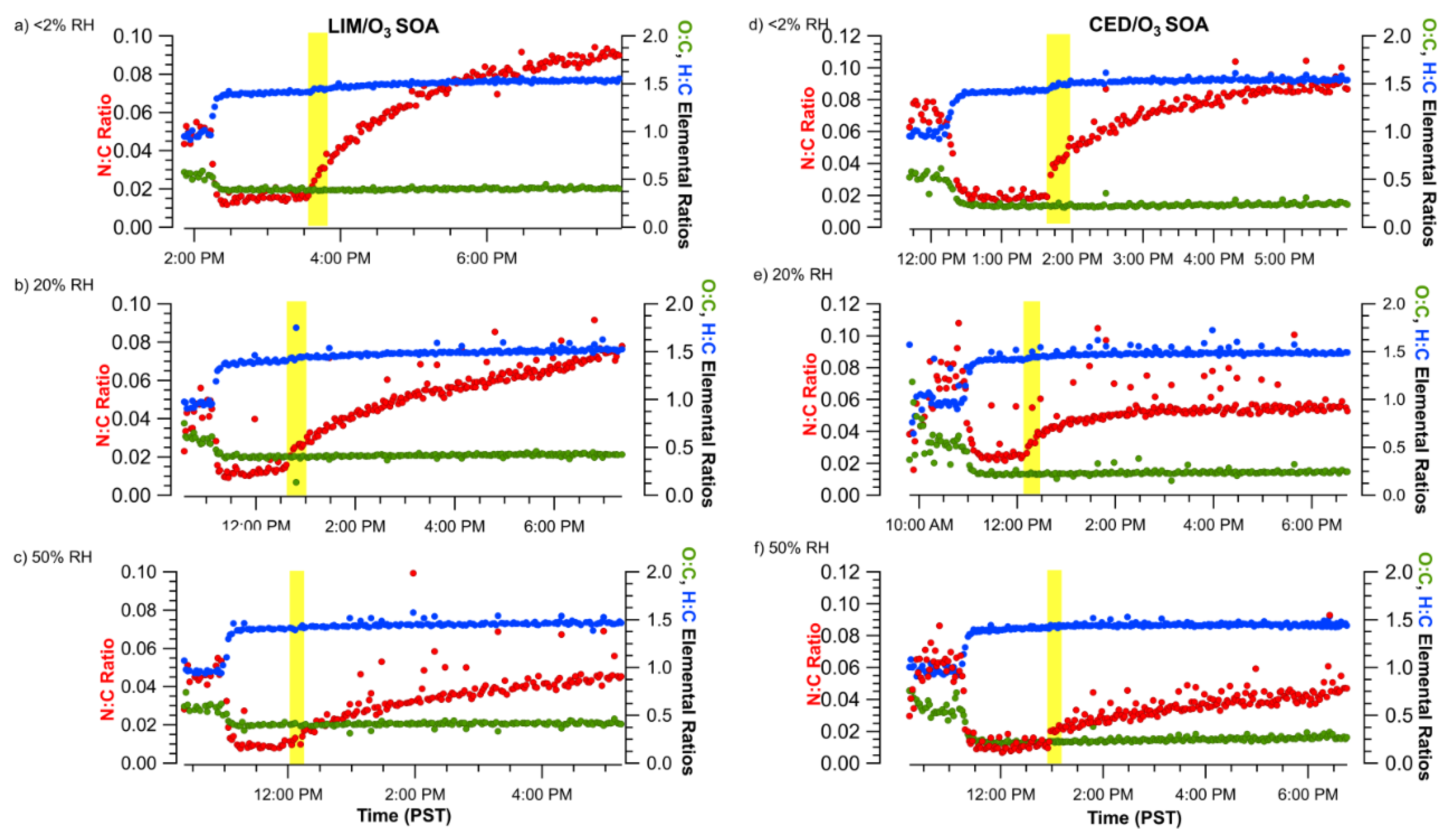
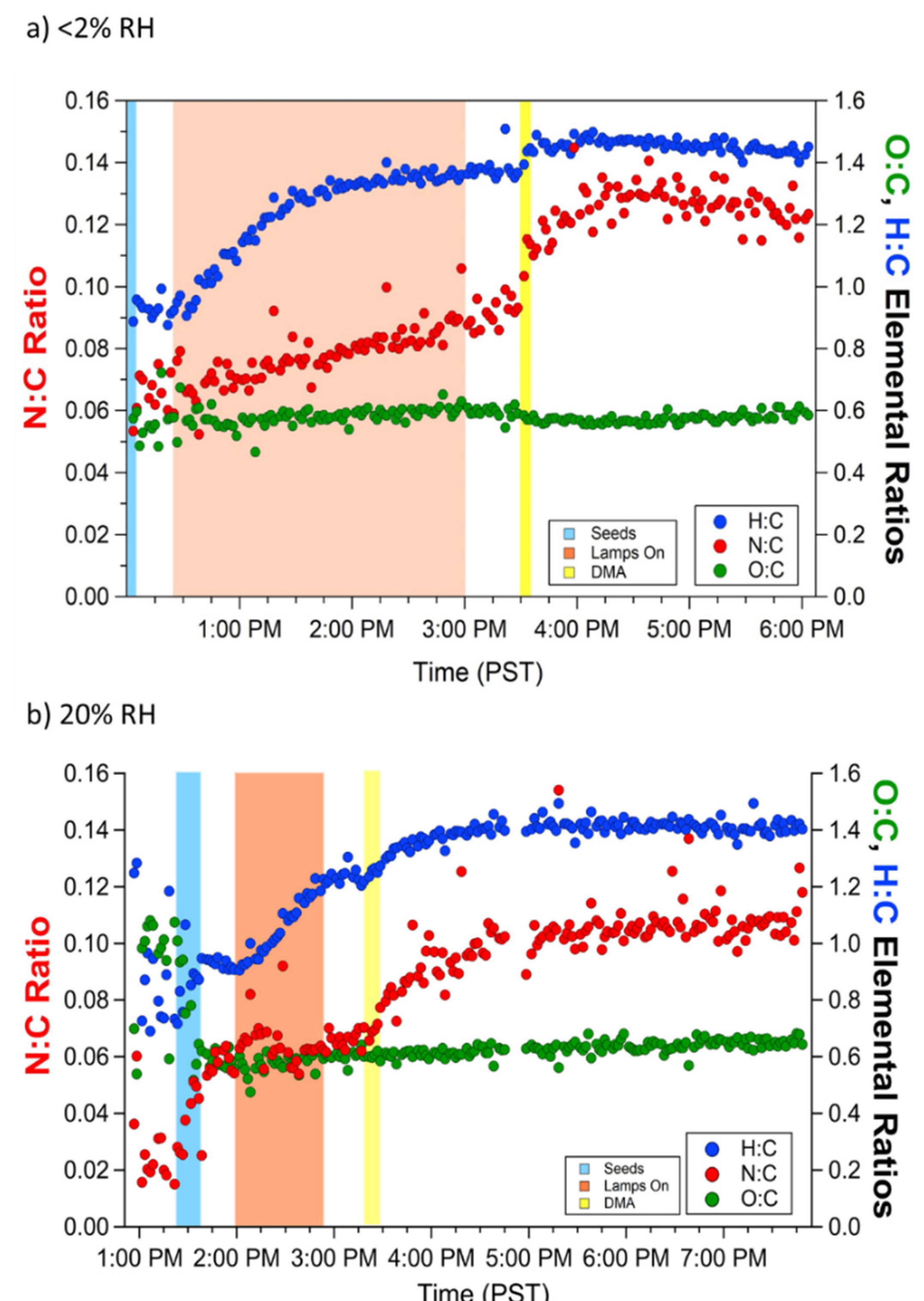
3.1. Effect of NH3 and DMA on SOA Concentration and Composition
3.2. Uptake Coefficients from NH3 Monitor Data
3.3. Uptake Coefficients from AMS Data
4. Atmospheric Implications
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Behera, S.N.; Sharma, M.; Aneja, V.P.; Balasubramanian, R. Ammonia in the atmosphere: A review on emission sources, atmospheric chemistry and deposition on terrestrial bodies. Environ. Sci. Pollut. Res. 2013, 20, 8092–8131. [Google Scholar] [CrossRef]
- Sheppard, L.J.; Leith, I.D.; Mizunuma, T.; Neil Cape, J.; Crossley, A.; Leeson, S.; Sutton, M.A.; Dijk, N.; Fowler, D. Dry deposition of ammonia gas drives species change faster than wet deposition of ammonium ions: Evidence from a long-term field manipulation. Glob. Chang. Biol. 2011, 17, 3589–3607. [Google Scholar] [CrossRef]
- Erisman, J.W.; Sutton, M.A.; Galloway, J.; Klimont, Z.; Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008, 1, 636–639. [Google Scholar] [CrossRef]
- Pinder, R.W.; Strader, R.; Davidson, C.I.; Adams, P.J. A temporally and spatially resolved ammonia emission inventory for dairy cows in the United States. Atmos. Environ. 2004, 38, 3747–3756. [Google Scholar] [CrossRef]
- Pinder, R.W.; Adams, P.J.; Pandis, S.N.; Gilliland, A.B. Temporally resolved ammonia emission inventories: Current estimates, evaluation tools, and measurement needs. J. Geophys. Res. 2006, 111, D16310. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Tian, H.; Pan, S.; Prior, S.A.; Feng, Y.; Batchelor, W.D.; Chen, J.; Yang, J. Global ammonia emissions from synthetic nitrogen fertilizer applications in agricultural systems: Empirical and process-based estimates and uncertainty. Glob. Chang. Biol. 2019, 25, 314–326. [Google Scholar] [CrossRef] [Green Version]
- Crippa, M.; Guizzardi, D.; Muntean, M.; Schaaf, E.; Oreggioni, G. EDGAR v5.0 Global Air Pollutant Emissions. Eur. Comm. Jt. Res. Cent. 2019. Available online: http//data.europa.eu/89h/377801af-b094-4943-8fdc-f79a7c0c2d19 (accessed on 8 November 2021).
- Amann, M.; Klimont, Z.; Wagner, F. Regional and Global Emissions of Air Pollutants: Recent Trends and Future Scenarios. Annu. Rev. Environ. Resour. 2013, 38, 31–35. [Google Scholar] [CrossRef]
- Galloway, J.N.; Townsend, A.R.; Erisman, J.W.; Bekunda, M.; Cai, Z.; Freney, J.R.; Martinelli, L.A.; Seitzinger, S.P.; Sutton, M.A. Transformation of the nitrogen cycle: Recent trends, questions, and potential solutions. Science 2008, 320, 889–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, J.X.; Dickerson, R.R.; Wei, Z.; Strow, L.L.; Wang, Y.; Liang, Q. Increased atmospheric ammonia over the world’s major agricultural areas detected from space. Geophys. Res. Lett. 2017, 44, 2875–2884. [Google Scholar] [CrossRef]
- Vayenas, D.V. Simulation of the thermodynamics and removal processes in the sulfate-ammonia-nitric acid system during winter: Implications for PM 2.5 control strategies. J. Geophys. Res. 2005, 110, D07S14. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Q.Q.; He, K.; Zhang, Q.; Chai, L. Sulfate-nitrate-ammonium aerosols over China: Response to 2000–2015 emission changes of sulfur dioxide, nitrogen oxides, and ammonia. Atmos. Chem. Phys. 2013, 13, 2635–2652. [Google Scholar] [CrossRef] [Green Version]
- West, J.J.; Ansari, A.S.; Pandis, S.N. Marginal pm25:nonlinear aerosol mass response to sulfate reductions in the eastern united states. J. Air Waste Manag. Assoc. 1999, 49, 1415–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behera, S.N.; Sharma, M. Investigating the potential role of ammonia in ion chemistry of fine particulate matter formation for an urban environment. Sci. Total Environ. 2010, 408, 3569–3575. [Google Scholar] [CrossRef]
- Gray, H.A.; Cass, G.R.; Huntzicker, J.J.; Heyerdahl, E.K.; Rau, J.A. Characteristics of Atmospheric Organic and Elemental Carbon Particle Concentrations in Los Angeles. Environ. Sci. Technol. 1986, 20, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Seigneur, C.; Seinfeld, J.H.; Jacobson, M.; Clegg, S.L.; Binkowski, F.S. A comparative review of inorganic aerosol thermodynamic equilibrium modules: Similarities, differences, and their likely causes. Atmos. Environ. 2000, 34, 117–137. [Google Scholar] [CrossRef]
- Lelieveld, J.; Evans, J.S.; Fnais, M.; Giannadaki, D.; Pozzer, A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature 2015, 525, 367–371. [Google Scholar] [CrossRef]
- Pope III, C.A.; Dockery, D.W. Health Effects of Fine Particulate Air Pollution: Lines that Connect. J. Air Waste Manag. 2006, 56, 709–742. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Ma, Z.; Jiachen, Z.; Huanhuan, D.; Jianmin, C.; Hui, C.; Xin, Y.; Wei, G.; Fuhai, G. Important role of ammonia on haze formation in Shanghai. Environ. Res. Lett. 2011, 6, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Park, R.S.; Lee, S.; Shin, S.-K.; Song, C.H. Contribution of ammonium nitrate to aerosol optical depth and direct radiative forcing by aerosols over East Asia. Atmos. Chem. Phys. 2014, 14, 2185–2201. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Penner, J.E. Global simulations of nitrate and ammonium aerosols and their radiative effects. Atmos. Chem. Phys. 2012, 12, 9479–9504. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Ma, Y.; Chen, M.; Zhang, Q.; Wang, L.; Khalizov, A.F.; Yao, L.; Wang, Z.; Wang, X.; Chen, L. Measurement of atmospheric amines and ammonia using the high resolution time-of-flight chemical ionization mass spectrometry. Atmos. Environ. 2015, 102, 249–259. [Google Scholar] [CrossRef]
- Ge, X.; Wexler, A.S.; Clegg, S.L. Atmospheric amines—Part I. A review. Atmos. Environ. 2011, 45, 524–546. [Google Scholar] [CrossRef]
- Hanson, D.R.; McMurry, P.H.; Jiang, J.; Tanner, D.; Huey, L.G. Ambient Pressure Proton Transfer Mass Spectrometry: Detection of Amines and Ammonia. Environ. Sci. Technol. 2011, 45, 8881–8888. [Google Scholar] [CrossRef]
- Hutchinson, G.L.; Mosier, A.R.; Andre, C.E. Ammonia and Amine Emissions from a Large Cattle Feedlot. J. Environ. Qual. 1982, 11, 288–293. [Google Scholar] [CrossRef]
- Schade, G.W.; Crutzen, P.J. Emission of aliphatic amines from animal husbandry and their reactions: Potential source of N2O and HCN. J. Atmos. Chem. 1995, 22, 319–346. [Google Scholar] [CrossRef]
- Chang, I.H.; Lee, C.G.; Lee, D.S. Development of an Automated Method for Simultaneous Determination of Low Molecular Weight Aliphatic Amines and Ammonia in Ambient Air by Diffusion Scrubber Coupled to Ion Chromatography. Anal. Chem. 2003, 75, 6141–6146. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.J.; Chen, M.L.; Di Ye, A.; Chou, M.S.; Shen, S.H.; Mao, I.F. The relationship of odor concentration and the critical components emitted from food waste composting plants. Atmos. Environ. 2008, 42, 8246–8251. [Google Scholar] [CrossRef]
- Yu, F.; Luo, G. Modeling of gaseous methylamines in the global atmosphere: Impacts of oxidation and aerosol uptake. Atmos. Chem. Phys. 2014, 14, 12455–12464. [Google Scholar] [CrossRef] [Green Version]
- Mäkelä, J.M.; Ylikoivisto, S.; Hiltunen, V.; Seidl, W.; Swietlicki, E.; Teinilä, K.; Sillanpää, M.; Koponen, I.K.; Paatero, J.; Rosman, K.; et al. Chemical composition of aerosol during particle formation events in boreal forest. Tellus Ser. B Chem. Phys. Meteorol. 2001, 53, 380–393. [Google Scholar] [CrossRef]
- Sorooshian, A.; Murphy, S.M.; Hersey, S.; Gates, H.; Padro, L.T.; Nenes, A.; Brechtel, F.J.; Jonsson, H.; Flagan, R.C.; Seinfeld, J.H. Comprehensive airborne characterization of aerosol from a major bovine source. Atmos. Chem. Phys. 2008, 8, 5489–5520. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.M.; Sorooshian, A.; Kroll, J.H.; Ng, N.L.; Chhabra, P.; Tong, C.; Surratt, J.D.; Knipping, E.; Flagan, R.C.; Seinfeld, J.H. Secondary aerosol formation from atmospheric reactions of aliphatic amines. Atmos. Chem. Phys. 2007, 7, 2313–2337. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Li, H.; Zhang, X.; Zeng, X.C.; Francisco, J.S. Nitric Acid-Amine Chemistry in the Gas Phase and at the Air-Water Interface. J. Am. Chem. Soc. 2018, 140, 6456–6466. [Google Scholar] [CrossRef] [PubMed]
- Kurtén, T.; Loukonen, V.; Vehkamäki, H.; Kulmala, M. Amines are likely to enhance neutral and ion-induced sulfuric acid-water nucleation in the atmosphere more effectively than ammonia. Atmos. Chem. Phys. 2008, 8, 4095–4103. [Google Scholar] [CrossRef] [Green Version]
- Kupiainen, O.; Ortega, I.K.; Kurtén, T.; Vehkamäki, H. Amine substitution into sulfuric acid-ammonia clusters. Atmos. Chem. Phys. 2012, 12, 3591–3599. [Google Scholar] [CrossRef] [Green Version]
- Paasonen, P.; Olenius, T.; Kupiainen, O.; Kurtén, T.; Petäjä, T.; Birmili, W.; Hamed, A.; Hu, M.; Huey, L.G.; Plass-Duelmer, C.; et al. On the formation of sulphuric acid-amine clusters in varying atmospheric conditions and its influence on atmospheric new particle formation. Atmos. Chem. Phys. 2012, 12, 9113–9133. [Google Scholar] [CrossRef] [Green Version]
- Olenius, T.; Halonen, R.; Kurtén, T.; Henschel, H.; Kupiainen-Määttä, O.; Ortega, I.K.; Jen, C.N.; Vehkamäki, H.; Riipinen, I. New particle formation from sulfuric acid and amines: Comparison of monomethylamine, dimethylamine, and trimethylamine. J. Geophys. Res. Atmos. 2017, 122, 7103–7118. [Google Scholar] [CrossRef]
- Yao, L.; Garmash, O.; Bianchi, F.; Zheng, J.; Yan, C.; Kontkanen, J.; Junninen, H.; Mazon, S.B.; Ehn, M.; Paasonen, P.; et al. Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity. Science 2018, 361, 278–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Price, D.; Praske, E.; Lee, S.A.; Shattuck, M.A.; Purvis-Roberts, K.; Silva, P.J.; Asa-Awuku, A.; Cocker, D.R. NO3 radical, OH radical and O3−initiated secondary aerosol formation from aliphatic amines. Atmos. Environ. 2013, 72, 105–112. [Google Scholar] [CrossRef]
- Duporté, G.; Riva, M.; Parshintsev, J.; Heikkinen, E.; Barreira, L.M.F.; Myllys, N.; Heikkinen, L.; Hartonen, K.; Kulmala, M.; Ehn, M.; et al. Chemical Characterization of Gas- and Particle-Phase Products from the Ozonolysis of α-Pinene in the Presence of Dimethylamine. Environ. Sci. Technol. 2017, 51, 5602–5610. [Google Scholar] [CrossRef]
- De Haan, D.O.; Hawkins, L.N.; Kononenko, J.A.; Turley, J.J.; Corrigan, A.L.; Tolbert, M.A.; Jimenez, J.L. Formation of nitrogen-containing oligomers by methylglyoxal and amines in simulated evaporating cloud droplets. Environ. Sci. Technol. 2011, 45, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Galloway, M.M.; Powelson, M.H.; Sedehi, N.; Wood, S.E.; Millage, K.D.; Kononenko, J.A.; Rynaski, A.D.; De Haan, D.O. Secondary organic aerosol formation during evaporation of droplets containing atmospheric aldehydes, amines, and ammonium sulfate. Environ. Sci. Technol. 2014, 48, 14417–14425. [Google Scholar] [CrossRef] [Green Version]
- Duporté, G.; Parshintsev, J.; Barreira, L.M.F.; Hartonen, K.; Kulmala, M.; Riekkola, M.L. Nitrogen-Containing Low Volatile Compounds from Pinonaldehyde-Dimethylamine Reaction in the Atmosphere: A Laboratory and Field Study. Environ. Sci. Technol. 2016, 50, 4693–4700. [Google Scholar] [CrossRef]
- Kampf, C.J.; Filippi, A.; Zuth, C.; Hoffmann, T.; Opatz, T. Secondary brown carbon formation: Via the dicarbonyl imine pathway: Nitrogen heterocycle formation and synergistic effects. Phys. Chem. Chem. Phys. 2016, 18, 18353–18364. [Google Scholar] [CrossRef] [Green Version]
- De Haan, D.O.; Hawkins, L.N.; Welsh, H.G.; Pednekar, R.; Casar, J.R.; Pennington, E.A.; De Loera, A.; Jimenez, N.G.; Symons, M.A.; Zauscher, M.; et al. Brown Carbon Production in Ammonium- or Amine-Containing Aerosol Particles by Reactive Uptake of Methylglyoxal and Photolytic Cloud Cycling. Environ. Sci. Technol. 2017, 51, 7458–7466. [Google Scholar] [CrossRef]
- De Haan, D.O.; Pajunoja, A.; Hawkins, L.N.; Welsh, H.G.; Jimenez, N.G.; De Loera, A.; Zauscher, M.; Andretta, A.D.; Joyce, B.W.; De Haan, A.C.; et al. Methylamine’s Effects on Methylglyoxal-Containing Aerosol: Chemical, Physical, and Optical Changes. ACS Earth Space Chem. 2019, 3, 1706–1716. [Google Scholar] [CrossRef]
- Bones, D.L.; Henricksen, D.K.; Mang, S.A.; Gonsior, M.; Bateman, A.P.; Nguyen, T.B.; Cooper, W.J.; Nizkorodov, S.A. Appearance of strong absorbers and fluorophores in limonene-O 3 secondary organic aerosol due to NH 4 + -mediated chemical aging over long time scales. J. Geophys. Res. 2010, 115, D05203. [Google Scholar] [CrossRef] [Green Version]
- Updyke, K.M.; Nguyen, T.B.; Nizkorodov, S.A. Formation of brown carbon via reactions of ammonia with secondary organic aerosols from biogenic and anthropogenic precursors. Atmos. Environ. 2012, 63, 22–31. [Google Scholar] [CrossRef]
- Lin, P.; Laskin, J.; Nizkorodov, S.A.; Laskin, A. Revealing Brown Carbon Chromophores Produced in Reactions of Methylglyoxal with Ammonium Sulfate. Environ. Sci. Technol. 2015, 49, 14257–14266. [Google Scholar] [CrossRef] [PubMed]
- Laskin, A.; Laskin, J.; Nizkorodov, S.A. Chemistry of Atmospheric Brown Carbon. Chem. Rev. 2015, 115, 4335–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Li, Y.J.; Wang, Y.; Bateman, A.P.; Zhang, Y.; Gong, Z.; Bertram, A.K.; Martin, S.T.; Paulson, J.A.; Chan, T.H. Highly Viscous States Affect the Browning of Atmospheric Organic Particulate Matter. ACS Cent. Sci. 2018, 4, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.J.; Liu, P.; Gong, Z.; Wang, Y.; Bateman, A.P.; Bergoend, C.; Bertram, A.K.; Martin, S.T. Chemical Reactivity and Liquid/Nonliquid States of Secondary Organic Material. Environ. Sci. Technol. 2015, 49, 13264–13274. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Liu, P.F.; Hanna, S.J.; Zaveri, R.A.; Potter, K.; You, Y.; Martin, S.T.; Bertram, A.K. Relative humidity-dependent viscosity of secondary organic material from toluene photo-oxidation and possible implications for organic particulate matter over megacities. Atmos. Chem. Phys. 2016, 16, 8817–8830. [Google Scholar] [CrossRef] [Green Version]
- Hinks, M.L.; Brady, M.V.; Lignell, H.; Song, M.; Grayson, J.W.; Bertram, A.K.; Lin, P.; Laskin, A.; Laskin, J.; Nizkorodov, S.A. Effect of viscosity on photodegradation rates in complex secondary organic aerosol materials. Phys. Chem. Chem. Phys. 2016, 18, 8785–8793. [Google Scholar] [CrossRef] [PubMed]
- Hinks, M.L.; Montoya-Aguilera, J.; Ellison, L.; Lin, P.; Laskin, A.; Laskin, J.; Shiraiwa, M.; Dabdub, D.; Nizkorodov, S.A. Effect of relative humidity on the composition of secondary organic aerosol from the oxidation of toluene. Atmos. Chem. Phys. 2018, 18, 1643–1652. [Google Scholar] [CrossRef] [Green Version]
- Canagaratna, M.R.; Jimenez, J.L.; Kroll, J.H.; Chen, Q.; Kessler, S.H.; Massoli, P.; Hildebrandt Ruiz, L.; Fortner, E.; Williams, L.R.; Wilson, K.R.; et al. Elemental ratio measurements of organic compounds using aerosol mass spectrometry: Characterization, improved calibration, and implications. Atmos. Chem. Phys. 2015, 15, 253–272. [Google Scholar] [CrossRef] [Green Version]
- McLafferty, F.W.; Turecek, F. Interpretation of Mass Spectra, 4th ed.; University Science Books: Sausalito, CA, USA, 1993. [Google Scholar]
- Price, D.J.; Clark, C.H.; Tang, X.; Cocker, D.R.; Purvis-Roberts, K.L.; Silva, P.J. Proposed chemical mechanisms leading to secondary organic aerosol in the reactions of aliphatic amines with hydroxyl and nitrate radicals. Atmos. Environ. 2014, 96, 135–144. [Google Scholar] [CrossRef]
- Moise, T.; Flores, J.M.; Rudich, Y. Optical Properties of Secondary Organic Aerosols and Their Changes by Chemical Processes. Chem. Rev. 2015, 115, 4400–4439. [Google Scholar] [CrossRef]
- Zhu, S.; Horne, J.R.; Montoya-Aguilera, J.; Hinks, M.L.; Nizkorodov, S.A.; Dabdub, D. Modeling reactive ammonia uptake by secondary organic aerosol in CMAQ: Application to the continental US. Atmos. Chem. Phys. 2018, 18, 3641–3657. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liggio, J.; Staebler, R.; Li, S.-M. Reactive uptake of ammonia to secondary organic aerosols: Kinetics of organonitrogen formation. Atmos. Chem. Phys. 2015, 15, 13569–13584. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
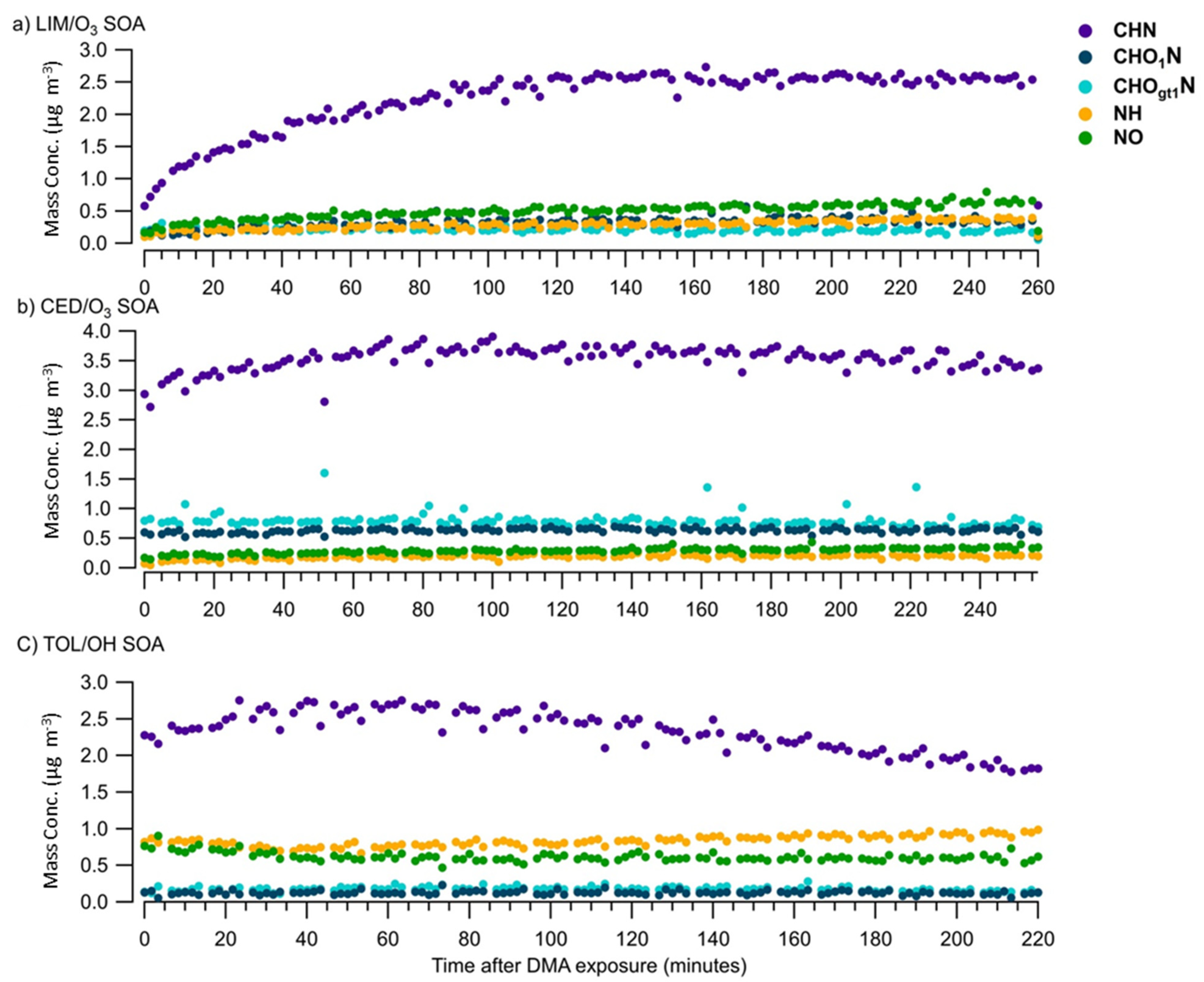{kind=link}
| Nitrogen Source | VOC/Oxidant | [VOC] (ppb) | [H2O2] (ppm) | [O3] (ppb) | RH (%) | ϒNH3 or DMA | Normalized ϒNH3 or DMA | NH3 Monitor | AMS | ∆N:C |
|---|---|---|---|---|---|---|---|---|---|---|
| nN Added (mol) | nN Added (mol) | |||||||||
| NH3 | LIM/OH | 50 | 1 | - | 50 | 3.9 × 10−4 | 9.7 × 10−6 | 1.7 × 10−5 | 4.2 × 10−7 | 0.005 |
| LIM/OH | 50 | 1 | - | 50 | 4.9 × 10−4 | 1.5 × 10−5 | 1.9 × 10−5 | 6.0 × 10−7 | 0.004 | |
| LIM/OH | 50 | 1 | - | 50 | 2.9 × 10−4 | 4.7 × 10−6 | 1.3 × 10−5 | 2.1 × 10−7 | 0.005 | |
| LIM/OH | 50 | 1 | - | 20 | 9.0 × 10−4 | 1.5 × 10−5 | 3.2 × 10−5 | 5.5 × 10−7 | 0.002 | |
| LIM/OH | 50 | 1 | - | <2 | 8.3 × 10−4 | 2.7 × 10−5 | 4.0 × 10−5 | 1.3 × 10−6 | 0.003 | |
| LIM/OH | 50 | 1 | - | <2 | 8.2 × 10−4 | 3.3 × 10−5 | 9.0 × 10−6 | 3.6 × 10−7 | 0.003 | |
| LIM/O3 | 50 | - | 500 | 50 | 2.0 × 10−4 | 2.5 × 10−6 | 2.6 × 10−5 | 3.3 × 10−7 | 0.008 | |
| LIM/O3 | 50 | - | 500 | 50 | 6.4 × 10−4 | 2.5 × 10−5 | 2.5 × 10−5 | 9.8 × 10−7 | 0.007 | |
| LIM/O3 | 50 | - | 500 | 50 | 5.0 × 10−4 | 3.2 × 10−5 | 1.7 × 10−5 | 1.1 × 10−6 | 0.011 | |
| LIM/O3 | 50 | - | 500 | 20 | 2.1 × 10−4 | 1.7 × 10−5 | 2.0 × 10−5 | 1.6 × 10−6 | 0.007 | |
| LIM/O3 | 50 | - | 500 | <2 | 2.0 × 10−4 | 1.2 × 10−5 | 1.3 × 10−5 | 8.0 × 10−7 | 0.004 | |
| DMA | LIM/O3 | 50 | - | 500 | 50 | 2.2 × 10−4 | 2.8 × 10−5 | 3.2 × 10−5 | 4.1 × 10−6 | 0.026 |
| LIM/O3 | 50 | - | 500 | 20 | 1.8 × 10−4 | 3.0 × 10−5 | 2.9 × 10−5 | 4.8 × 10−6 | 0.039 | |
| LIM/O3 | 50 | - | 500 | <2 | 3.4 × 10−4 | 8.0 × 10−5 | 3.6 × 10−5 | 8.5 × 10−6 | 0.073 | |
| CED/O3 | 50 | - | 500 | 50 | 3.3 × 10−4 | 1.5 × 10−5 | 3.1 × 10−5 | 1.4 × 10−6 | 0.017 | |
| CED/O3 | 50 | - | 500 | 20 | 2.0 × 10−4 | 1.2 × 10−5 | 3.6 × 10−5 | 2.1 × 10−6 | 0.021 | |
| CED/O3 | 50 | - | 500 | <2 | 2.1 × 10−4 | 3.2 × 10−5 | 3.2 × 10−5 | 4.8 × 10−6 | 0.048 | |
| TOL/OH | 1000 | 2 | - | 20 | 3.5 × 10−3 | 1.0 × 10−4 | 4.0 × 10−5 | 1.2 × 10−6 | 0.036 | |
| TOL/OH | 1000 | 2 | - | 20 | 2.5 × 10−3 | 7.6 × 10−5 | 3.6 × 10−5 | 1.1 × 10−6 | 0.040 | |
| TOL/OH | 1000 | 2 | - | <2 | 2.2 × 10−3 | 3.4 × 10−5 | 3.9 × 10−5 | 6.0 × 10−7 | 0.037 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, N.R.; Montoya-Aguilera, J.; Dabdub, D.; Nizkorodov, S.A. Effect of Humidity on the Reactive Uptake of Ammonia and Dimethylamine by Nitrogen-Containing Secondary Organic Aerosol. Atmosphere 2021, 12, 1502. https://0-doi-org.brum.beds.ac.uk/10.3390/atmos12111502
Smith NR, Montoya-Aguilera J, Dabdub D, Nizkorodov SA. Effect of Humidity on the Reactive Uptake of Ammonia and Dimethylamine by Nitrogen-Containing Secondary Organic Aerosol. Atmosphere. 2021; 12(11):1502. https://0-doi-org.brum.beds.ac.uk/10.3390/atmos12111502
Chicago/Turabian StyleSmith, Natalie R., Julia Montoya-Aguilera, Donald Dabdub, and Sergey A. Nizkorodov. 2021. "Effect of Humidity on the Reactive Uptake of Ammonia and Dimethylamine by Nitrogen-Containing Secondary Organic Aerosol" Atmosphere 12, no. 11: 1502. https://0-doi-org.brum.beds.ac.uk/10.3390/atmos12111502