
New Facile One-Pot Synthesis of Isobutyl Thiocarbamate in Recycling Solvent Mixture

 ,
,
Abstract
:1. Introduction
2. Experimental Part
2.1. Materials
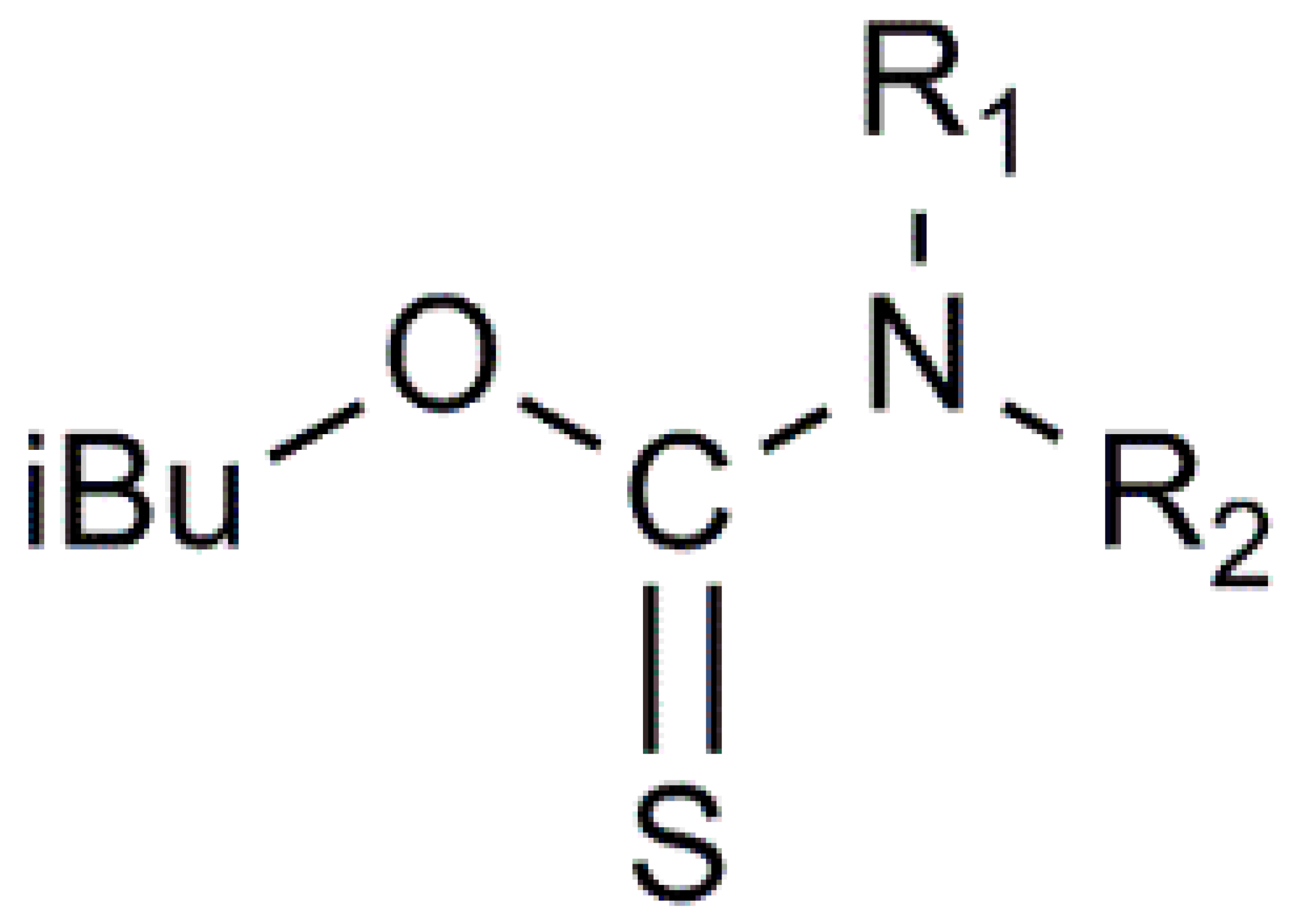
2.2. Thiocarbamate Synthesis
2.2.1. Optimal Laboratory Procedure for the Synthesis of N-Ethyl-O-Isobutyl Thiocarbamate (iBuOC(= S)NHEt) (Method A)
2.2.2. An Optimal Laboratory Procedure for the Synthesis of N-Ethyl-O-Ethyl Thiocarbamate (EtOC(= S)NHEt) (Method A)
2.2.3. Optimal Laboratory Procedure for the Synthesis of N-Ethyl-O-Isobutyl Thiocarbamates (Method B)
2.2.4. An Optimal Laboratory Procedure for the Synthesis of N-Ethyl-O-Ethyl Thiocarbamate (Method B)
2.2.5. Optimal Laboratory Procedure for the Synthesis of N-Ethyl-O-Isobutyl Thiocarbamate (Method C)
2.2.6. An Optimal Laboratory Procedure for the Synthesis of N-Ethyl-O-Ethyl Thiocarbamate (Method C)
2.2.7. Laboratory Procedure for the Synthesis of N-Ethyl-O-Isobutyl Thiocarbamates Using a Recycled Xylene
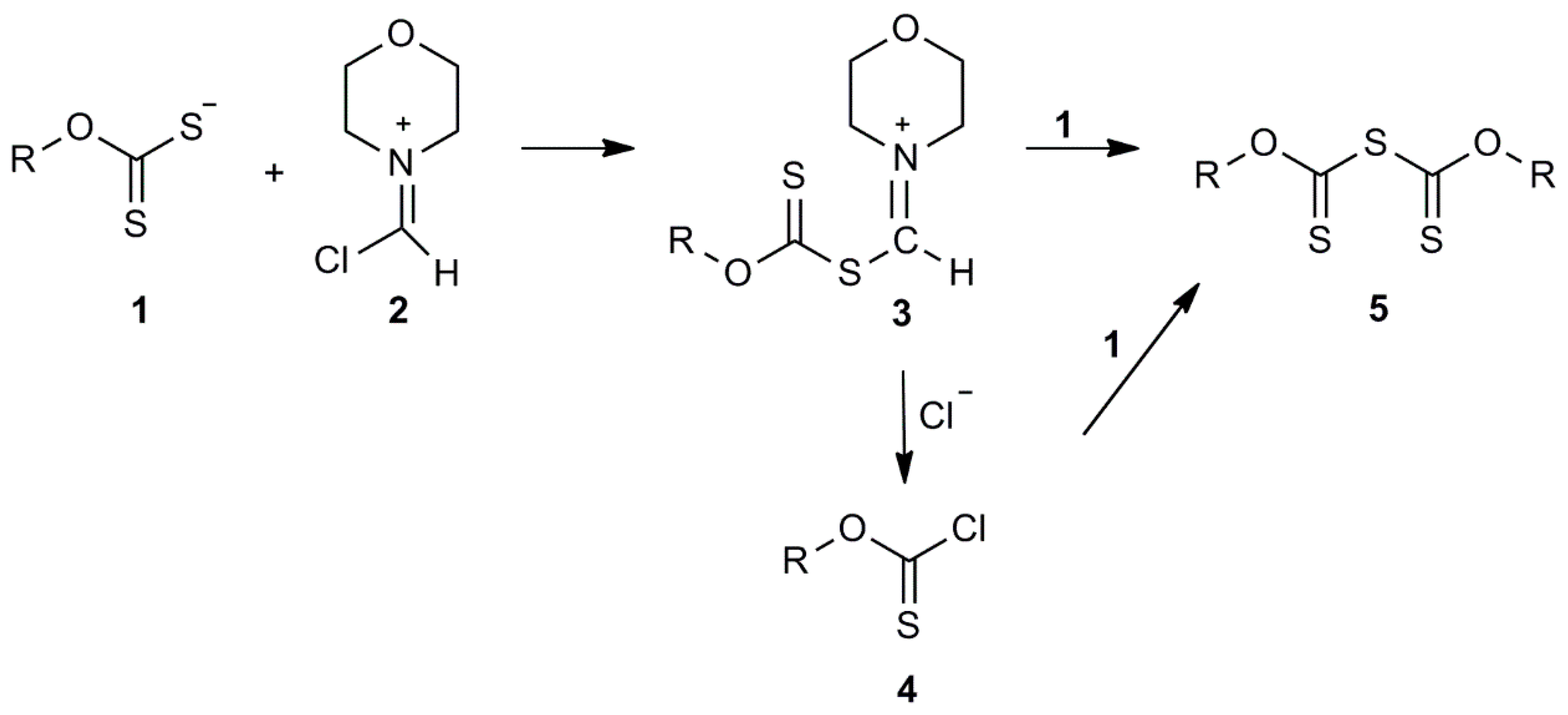
2.3. Laboratory Method for Defining the Reaction Mechanism of the Described Method for the Synthesis of O-Isobutyl Thiocarbamate
2.4. Structural Instrumental Analysis
- Injector temperature, 250 °C;
- Detector temperature, 270 °C;
- Column temperature program mode, 50 °C (5 min) → 10 °C/min → 130 °C (15 min);
- Carrier gas, nitrogen (purity 99.99%), flow of 1 cm3/min;
- Air flow, 250 cm3/min (purity 99.99%); and
- Hydrogen flow, 25 cm3/min (purity 99.99%).
3. Results
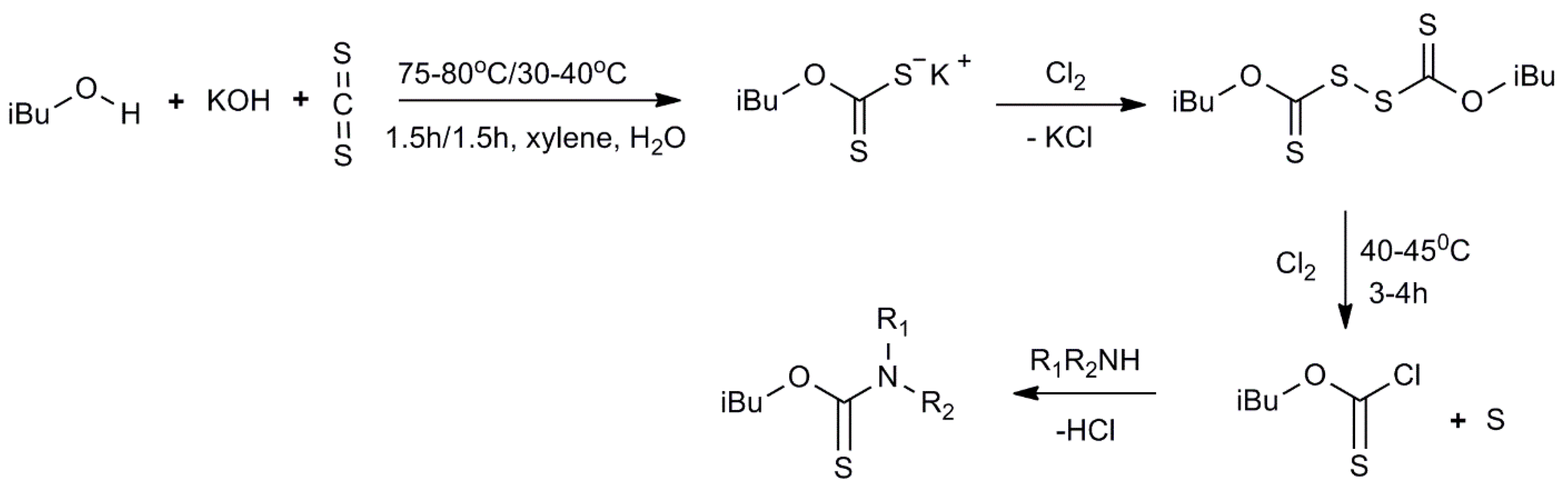
3.1. Synthesis of Thiocarbamates
3.2. Semi-Industrial Synthesis
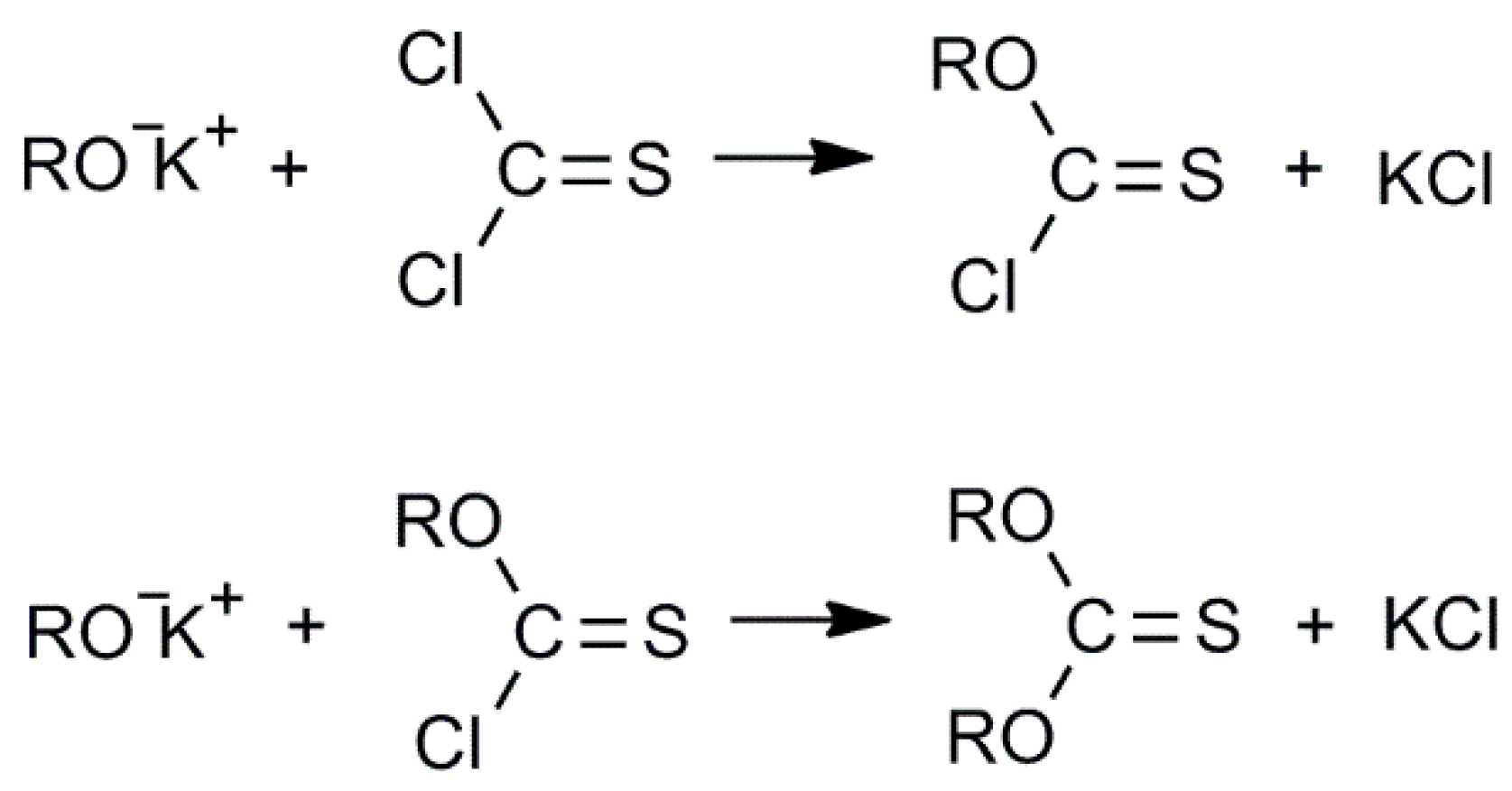
3.3. Confirmation of the Reaction Mechanism and Formation of the Intermediates
3.4. Application of The Obtained N-Ethyl-O-Isobutyl Thiocarbamate at the Laboratory Level of Flotation of Ore Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Walter, W.; Bode, K.D. Syntheses of thiocarbamates. Angew. Chem. Int. Ed. Engl. 1967, 79, 281–293. [Google Scholar] [CrossRef]
- Jamira, L.; Yella, R.; Patel, B.K. Efficient one-pot preparation of bisalkyl xanthogen disulfides from alcohols. J. Sulfur Chem. 2009, 30, 128–134. [Google Scholar] [CrossRef]
- Ma, Z.; Cheng, L.; Weng, X.; Gao, Y.; Huang, J. Study on Phase Transfer Catalyst Used in the Synthesis of Sodium Isobutyl Xanthate. Minerals 2021, 11, 850. [Google Scholar] [CrossRef]
- Zhang, J. An Investigation of the Adsorption of Xanthate on Bornite in Aqueous Solutions Using an Atomic Force Microscope. Minerals 2021, 11, 906. [Google Scholar] [CrossRef]
- Glazsrin, A.B.; Denisov, A.N.; Talzi, V.P.; Savin, B.P. Prommiilennostm po proizvodstvu mineralmnih udobreniy, seriz, Himicheskie sredstva zaoitmi rasteniy. In Tiokarbamats; Nauchno-Isledovalteski Institut Tehniko-Ekonomicheskii Isledovanii: Moskva, Russia, 1988; pp. 1–32. [Google Scholar]
- Weien, P.; Tao, S.; Weiping, L. Preparation Method of Xanthate. CN104774166A, 28 April 2015. [Google Scholar]
- Kemppinen, J.; Aaltonen, A.; Sihvonen, T.; Leppinen, J.; Sirén, H. Xanthate degradation occurring in flotation process waters of a gold concentrator plant. Miner. Eng. 2015, 80, 1–7. [Google Scholar] [CrossRef]
- Özün, S.; Ergen, G. Determination of optimum parameters for flotation of galena: Effect of chain length and chain structure of xanthates on flotation recovery. ACS Omega 2019, 4, 1516–1524. [Google Scholar] [CrossRef]
- Bai, L.; Liu, J.; Han, Y.; Jiang, K.; Zhao, W. Effects of xanthate on flotation kinetics of chalcopyrite and talc. Minerals 2018, 8, 369. [Google Scholar] [CrossRef] [Green Version]
- Abramov, A.A. Physico-chemical modeling of flotation systems. Miner. Procesing Extr. Metall. Rev. 1998, 19, 409–459. [Google Scholar] [CrossRef]
- Abramov, A.A.; Forssberg, K.S.E. Chemistry and optimal conditions for copper minerals flotation: Theory and practice. Miner. Process. Extr. Metall. Rev. 2005, 26, 77–143. [Google Scholar] [CrossRef]
- Li, Z.; Rao, F.; Song, S.; Uribe-Salas, A.; López-Valdivieso, A. Reexamining the Adsorption of Octyl Hydroxamate on Malachite Surface: Forms of Molecules and Anions. Miner. Process. Extr. Metall. Rev. 2020, 41, 178–186. [Google Scholar] [CrossRef]
- Bakalarz, A.; Gloy, G.; Luszczkiewicz, A. Flotation of sulfide components of copper ore in the presence of n-dodecane. Miner. Process. Extr. Metall. Rev. 2015, 36, 103–111. [Google Scholar] [CrossRef]
- Rao, S.R.; Finch, J.A. Base metal oxide flotation using long chain xanthates. Int. J. Miner. Process. 2003, 69, 251–258. [Google Scholar] [CrossRef]
- Lin, S.; Liu, R.; Bu, Y.; Wang, C.; Wang, L.; Sun, W.; Hu, Y. Oxidative depression of arsenopyrite by using calcium hypochlorite and sodium humate. Minerals 2018, 8, 463. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shao, Y.; Zhang, R.; Li, D.; Liu, Z.; Chen, H. Dating ore deposit using garnet U–Pb geochronology: Example from the xinqiao Cu–S–Fe–Au deposit, eastern china. Minerals 2018, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Niu, X.; Chen, J.; Li, Y.; Xia, L.; Li, L.; Sun, H.; Ruan, R. Correlation of surface oxidation with xanthate adsorption and pyrite flotation. Appl. Surf. Sci. 2019, 495, 143411–143419. [Google Scholar] [CrossRef]
- Nava-Alonso, F.; Pecina-Treviño, T.; Perez-Garibay, R.; Uribe-Salas, A. Pulp potential control in flotation: The effect of hydrogen peroxide addition on the extent of xanthate oxidation. Can. Metall. Q. 2002, 41, 391–397. [Google Scholar] [CrossRef]
- Kim, D.S.; Kuh, S.E.; Moon, K.S. Characteristics of xanthates related to hydrocarbon chain length. Geosystem Eng. 2000, 3, 30–34. [Google Scholar] [CrossRef]
- Chanturiya, V.A.; Kondratiev, S.A. Contemporary Understanding and Developments in the Flotation Theory of Non-Ferrous Ores. Miner. Process. Extr. Metall. Rev. 2019, 40, 390–401. [Google Scholar] [CrossRef]
- Minić, D. Hemija Pesticida; Panda Graf: Beograd, Serbia, 1994; pp. 282–286. [Google Scholar]
- Chisholm, M.H.; Extine, M.W. Reactions of transition metal-nitrogen σ bonds. 3. Early transition metal N,N-dimethylcarbamates. Preparation, properties, and carbon dioxide exchange reactions. J. Am. Chem. Soc. 1977, 99, 782–792. [Google Scholar] [CrossRef]
- Savin, V.P.; Tolzi, V.P.; Bek, N.O. Carbon-13 and proton NMR studies of salts formed as a result of the reaction of heterocumulenes: Carbon dioxide, carbonyl sulfide, and carbon disulfide with secondary amines. Zh. Org. Khim. 1984, 20, 1842–1851. [Google Scholar]
- Ewing, S.P.; Lochson, D.; Jencks, W.P. Mechanism of cleavage of carbamate anions. J. Am. Chem. Soc. 1980, 102, 3072–3084. [Google Scholar] [CrossRef]
- Milosavljević, M.M.; Ražić, S. Synthesis of N-alkyl O-ethylthioncarbamate and N,N-dialkylthioncarbamate and kinetics investigation by the integral mode of the tangential method. Vaprosy Him. I Him. Tehnol. 2005, 3, 50. [Google Scholar]
- Fuerst, E.P. Understanding the Mode of Action of the Chloroacetamide and Thiocarbamate Herbicides. Weed Technol. 1987, 1, 270–277. [Google Scholar] [CrossRef]
- Kunihiko, F.; Kuniak, S. Germicidal and Acaricidal Compositions. U.S. Patent 4101670, 18 July 1978. [Google Scholar]
- Rinehart, J.K. S-Naphthyl N-Alkylthiolcarbamates. U.S. Patent 4059609A, 22 November 1977. [Google Scholar]
- Rinehart, J.K. Method of Controlling Pea Aphids. U.S. Patent 4055656, 25 October 1977. [Google Scholar]
- Ruminski, P.G. Fluoroalkenyl Compounds and Their Use as Pest Repellents. U.S. Patent 5457134, 10 October 1995. [Google Scholar]
- Crudden, J.J. Bioactive Acid Agrichemical Compositions and Use Thereof. U.S. Patent 9241492B2, 26 January 2016. [Google Scholar]
- Kisida, H.; Hatakoshi, M. Thiocarbamate Compounds, and Their Use. U.S. Patent 4486449, 4 December 1984. [Google Scholar]
- Milosavljević, M. Kinetika i Optimizacija Reakcija Sinteze Tion-i Tiolkarbamata. Ph.D. Thesis, Faculty of Technology, University of Niš, Leskovac, Serbia, 2006; pp. 1–5. [Google Scholar]
- Millauer, H.; Edelmann, G. Process for the Preparation of Thiocarbamic Acid O-Esters. U.S. Patent 3963768, 15 June 1976. [Google Scholar]
- Calcagno, G. Preparation of Thion- and Thiol-Carbamic Esters. U.S. Patent 4298524, 3 November 1981. [Google Scholar]
- Movassagh, B.; Soleiman-Beigi, M. Synthesis of thiocarbamates from thiols and isocyanates under catalyst-and solvent-free conditions. Mon. Für Chem. Chem. Mon. 2008, 139, 137–140. [Google Scholar] [CrossRef]
- Hall, V.J.; Siasios, G.; Tiekink, E.R.T. Triorganophosphinegold(I) carbonimidothioates. Aust. J. Chem. 1993, 46, 561–570. [Google Scholar] [CrossRef]
- Milosavljević, M.; Marinković, A.D.; Đorđević, S. Sinteza N-i N, N-dialkil-S-alkiltiol-karbamata premeštanjem N-i N,N-dialkil-O-alkiltionkarbamata. Hem. Ind. 2006, 60, 27–32. [Google Scholar]
- Movassagh, B.; Zakinezhad, Y. A New one-pot synthesis of thiocarbamates from isocyanates and disulfides in the presence of Zn/AlCl3 System. Chem. Lett. 2005, 34, 1330–1331. [Google Scholar] [CrossRef]
- Karrer, F. Phenoxy-Phenoxy-Alkyl-Thionocarbamate Compounds. U.S. Patent 4060629, 29 November 1977. [Google Scholar]
- Reich, P.; Martin, D. Cyanic acid esters. IV. Molecule spectroscopic investigations of cyanic acid esters. Chem. Ber. 1965, 98, 2063–2069. [Google Scholar] [CrossRef]
- Walter, W.; Bode, K.D. Oxidation products of thiocarboxylic acid amides. XV: Oxidation of thiocarbamic acid O-aryl esters to ortho-substituted aryloxyiminomethanesulfenic acids. Justus Liebigs Ann. Der Chem. 1966, 698, 122–130. [Google Scholar] [CrossRef]
- Jonhson, G.; Rafferty, M.F. Amide, Sulfonamide, Urea, Carbamate, Thiocarbamate, and Thiourea Derivatives of 4-hydroxybenzylamine Having Anti-Inflammatory and Analgesic Activity. U.S. Patent 4980366, 25 December 1990. [Google Scholar]
- Milisavljević, S.S.; Marinković, A.D.; Milosavljević, M.M. Novi postupak sinteze N-alkil-i N,N-dialkil-O-etil-.i O-izopropiltiokarbamata oksidacijom aminskih soli ksantogene kiseline. Hem. Ind. 2010, 64, 401–409. [Google Scholar]
- Milosavljevic, M.; Sovrlić, M.; Marinkovic, A.D.; Milenković, D.D. A synthesis of N-alkyl and N,N-dialkyl O-ethyl thiocarbamates from diethyl dixanthogenate using different oxidants. Mon. Für Chem. Chem. Mon. 2010, 141, 749. [Google Scholar] [CrossRef]
- Newman, M.S.; Hetzel, F.W. Thiophenols from Phenols: 2-Naphthalenethiol. Org. Synth. 2003, 51, 139. [Google Scholar]
- Harayama, H.; Nagahama, T.; Kozera, T.; Kimura, M.; Fugami, K.; Tanaka, S.; Tamaru, Y. Aromatic Allylsulfenylation with in situ Generated Allylic Thiols under the Heck Conditions. Bull. Chem. Soc. Jpn. 1997, 70, 445–456. [Google Scholar] [CrossRef]
- Dzurilla, M.; Kutschy, P.; Koscik, D.; Toma, S. Intramolecular cyclization of O-alkyl-N-(3-phenyl-propenoyl)-thiocarbametes catalyzed by boron trifluoride. Coll Czech Chem Commun. 1990, 55, 710. [Google Scholar] [CrossRef]
- Böhme, A.; Gais, H.J. Palladium (0) catalyzed enantioselective rearrangement of O-allylic thiocarbamates to S-allylic thiocarbamates: Asymmetric synthesis of allylic thiols. Tetrahedron Asymmetry 1999, 10, 2510. [Google Scholar] [CrossRef]
- Sovrlić, M.Ž.; Milosavljević, M.M.; Marinković, A.D.; Đukanović, J.S.; Brković, D.V.; Konstantinović, S.S. Uporedna analiza oksidativnih postupaka sinteze N-alkil, N,N-dialkil i N-cikloalkil-O-izobutil tionkarbamata. Hem. Ind. 2011, 65, 541–549. [Google Scholar]
- Shrivash, M.K.; Adeppa, K.; Singh, R.; Pandey, J.; Misra, K. A Novel, Efficient and Multigram Scale Synthesis of S-Alkyl thiocarbamates via Newman Kwart Rearrangement. Proc. Natl. Acad. Sci. India Sect. A Phys. Sci. 2017, 87, 189–193. [Google Scholar] [CrossRef]
- Milosavljević, M.M.; Marinković, A.D.; Rančić, M.; Milentijević, G.; Bogdanović, A.; Cvijetić, I.N.; Gurešić, D. New Eco-Friendly Xanthate-Based Flotation Agents. Minerals 2020, 10, 350. [Google Scholar] [CrossRef] [Green Version]
- Hudson, H.R.; Koplick, A.J. The Thermal and Mass Spectral Fragmentation of N-Butyl Thiolo-, Thiono-, and Dithio-Chloroformate. Phosphorus Sulfur Silicon Relat. Elem. 2011, 186, 1635–1649. [Google Scholar] [CrossRef]
- Martinez, M.A.; Vega, J.C. Synthesis of O-alkyl carbonochloridothioates. Synthesis 1986, 9, 760–761. [Google Scholar] [CrossRef]
- Megan, A.; William, F.; Bylund, E.; Holubowitch, N.E.; Abelt, C.J. Synthesis of Chlorothioformates from Xanthates. Synthesis 2006, 24, 4118–4120. [Google Scholar]
- Borths, C.J.; Chan, J.; Burke, B.J.; Larsen, R.D. Synthesis of O-Ethyl Thioformate: A Useful Reagent for the Thioformylation of Amines. Synlett 2009, 3139–3142. [Google Scholar] [CrossRef]
- Mueller, W.; Butler, P.E. The Reaction of Sylfenyl shlorides with Allone. J. Org. Chem. 1968, 33, 1533–1537. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Reaction Conditions | GC % | Yield d % | |||
|---|---|---|---|---|---|---|
| Time c h | Temperature d °C | Mols ratio/mol b iBuOH/KOH/CS2/Cl2/EtNH2:KOH | ||||
| iBuOC(S)NHEt | A | 1.5/1.5/3.0–4.0/1.5 | 75–80/35–40/40–45/30–35 | 0.2/0.2/0.2/0.21/0.4:0.0 | 98.5 | 81.2 |
| B e | 1.5/1.5/3.0–4.0/2.0 | 75–80/35–40/40–45/30–35 | 0.2/0.2/0.2/0.21/0.2:0.2 | 99.2 | 80.0 | |
| C f | 1.5/1.5/3.0–4.0/1.5 | 75–80/35–40/40–45/20–25 | 0.2/0.2/0.2/0.21/0.4:0.0 | 98.9 | 82.5 | |
| EtOC(S)NHEt | A | 1.5/1.5/3.0–4.0/1.5 | 75–80/35–40/40–45/30–35 | 0.2/0.2/0.2/0.21/0.4:0.0 | 98.7 | 82.5 |
| B e | 1.5/1.5/3.0–4.0/2.0 | 75–80/35–40/40–45/30–35 | 0.2/0.2/0.2/0.21/0.2:0.2 | 99.4 | 81.0 | |
| C f | 1.5/1.5/3.0–4.0/1.5 | 75–80/35–40/40–45/20–25 | 0.2/0.2/0.2/0.21/0.4:0.0 | 98.8 | 84.5 | |
| R1 | R2 | Boiling Point b/°C | GC Analysis | Yield c/% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Method a | Lit. [50] | Method a | |||||||||
| A | B | C | A | B | C | A | B | C | |||
| Et | H | 114–124 | 120–123 | 119–123 | 120–122 | 98.5 | 99.2 | 98.9 | 81.2 | 80.0 | 82.5 |
| Pr | H | 129–131 | 128–131 | 128–129 | 130–132 | 98.4 | 98.6 | 98.9 | 81.8 | 80.8 | 83.0 |
| Bu | H | 139–140 | 138–140 | 139–142 | 140–142 | 99.0 | 98.9 | 99.1 | 81.5 | 80.5 | 82.6 |
| iPr | H | 127–129 | 127–130 | 127–130 | 128–130 | 98.7 | 98.7 | 98.8 | 80.9 | 71.8 | 81.5 |
| iBu | H | 147–149 | 147–149 | 147–149 | 148–150 | 98.7 | 98.6 | 98.8 | 82.0 | 81.8 | 82.7 |
| iPe | H | 190–192 | 190–192 | 191–193 | 192–193 | 98.7 | 98.7 | 98.8 | 82.8 | 81.9 | 83.4 |
| sBu | H | 136–138 | 136–138 | 137–139 | 137–139 | 98.6 | 98.4 | 98.8 | 81.4 | 80.4 | 82.3 |
| cycPr | H | 133–136 | 133–136 | 134–137 | 134–136 | 98.6 | 98.6 | 98.8 | 78.9 | 77.0 | 79.3 |
| cycPe | H | 178–180 | 178–181 | 179–181 | 180–182 | 98.7 | 98.7 | 98.8 | 80.6 | 80.0 | 82.1 |
| cycHe | H | 177–179 | 177–180 | 178–180 | 179–180 | 99.0 | 98.9 | 99.2 | 81.4 | 81.0 | 82.8 |
| Et | Et | 142–145 | 143–145 | 144–147 | 144–146 | 98.7 | 98.9 | 99.1 | 82.4 | 81.9 | 83.0 |
| Pr | Pr | 176–178 | 176–179 | 178–180 | 178–179 | 99.1 | 99.0 | 99.2 | 83.3 | 82.5 | 83.9 |
| Bu | Bu | 190–192 | 190–192 | 191–193 | 192–194 | 99.2 | 99.0 | 99.3 | 83.6 | 83.0 | 83.8 |
| EXAMPLE | Reactants a | Reaction Conditions | Byproducts | Product | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KOH | iBuOH | CS2 | Amine | Time b | Temp c | Sulphur | Yield | GC | ||||||
| kg | kmol | kg | kmol | kg | kmol | m3 | kmol | h | °C | kg | kg | % | % | |
| 1 | 60.0 | 1.1 | 81.2 | 1.1 | 77.5 | 1.0 | 0.83 | 1.0 | 1.5/1.5 | 75/35 | 28.5 | 128.9 | 80.1 | 99.2 |
| 2 | 60.0 | 1,1 | 81.2 | 1.1 | 85.2 | 1.1 | 0.83 | 1.0 | 1.5/1.5 | 80/35 | 29.0 | 132.3 | 82.2 | 98.9 |
| 3 | 54.5 | 1.0 | 73.8 | 1.0 | 85.2 | 1.1 | 1.66 | 2.0 | 1.5/2.0 | 80/35 | 29.1 | 132.8 | 82.5 | 99.1 |
| Components | OKCu [%Cu] | KKCu [%Cu] | UKCu [%Cu] | Tailing [%Cu] |
|---|---|---|---|---|
| iButOC(S)NHEt (method A) | 68.12 | 23.55 | 91.67 | 8.33 |
| iButOC(S)NHEt (method B) | 68.11 | 23.96 | 92.07 | 7.93 |
| iButOC(S)NHEt (method C) | 68.25 | 24.89 | 93.14 | 6.86 |
| EtOC(S)NHEt (method C) | 67.10 | 23.00 | 90.10 | 9.90 |
| iPrOC(S)NHEt | 68.00 | 23.54 | 91.54 | 8.46 |
| KiBuX | 66.85 | 24.07 | 90.92 | 9.08 |
| KAmX | 66.02 | 23.96 | 89.98 | 10.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milentijević, G.; Marinković, A.D.; Rančić, M.; Bogdanović, A.; Prlainović, N.; Marković, S.; Milosavljević, M. New Facile One-Pot Synthesis of Isobutyl Thiocarbamate in Recycling Solvent Mixture. Minerals 2021, 11, 1346. https://0-doi-org.brum.beds.ac.uk/10.3390/min11121346
Milentijević G, Marinković AD, Rančić M, Bogdanović A, Prlainović N, Marković S, Milosavljević M. New Facile One-Pot Synthesis of Isobutyl Thiocarbamate in Recycling Solvent Mixture. Minerals. 2021; 11(12):1346. https://0-doi-org.brum.beds.ac.uk/10.3390/min11121346
Chicago/Turabian StyleMilentijević, Goran, Aleksandar D. Marinković, Milica Rančić, Aleksandra Bogdanović, Nevena Prlainović, Smiljana Marković, and Milutin Milosavljević. 2021. "New Facile One-Pot Synthesis of Isobutyl Thiocarbamate in Recycling Solvent Mixture" Minerals 11, no. 12: 1346. https://0-doi-org.brum.beds.ac.uk/10.3390/min11121346