
Mitochondria May Mediate Prenatal Environmental Influences in Autism Spectrum Disorder

 ,
, 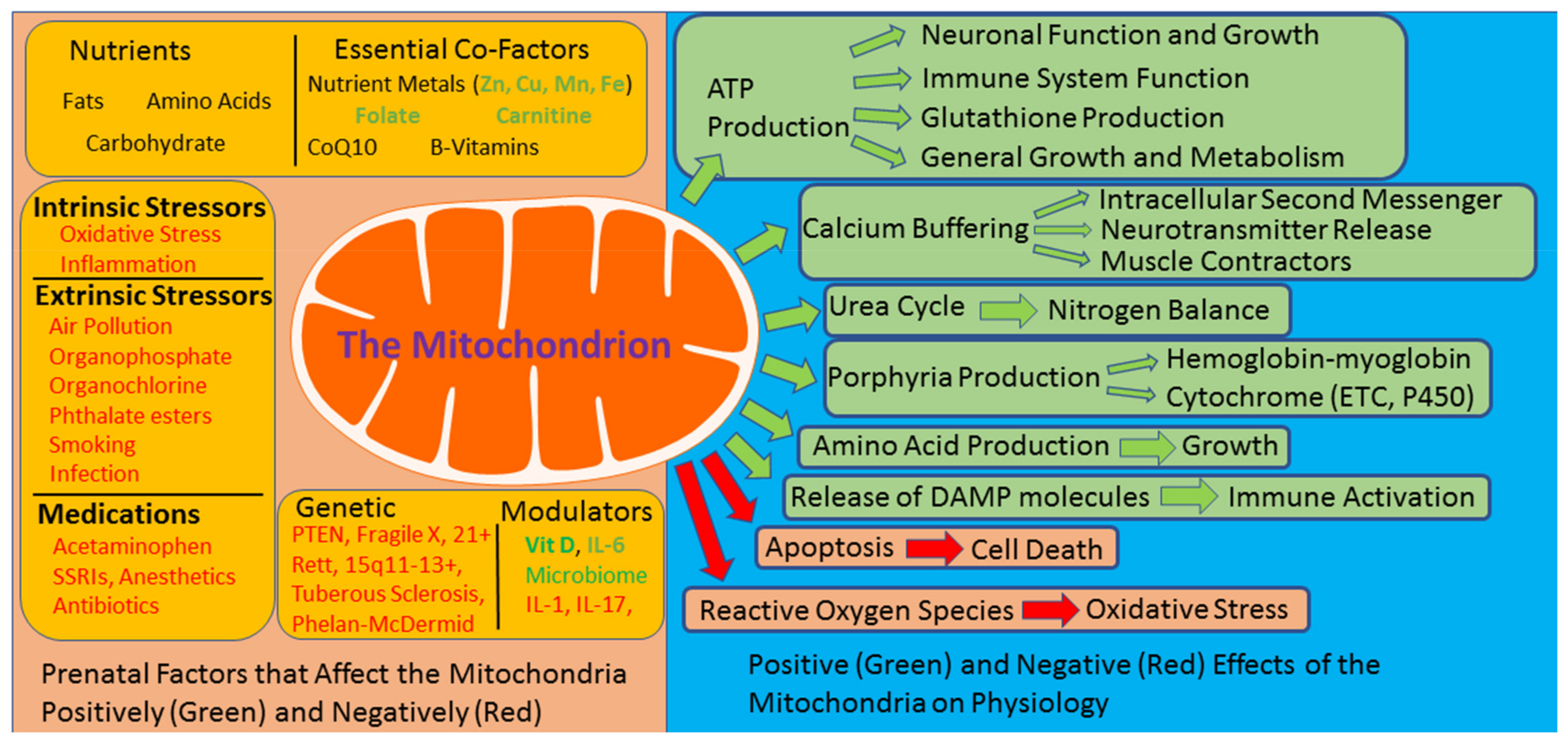{kind=link}
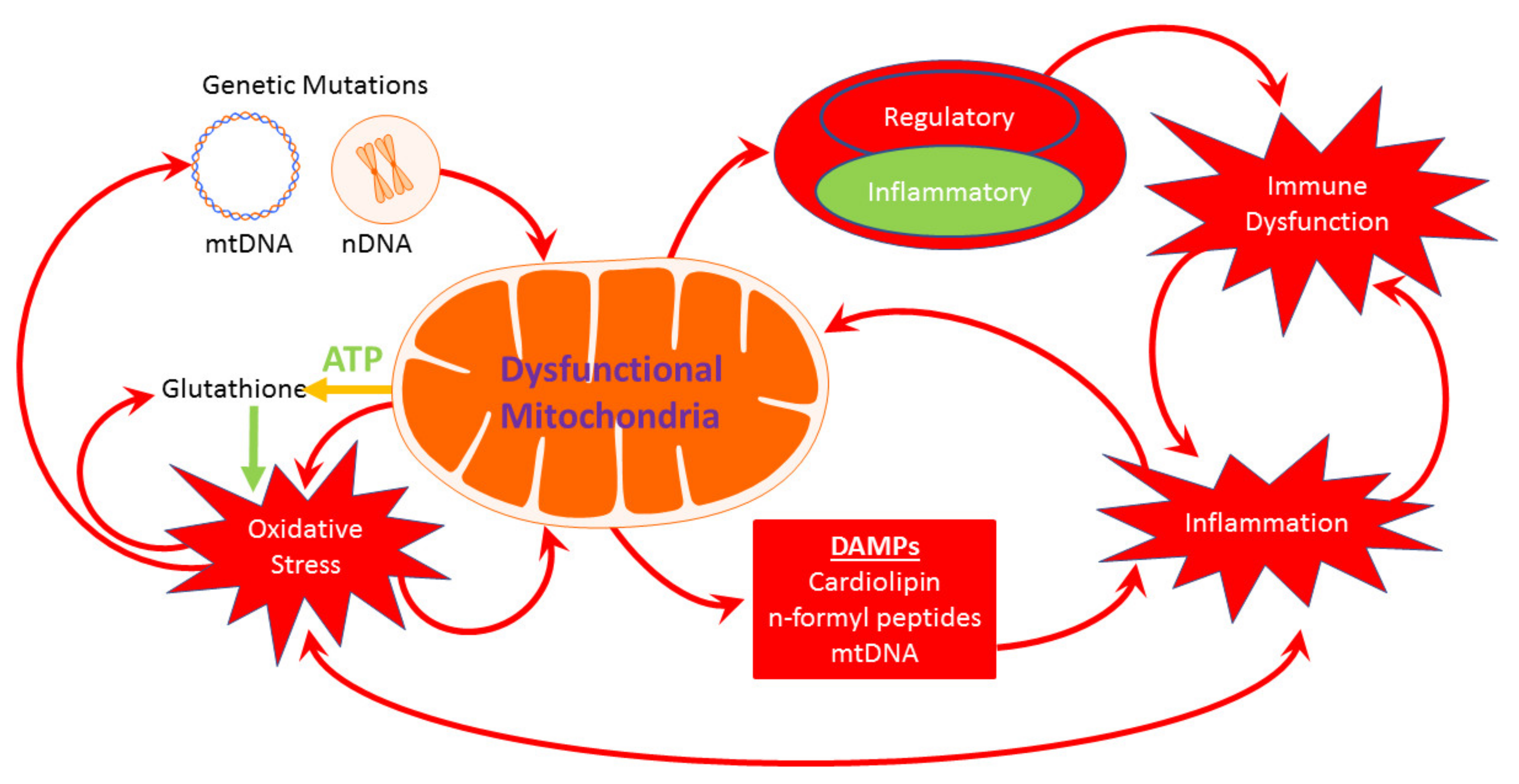{kind=link}
Abstract
:1. Introduction
2. The Mitochondria: Dysfunction Can Be Self-Perpetuating
3. Prenatal Risk Factors for ASD Modulate Mitochondrial Function
4. Unique Abnormalities in Mitochondrial Function Are Prevalent in ASD
5. The Significance of Mitochondrial Dysfunction in ASD: Sensitivity to Physiological Stress
6. Unique ASD Mitochondrial Abnormalities May Be Linked to Both Environmental and Genetic Factors
7. Long-Term Induced Changes in Mitochondrial Function: Adaptive or Maladaptive
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2016. Morb. Mortal. Wkly. Rep. Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, G.B.; Mendelsohn, N.J. Professional Practice Guidelines Committee. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet. Med. 2013, 15, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef]
- Hertz-Picciotto, I.; Schmidt, R.J.; Krakowiak, P. Understanding environmental contributions to autism: Causal concepts and the state of science. Autism Res. Off. J. Int. Soc. Autism Res. 2018, 11, 554–586. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Genuis, S.J.; Frye, R.E. Environmental toxicants and autism spectrum disorders: A systematic review. Transl. Psychiatry 2014, 4, e360. [Google Scholar] [CrossRef] [PubMed]
- Panisi, C.; Guerini, F.R.; Abruzzo, P.M.; Balzola, F.; Biava, P.M.; Bolotta, A.; Brunero, M.; Burgio, E.; Chiara, A.; Clerici, M.; et al. Autism spectrum disorder from the womb to adulthood: Suggestions for a paradigm shift. J. Pers. Med. 2021, 11, 70. [Google Scholar] [CrossRef]
- Jose, C.; Melser, S.; Benard, G.; Rossignol, R. Mitoplasticity: Adaptation biology of the mitochondrion to the cellular redox state in physiology and carcinogenesis. Antioxid. Redox Signal. 2013, 18, 808–849. [Google Scholar] [CrossRef]
- Frye, R.E.; Rossignol, D.A. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr. Res. 2011, 69, 41R–47R. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, R.E.; Rossignol, D. Mitochondrial physiology and autism spectrum disorder. OA Autism 2013, 1. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef] [Green Version]
- Banoth, B.; Cassel, S.L. Mitochondria in innate immune signaling. Transl. Res. 2018, 202, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 2014, 5, 150. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frustaci, A.; Neri, M.; Cesario, A.; Adams, J.B.; Domenici, E.; Dalla Bernardina, B.; Bonassi, S. Oxidative stress-related biomarkers in autism: Systematic review and meta-analyses. Free Radic. Biol. Med. 2012, 52, 2128–2141. [Google Scholar] [CrossRef]
- Napoli, E.; Wong, S.; Giulivi, C. Evidence of reactive oxygen species-mediated damage to mitochondrial DNA in children with typical autism. Mol. Autism 2013, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jyonouchi, H.; Geng, L.; Rose, S.; Bennuri, S.C.; Frye, R.E. Variations in mitochondrial respiration differ in IL-1ß/IL-10 ratio based subgroups in autism spectrum disorders. Front. Psychiatry 2019, 10, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delhey, L.M.; Nur Kilinc, E.; Yin, L.; Slattery, J.C.; Tippett, M.L.; Rose, S.; Bennuri, S.C.; Kahler, S.G.; Damle, S.; Legido, A.; et al. The effect of mitochondrial supplements on mitochondrial activity in children with autism spectrum disorder. J. Clin. Med. 2017, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, S.J.; Melnyk, S.; Jernigan, S.; Pavliv, O.; Trusty, T.; Lehman, S.; Seidel, L.; Gaylor, D.W.; Cleves, M.A. A functional polymorphism in the reduced folate carrier gene and DNA hypomethylation in mothers of children with autism. Am. J. Med Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2010, 153b, 1209–1220. [Google Scholar] [CrossRef] [Green Version]
- Quadros, E.V.; Sequeira, J.M.; Brown, W.T.; Mevs, C.; Marchi, E.; Flory, M.; Jenkins, E.C.; Velinov, M.T.; Cohen, I.L. Folate receptor autoantibodies are prevalent in children diagnosed with autism spectrum disorder, their normal siblings and parents. Autism Res. Off. J. Int. Soc. Autism Res. 2018, 11, 707–712. [Google Scholar] [CrossRef]
- Sequeira, J.M.; Desai, A.; Berrocal-Zaragoza, M.I.; Murphy, M.M.; Fernandez-Ballart, J.D.; Quadros, E.V. Exposure to folate receptor alpha antibodies during gestation and weaning leads to severe behavioral deficits in rats: A pilot study. PLoS ONE 2016, 11, e0152249. [Google Scholar] [CrossRef]
- Curtin, P.; Austin, C.; Curtin, A.; Gennings, C.; Arora, M.; Tammimies, K.; Willfors, C.; Berggren, S.; Siper, P.; Rai, D.; et al. Dynamical features in fetal and postnatal zinc-copper metabolic cycles predict the emergence of autism spectrum disorder. Sci. Adv. 2018, 4, eaat1293. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; Cakir, J.; Rose, S.; Delhey, L.; Bennuri, S.C.; Tippett, M.; Melnyk, S.; James, S.J.; Palmer, R.F.; Austin, C.; et al. Prenatal air pollution influences neurodevelopment and behavior in autism spectrum disorder by modulating mitochondrial physiology. Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Zhong, C.; Tessing, J.; Lee, B.K.; Lyall, K. Maternal dietary factors and the risk of autism spectrum disorders: A systematic review of existing evidence. Autism Res. Off. J. Int. Soc. Autism Res. 2020. [Google Scholar] [CrossRef]
- Frye, R.E. Mitochondrial dysfunction in autism spectrum disorder: Unique abnormalities and targeted treatments. Semin. Pediatr. Neurol. 2020, 35, 100829. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and molecular characteristics of mitochondrial dysfunction in autism spectrum disorder. Mol. Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Zhao, B.; He, S.; Weng, R.; Wang, B.; Ding, Y.; Huang, X.; Luo, Q. The alteration of carnitine metabolism in second trimester in GDM and a nomogram for predicting macrosomia. J. Diabetes Res. 2020, 2020, 4085757. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Zhang, C.; Li, H.; Luan, S.; Liu, C. Association of maternal diabetes with autism spectrum disorders in offspring: A systemic review and meta-analysis. Medicine 2018, 97, e9438. [Google Scholar] [CrossRef] [PubMed]
- Celestino-Soper, P.B.; Violante, S.; Crawford, E.L.; Luo, R.; Lionel, A.C.; Delaby, E.; Cai, G.; Sadikovic, B.; Lee, K.; Lo, C.; et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc. Natl. Acad. Sci. USA 2012, 109, 7974–7981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Kim, H.K.; Kwon, J.T.; Park, S.; Park, H.J.; Kim, S.K.; Park, J.K.; Kang, W.S.; Kim, Y.J.; Chung, J.H.; et al. BBOX1 is down-regulated in maternal immune-activated mice and implicated in genetic susceptibility to human schizophrenia. Psychiatry Res. 2018, 259, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Bankaitis, V.A.; Xie, Z. The neural stem cell/carnitine malnutrition hypothesis: New prospects for effective reduction of autism risk? J. Biol. Chem. 2019, 294, 19424–19435. [Google Scholar] [CrossRef] [Green Version]
- Hollowood, K.; Melnyk, S.; Pavliv, O.; Evans, T.; Sides, A.; Schmidt, R.J.; Hertz-Picciotto, I.; Elms, W.; Guerrero, E.; Kruger, U.; et al. Maternal metabolic profile predicts high or low risk of an autism pregnancy outcome. Res. Autism Spectr. Disord. 2018, 56, 72–82. [Google Scholar] [CrossRef]
- James, S.J.; Melnyk, S.; Jernigan, S.; Hubanks, A.; Rose, S.; Gaylor, D.W. Abnormal transmethylation/transsulfuration metabolism and DNA hypomethylation among parents of children with autism. J. Autism Dev. Disord. 2008, 38, 1966–1975. [Google Scholar] [CrossRef]
- Bauman, M.D.; Van de Water, J. Translational opportunities in the prenatal immune environment: Promises and limitations of the maternal immune activation model. Neurobiol. Dis. 2020, 141, 104864. [Google Scholar] [CrossRef]
- Naviaux, R.K.; Zolkipli, Z.; Wang, L.; Nakayama, T.; Naviaux, J.C.; Le, T.P.; Schuchbauer, M.A.; Rogac, M.; Tang, Q.; Dugan, L.L.; et al. Antipurinergic therapy corrects the autism-like features in the poly(IC) mouse model. PLoS ONE 2013, 8, e57380. [Google Scholar] [CrossRef]
- Giulivi, C.; Napoli, E.; Schwartzer, J.; Careaga, M.; Ashwood, P. Gestational exposure to a viral mimetic poly(i:C) results in long-lasting changes in mitochondrial function by leucocytes in the adult offspring. Mediat. Inflamm. 2013, 2013, 609602. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.L.; Croen, L.A.; Yoshida, C.K.; Heuer, L.; Hansen, R.; Zerbo, O.; DeLorenze, G.N.; Kharrazi, M.; Yolken, R.; Ashwood, P.; et al. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol. Psychiatry 2017, 22, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, H.; Hoeffer, C. Maternal IL-17A in autism. Exp. Neurol. 2018, 299, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Kwon, J.E.; Lee, S.Y.; Lee, E.J.; Kim, D.S.; Moon, S.J.; Lee, J.; Kwok, S.K.; Park, S.H.; Cho, M.L. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis. 2017, 8, e2565. [Google Scholar] [CrossRef]
- Kakareko, M.; Jabłońska, E.; Ratajczak-Wrona, W. Role of rhIL-17 in regulating the mitochondrial pathway proteins in peripheral blood neutrophils. Clin. Lab. 2015, 61, 345–351. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, H.; Yu, M.; Schugar, R.C.; Qian, W.; Tang, F.; Liu, W.; Yang, H.; McDowell, R.E.; Zhao, J.; et al. IL-1 induces mitochondrial translocation of IRAK2 to suppress oxidative metabolism in adipocytes. Nat. Immunol. 2020, 21, 1219–1231. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; Ye, J. IL-6: A potential role in cardiac metabolic homeostasis. Int. J. Mol. Sci. 2018, 19, 2474. [Google Scholar] [CrossRef] [Green Version]
- Karami-Mohajeri, S.; Abdollahi, M. Toxic influence of organophosphate, carbamate, and organochlorine pesticides on cellular metabolism of lipids, proteins, and carbohydrates: A systematic review. Hum. Exp. Toxicol. 2011, 30, 1119–1140. [Google Scholar] [CrossRef]
- Posnack, N.G.; Swift, L.M.; Kay, M.W.; Lee, N.H.; Sarvazyan, N. Phthalate exposure changes the metabolic profile of cardiac muscle cells. Environ. Health Perspect. 2012, 120, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Pant, N.; Shukla, M.; Kumar Patel, D.; Shukla, Y.; Mathur, N.; Kumar Gupta, Y.; Saxena, D.K. Correlation of phthalate exposures with semen quality. Toxicol. Appl. Pharmacol. 2008, 231, 112–116. [Google Scholar] [CrossRef]
- Breton, C.V.; Song, A.Y.; Xiao, J.; Kim, S.J.; Mehta, H.H.; Wan, J.; Yen, K.; Sioutas, C.; Lurmann, F.; Xue, S.; et al. Effects of air pollution on mitochondrial function, mitochondrial DNA methylation, and mitochondrial peptide expression. Mitochondrion 2019, 46, 22–29. [Google Scholar] [CrossRef]
- Aghapour, M.; Remels, A.H.V.; Pouwels, S.D.; Bruder, D.; Hiemstra, P.S.; Cloonan, S.M.; Heijink, I.H. Mitochondria: At the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L149–l164. [Google Scholar] [CrossRef]
- Bauer, A.Z.; Kriebel, D.; Herbert, M.R.; Bornehag, C.G.; Swan, S.H. Prenatal paracetamol exposure and child neurodevelopment: A review. Horm. Behav. 2018, 101, 125–147. [Google Scholar] [CrossRef]
- Andalib, S.; Emamhadi, M.R.; Yousefzadeh-Chabok, S.; Shakouri, S.K.; Høilund-Carlsen, P.F.; Vafaee, M.S.; Michel, T.M. Maternal SSRI exposure increases the risk of autistic offspring: A meta-analysis and systematic review. Eur. Psychiatry J. Assoc. Eur. Psychiatr. 2017, 45, 161–166. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Acetaminophen hepatotoxicity: A mitochondrial perspective. Adv. Pharmacol. 2019, 85, 195–219. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.R. Fluoxetine and the mitochondria: A review of the toxicological aspects. Toxicol. Lett. 2016, 258, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Da L.D. Barros, M.; Manhães-de-Castro, R.; Alves, D.T.; Quevedo, O.G.; Toscano, A.E.; Bonnin, A.; Galindo, L. Long term effects of neonatal exposure to fluoxetine on energy balance: A systematic review of experimental studies. Eur. J. Pharmacol. 2018, 833, 298–306. [Google Scholar] [CrossRef]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 2013, 5, 192ra85. [Google Scholar] [CrossRef] [Green Version]
- Bodolea, C. Anaesthesia in early childhood—Is the development of the immature brain in danger? Rom. J. Anaesth. Intensive Care 2016, 23, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Napoli, E.; Ross-Inta, C.; Wong, S.; Hung, C.; Fujisawa, Y.; Sakaguchi, D.; Angelastro, J.; Omanska-Klusek, A.; Schoenfeld, R.; Giulivi, C. Mitochondrial dysfunction in Pten haplo-insufficient mice with social deficits and repetitive behavior: Interplay between Pten and p53. PLoS ONE 2012, 7, e42504. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi-Fakhari, D.; Saffari, A.; Wahlster, L.; Sahin, M. Using tuberous sclerosis complex to understand the impact of MTORC1 signaling on mitochondrial dynamics and mitophagy in neurons. Autophagy 2017, 13, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Bennuri, S.C.; Rose, S.; Frye, R.E. Mitochondrial dysfunction is inducible in lymphoblastoid cell lines from children with autism and may involve the TORC1 pathway. Front. Psychiatry Mol. Psychiatry 2019, 10, 269. [Google Scholar] [CrossRef] [Green Version]
- D’Antoni, S.; de Bari, L.; Valenti, D.; Borro, M.; Bonaccorso, C.M.; Simmaco, M.; Vacca, R.A.; Catania, M.V. Aberrant mitochondrial bioenergetics in the cerebral cortex of the Fmr1 knockout mouse model of fragile X syndrome. Biol. Chem. 2020, 401, 497–503. [Google Scholar] [CrossRef]
- Griffiths, K.K.; Wang, A.; Wang, L.; Tracey, M.; Kleiner, G.; Quinzii, C.M.; Sun, L.; Yang, G.; Perez-Zoghbi, J.F.; Licznerski, P.; et al. Inefficient thermogenic mitochondrial respiration due to futile proton leak in a mouse model of fragile X syndrome. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 7404–7426. [Google Scholar] [CrossRef] [Green Version]
- Grosser, E.; Hirt, U.; Janc, O.A.; Menzfeld, C.; Fischer, M.; Kempkes, B.; Vogelgesang, S.; Manzke, T.U.; Opitz, L.; Salinas-Riester, G.; et al. Oxidative burden and mitochondrial dysfunction in a mouse model of Rett syndrome. Neurobiol. Dis. 2012, 48, 102–114. [Google Scholar] [CrossRef]
- Gibson, J.H.; Slobedman, B.; KN, H.; Williamson, S.L.; Minchenko, D.; El-Osta, A.; Stern, J.L.; Christodoulou, J. Downstream targets of methyl CpG binding protein 2 and their abnormal expression in the frontal cortex of the human Rett syndrome brain. BMC Neurosci. 2010, 11, 53. [Google Scholar] [CrossRef] [Green Version]
- Condie, J.; Goldstein, J.; Wainwright, M.S. Acquired microcephaly, regression of milestones, mitochondrial dysfunction, and episodic rigidity in a 46,XY male with a de novo MECP2 gene mutation. J. Child Neurol. 2010, 25, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E. Mitochondrial disease in 22q13 duplication syndrome. J. Child Neurol. 2012, 27, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E. 15q11.2-13 duplication, mitochondrial dysfunction, and developmental disorders. J. Child Neurol. 2009, 24, 1316–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipek, P.A.; Juranek, J.; Smith, M.; Mays, L.Z.; Ramos, E.R.; Bocian, M.; Masser-Frye, D.; Laulhere, T.M.; Modahl, C.; Spence, M.A.; et al. Mitochondrial dysfunction in autistic patients with 15q inverted duplication. Ann. Neurol. 2003, 53, 801–804. [Google Scholar] [CrossRef]
- Su, H.; Fan, W.; Coskun, P.E.; Vesa, J.; Gold, J.A.; Jiang, Y.H.; Potluri, P.; Procaccio, V.; Acab, A.; Weiss, J.H.; et al. Mitochondrial dysfunction in CA1 hippocampal neurons of the UBE3A deficient mouse model for Angelman syndrome. Neurosci. Lett. 2011, 487, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Pagano, G.; Castello, G. Oxidative stress and mitochondrial dysfunction in Down syndrome. Adv. Exp. Med. Biol. 2012, 724, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Pallardo, F.V.; Lloret, A.; Lebel, M.; d’Ischia, M.; Cogger, V.C.; Le Couteur, D.G.; Gadaleta, M.N.; Castello, G.; Pagano, G. Mitochondrial dysfunction in some oxidative stress-related genetic diseases: Ataxia-telangiectasia, Down syndrome, Fanconi anaemia and Werner syndrome. Biogerontology 2010, 11, 401–419. [Google Scholar] [CrossRef]
- Schuelke, M.; Krude, H.; Finckh, B.; Mayatepek, E.; Janssen, A.; Schmelz, M.; Trefz, F.; Trijbels, F.; Smeitink, J. Septo-optic dysplasia associated with a new mitochondrial cytochrome b mutation. Ann. Neurol. 2002, 51, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xin, K.; Wei, J.; Zhang, K.; Xiao, H. Lower maternal serum 25(OH) D in first trimester associated with higher autism risk in Chinese offspring. J. Psychosom. Res. 2016, 89, 98–101. [Google Scholar] [CrossRef]
- Vinkhuyzen, A.A.E.; Eyles, D.W.; Burne, T.H.J.; Blanken, L.M.E.; Kruithof, C.J.; Verhulst, F.; White, T.; Jaddoe, V.W.; Tiemeier, H.; McGrath, J.J. Gestational vitamin D deficiency and autism spectrum disorder. Bjpsych. Open 2017, 3, 85–90. [Google Scholar] [CrossRef]
- Magnusson, C.; Lundberg, M.; Lee, B.K.; Rai, D.; Karlsson, H.; Gardner, R.; Kosidou, K.; Arver, S.; Dalman, C. Maternal vitamin D deficiency and the risk of autism spectrum disorders: Population-based study. Bjpsych. Open 2016, 2, 170–172. [Google Scholar] [CrossRef] [Green Version]
- Dzik, K.P.; Kaczor, J.J. Mechanisms of vitamin D on skeletal muscle function: Oxidative stress, energy metabolism and anabolic state. Eur. J. Appl. Physiol. 2019, 119, 825–839. [Google Scholar] [CrossRef] [Green Version]
- Silvagno, F.; Pescarmona, G. Spotlight on vitamin D receptor, lipid metabolism and mitochondria: Some preliminary emerging issues. Mol. Cell. Endocrinol. 2017, 450, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Slattery, J.; MacFabe, D.F.; Kahler, S.G.; Frye, R.E. Enteric ecosystem disruption in autism spectrum disorder: Can the microbiota and macrobiota be restored? Curr. Pharm. Des. 2016, 22, 6107–6121. [Google Scholar] [CrossRef] [Green Version]
- Lammert, C.R.; Frost, E.L.; Bolte, A.C.; Paysour, M.J.; Shaw, M.E.; Bellinger, C.E.; Weigel, T.K.; Zunder, E.R.; Lukens, J.R. Cutting edge: Critical roles for microbiota-mediated regulation of the immune system in a prenatal immune activation model of autism. J. Immunol. 2018, 201, 845–850. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Horton-Sparks, K.; Hull, V.; Li, R.W.; Martínez-Cerdeño, V. The valproic acid rat model of autism presents with gut bacterial dysbiosis similar to that in human autism. Mol. Autism 2018, 9, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.P.; Xu, Y.C.; Hou, J.Q.; Li, J.Y.; Xing, J.; Yang, B.X.; Zhang, Z.H.; Zhang, B.L.; Li, H.H.; Li, P. Effects of dietary fat profile on gut microbiota in valproate animal model of autism. Front. Med. 2020, 7, 151. [Google Scholar] [CrossRef]
- De Theije, C.G.; Wopereis, H.; Ramadan, M.; van Eijndthoven, T.; Lambert, J.; Knol, J.; Garssen, J.; Kraneveld, A.D.; Oozeer, R. Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain Behav. Immun. 2014, 37, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.; Narous, M.; Tobias, R.; Rho, J.M.; Mychasiuk, R. The ketogenic diet modifies social and metabolic alterations identified in the prenatal valproic acid model of autism spectrum disorder. Dev. Neurosci. 2014, 36, 371–380. [Google Scholar] [CrossRef]
- Castro, K.; Baronio, D.; Perry, I.S.; Riesgo, R.D.S.; Gottfried, C. The effect of ketogenic diet in an animal model of autism induced by prenatal exposure to valproic acid. Nutr. Neurosci. 2017, 20, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [Green Version]
- Vallès, Y.; Francino, M.P. Air pollution, early life microbiome, and development. Curr. Environ. Health Rep. 2018, 5, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, Y.; Yang, J.; Chang, L.; Qu, Y.; Wang, S.; Zhang, K.; Xiong, Z.; Zhang, J.; Tan, Y.; Wang, X.; et al. Maternal glyphosate exposure causes autism-like behaviors in offspring through increased expression of soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA 2020, 117, 11753–11759. [Google Scholar] [CrossRef]
- Hamad, A.F.; Alessi-Severini, S.; Mahmud, S.M.; Brownell, M.; Kuo, I.F. Prenatal antibiotics exposure and the risk of autism spectrum disorders: A population-based cohort study. PLoS ONE 2019, 14, e0221921. [Google Scholar] [CrossRef] [Green Version]
- Beversdorf, D.Q.; Stevens, H.E.; Margolis, K.G.; Van de Water, J. Prenatal stress and maternal immune dysregulation in autism spectrum disorders: Potential points for intervention. Curr. Pharm. Des. 2019, 25, 4331–4343. [Google Scholar] [CrossRef]
- Dong, T.; Guan, Q.; Hu, W.; Zhang, M.; Zhang, Y.; Chen, M.; Wang, X.; Xia, Y. Prenatal exposure to glufosinate ammonium disturbs gut microbiome and induces behavioral abnormalities in mice. J. Hazard. Mater. 2020, 389, 122152. [Google Scholar] [CrossRef]
- Frye, R.E.; Rose, S.; Slattery, J.; MacFabe, D.F. Gastrointestinal dysfunction in autism spectrum disorder: The role of the mitochondria and the enteric microbiome. Microb. Ecol. Health Dis. 2015, 26, 27458. [Google Scholar] [CrossRef]
- Delhey, L.; Kilinc, E.N.; Yin, L.; Slattery, J.; Tippett, M.; Wynne, R.; Rose, S.; Kahler, S.; Damle, S.; Legido, A.; et al. Bioenergetic variation is related to autism symptomatology. Metab. Brain Dis. 2017, 32, 2021–2031. [Google Scholar] [CrossRef] [Green Version]
- Goldenthal, M.J.; Damle, S.; Sheth, S.; Shah, N.; Melvin, J.; Jethva, R.; Hardison, H.; Marks, H.; Legido, A. Mitochondrial enzyme dysfunction in autism spectrum disorders; a novel biomarker revealed from buccal swab analysis. Biomark. Med. 2015, 9, 957–965. [Google Scholar] [CrossRef]
- Giulivi, C.; Zhang, Y.F.; Omanska-Klusek, A.; Ross-Inta, C.; Wong, S.; Hertz-Picciotto, I.; Tassone, F.; Pessah, I.N. Mitochondrial dysfunction in autism. JAMA 2010, 304, 2389–2396. [Google Scholar] [CrossRef] [Green Version]
- Napoli, E.; Wong, S.; Hertz-Picciotto, I.; Giulivi, C. Deficits in bioenergetics and impaired immune response in granulocytes from children with autism. Pediatrics 2014, 133, e1405–e1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, W.D.; Marin-Garcia, J.; Gao, H.G.; Pizzo, S.; Naviaux, R.K.; Markusic, D.; Barshop, B.A.; Courchesne, E.; Haas, R.H. Autism associated with the mitochondrial DNA G8363A transfer RNA(Lys) mutation. J. Child Neurol. 2000, 15, 357–361. [Google Scholar] [CrossRef]
- Frye, R.E.; Naviaux, R.K. Autistic disorder with complex IV overactivity: A new mitochondrial syndrome. J. Pediatr. Neurol. 2011, 9, 427–434. [Google Scholar]
- Palmieri, L.; Papaleo, V.; Porcelli, V.; Scarcia, P.; Gaita, L.; Sacco, R.; Hager, J.; Rousseau, F.; Curatolo, P.; Manzi, B.; et al. Altered calcium homeostasis in autism-spectrum disorders: Evidence from biochemical and genetic studies of the mitochondrial aspartate/glutamate carrier AGC1. Mol. Psychiatry 2010, 15, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, H.; Gnaiger, E.; Zakaria, F.; Makpol, S.; Karim, N.A. Alterations in mitocohndrial respiratiory capacity and membrane potential: A link between mitochondrial dysregulation and autism. MitoFit 2020, in press. [Google Scholar]
- Fowler, B.A.; Woods, J.S. Ultrastructural and biochemical changes in renal mitochondria during chronic oral methyl mercury exposure: The relationship to renal function. Exp. Mol. Pathol. 1977, 27, 403–412. [Google Scholar] [CrossRef]
- Shenker, B.J.; Guo, T.L.; O, I.; Shapiro, I.M. Induction of apoptosis in human T-cells by methyl mercury: Temporal relationship between mitochondrial dysfunction and loss of reductive reserve. Toxicol. Appl. Pharmacol. 1999, 157, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Goyer, R.A. Toxic and essential metal interactions. Annu. Rev. Nutr. 1997, 17, 37–50. [Google Scholar] [CrossRef]
- Pourahmad, J.; Mihajlovic, A.; O’Brien, P.J. Hepatocyte lysis induced by environmental metal toxins may involve apoptotic death signals initiated by mitochondrial injury. Adv. Exp. Med. Biol. 2001, 500, 249–252. [Google Scholar] [CrossRef]
- Hiura, T.S.; Li, N.; Kaplan, R.; Horwitz, M.; Seagrave, J.C.; Nel, A.E. The role of a mitochondrial pathway in the induction of apoptosis by chemicals extracted from diesel exhaust particles. J. Immunol. 2000, 165, 2703–2711. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.W.; Garcia, E.F.; Pessah, I.N. Ortho-substituted PCB95 alters intracellular calcium signaling and causes cellular acidification in PC12 cells by an immunophilin-dependent mechanism. J. Neurochem. 2001, 76, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Richardson, J.R.; Testa, C.M.; Seo, B.B.; Panov, A.V.; Yagi, T.; Matsuno-Yagi, A.; Miller, G.W.; Greenamyre, J.T. Mechanism of toxicity of pesticides acting at complex I: Relevance to environmental etiologies of Parkinson’s disease. J. Neurochem. 2007, 100, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Yamano, T.; Morita, S. Effects of pesticides on isolated rat hepatocytes, mitochondria, and microsomes II. Arch. Environ. Contam. Toxicol. 1995, 28, 1–7. [Google Scholar] [CrossRef]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Pavliv, O.; Melnyk, S.; James, S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS ONE 2014, 9, e85436. [Google Scholar] [CrossRef]
- Hill, B.G.; Higdon, A.N.; Dranka, B.P.; Darley-Usmar, V.M. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim. Biophys. Acta 2010, 1797, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; Rose, S.; Wynne, R.; Bennuri, S.C.; Blossom, S.; Gilbert, K.M.; Heilbrun, L.; Palmer, R.F. Oxidative stress challenge uncovers trichloroacetaldehyde hydrate-induced mitoplasticity in autistic and control lymphoblastoid cell lines. Sci. Rep. 2017, 7, 4478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Melnyk, S.; James, S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autistic lymphoblastoid cell lines. Transl. Psychiatry 2015, 5, e526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, S.; Wynne, R.; Frye, R.E.; Melnyk, S.; James, S.J. Increased susceptibility to ethylmercury-induced mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines. J. Toxicol. 2015, 2015, 573701. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Rose, S.; Chacko, J.; Wynne, R.; Bennuri, S.C.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; et al. Modulation of mitochondrial function by the microbiome metabolite propionic acid in autism and control cell lines. Transl. Psychiatry 2016, 6, e927. [Google Scholar] [CrossRef]
- Rose, S.; Bennuri, S.C.; Davis, J.E.; Wynne, R.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; MacFabe, D.F.; et al. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl. Psychiatry 2018, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Bennuri, S.C.; Wynne, R.; Melnyk, S.; James, S.J.; Frye, R.E. Mitochondrial and redox abnormalities in autism lymphoblastoid cells: A sibling control study. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 904–909. [Google Scholar] [CrossRef] [Green Version]
- Melnyk, S.; Fuchs, G.J.; Schulz, E.; Lopez, M.; Kahler, S.G.; Fussell, J.J.; Bellando, J.; Pavliv, O.; Rose, S.; Seidel, L.; et al. Metabolic imbalance associated with methylation dysregulation and oxidative damage in children with autism. J. Autism Dev. Disord. 2012, 42, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Canitano, R.; Zappella, M. Autistic epileptiform regression. Funct. Neurol. 2006, 21, 97–101. [Google Scholar]
- Shoffner, J.; Hyams, L.; Langley, G.N.; Cossette, S.; Mylacraine, L.; Dale, J.; Ollis, L.; Kuoch, S.; Bennett, K.; Aliberti, A.; et al. Fever plus mitochondrial disease could be risk factors for autistic regression. J. Child Neurol. 2010, 25, 429–434. [Google Scholar] [CrossRef]
- Edmonds, J.L.; Kirse, D.J.; Kearns, D.; Deutsch, R.; Spruijt, L.; Naviaux, R.K. The otolaryngological manifestations of mitochondrial disease and the risk of neurodegeneration with infection. Arch. Otolaryngol. Head Neck Surg. 2002, 128, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Singh, I.N.; Diggins, E.; Connors, S.L.; Karim, M.A.; Lee, D.; Zimmerman, A.W.; Frye, R.E. Developmental regression and mitochondrial function in children with autism. Ann. Clin. Transl. Neurol. 2020, 7, 683–694. [Google Scholar] [CrossRef]
- Frye, R.E.; Cox, D.; Slattery, J.; Tippett, M.; Kahler, S.; Granpeesheh, D.; Damle, S.; Legido, A.; Goldenthal, M.J. Mitochondrial dysfunction may explain symptom variation in Phelan-McDermid syndrome. Sci. Rep. 2016, 6, 19544. [Google Scholar] [CrossRef] [Green Version]
- Kanellopoulos, A.K.; Mariano, V.; Spinazzi, M.; Woo, Y.J.; McLean, C.; Pech, U.; Li, K.W.; Armstrong, J.D.; Giangrande, A.; Callaerts, P.; et al. Aralar sequesters GABA into hyperactive mitochondria, causing social behavior deficits. Cell 2020, 180, 1178–1197.e20. [Google Scholar] [CrossRef]
- Curtin, P.; Curtin, A.; Austin, C.; Gennings, C.; Tammimies, K.; Bolte, S.; Arora, M. Recurrence quantification analysis to characterize cyclical components of environmental elemental exposures during fetal and postnatal development. PLoS ONE 2017, 12, e0187049. [Google Scholar] [CrossRef] [Green Version]
- Arora, M.; Bradman, A.; Austin, C.; Vedar, M.; Holland, N.; Eskenazi, B.; Smith, D.R. Determining fetal manganese exposure from mantle dentine of deciduous teeth. Environ. Sci. Technol. 2012, 46, 5118–5125. [Google Scholar] [CrossRef] [Green Version]
- Arora, M.; Hare, D.; Austin, C.; Smith, D.R.; Doble, P. Spatial distribution of manganese in enamel and coronal dentine of human primary teeth. Sci. Total Environ. 2011, 409, 1315–1319. [Google Scholar] [CrossRef]
- Arora, M.; Reichenberg, A.; Willfors, C.; Austin, C.; Gennings, C.; Berggren, S.; Lichtenstein, P.; Anckarsater, H.; Tammimies, K.; Bolte, S. Fetal and postnatal metal dysregulation in autism. Nat. Commun. 2017, 8, 15493. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Melnyk, S.; Trusty, T.A.; Pavliv, O.; Seidel, L.; Li, J.; Nick, T.; James, S.J. Intracellular and extracellular redox status and free radical generation in primary immune cells from children with autism. Autism Res. Treat. 2012, 2012, 986519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandewalle, J.; Bauters, M.; Van Esch, H.; Belet, S.; Verbeeck, J.; Fieremans, N.; Holvoet, M.; Vento, J.; Spreiz, A.; Kotzot, D.; et al. The mitochondrial solute carrier SLC25A5 at Xq24 is a novel candidate gene for non-syndromic intellectual disability. Hum. Genet. 2013, 132, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M.; et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licznerski, P.; Park, H.A.; Rolyan, H.; Chen, R.; Mnatsakanyan, N.; Miranda, P.; Graham, M.; Wu, J.; Cruz-Reyes, N.; Mehta, N.; et al. ATP Synthase c-subunit leak causes aberrant cellular metabolism in fragile X syndrome. Cell 2020, 182, 1170–1185.e9. [Google Scholar] [CrossRef]
- Napoli, E.; Song, G.; Panoutsopoulos, A.; Riyadh, M.A.; Kaushik, G.; Halmai, J.; Levenson, R.; Zarbalis, K.S.; Giulivi, C. Beyond autophagy: A novel role for autism-linked Wdfy3 in brain mitophagy. Sci. Rep. 2018, 8, 11348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.-L.; Chen, H.-I.; Li, L.-H.; Chien, Y.-L.; Liao, H.-M.; Chou, M.C.; Chou, W.-J.; Tsai, W.-C.; Chiu, Y.-N.; Wu, Y.-Y.; et al. Genome-wide analysis of copy number variations identifies PARK2 as a candidate gene for autism spectrum disorder. Mol. Autism 2016, 7, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Gutierrez Rios, P.; Kuo, S.-H.; Akman, H.O.; Rosoklija, G.; Tanji, K.; Dwork, A.; Schon, E.A.; Dimauro, S.; Goldman, J.; et al. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol. Dis. 2013, 54, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi-Fakhari, D.; Saffari, A.; Wahlster, L.; Di Nardo, A.; Turner, D.; Lewis, T.L., Jr.; Conrad, C.; Rothberg, J.M.; Lipton, J.O.; Kölker, S.; et al. Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep. 2016, 17, 1053–1070. [Google Scholar] [CrossRef] [Green Version]
- Pecorelli, A.; Ferrara, F.; Messano, N.; Cordone, V.; Schiavone, M.L.; Cervellati, F.; Woodby, B.; Cervellati, C.; Hayek, J.; Valacchi, G. Alterations of mitochondrial bioenergetics, dynamics, and morphology support the theory of oxidative damage involvement in autism spectrum disorder. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 6521–6538. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Deen, N.; Zamani, S.; Hasnat, M.A. Mitophagy and the release of inflammatory cytokines. Mitochondrion 2018, 41, 2–8. [Google Scholar] [CrossRef]
- Meeking, M.M.; MacFabe, D.F.; Mepham, J.R.; Foley, K.A.; Tichenoff, L.J.; Boon, F.H.; Kavaliers, M.; Ossenkopp, K.P. Propionic acid induced behavioural effects of relevance to autism spectrum disorder evaluated in the hole board test with rats. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2020, 97, 109794. [Google Scholar] [CrossRef] [PubMed]
- Shams, S.; Foley, K.A.; Kavaliers, M.; MacFabe, D.F.; Ossenkopp, K.P. Systemic treatment with the enteric bacterial metabolic product propionic acid results in reduction of social behavior in juvenile rats: Contribution to a rodent model of autism spectrum disorder. Dev. Psychobiol. 2019, 61, 688–699. [Google Scholar] [CrossRef]
- Foley, K.A.; MacFabe, D.F.; Kavaliers, M.; Ossenkopp, K.P. Sexually dimorphic effects of prenatal exposure to lipopolysaccharide, and prenatal and postnatal exposure to propionic acid, on acoustic startle response and prepulse inhibition in adolescent rats: Relevance to autism spectrum disorders. Behav. Brain Res. 2015, 278, 244–256. [Google Scholar] [CrossRef]
- Foley, K.A.; MacFabe, D.F.; Vaz, A.; Ossenkopp, K.P.; Kavaliers, M. Sexually dimorphic effects of prenatal exposure to propionic acid and lipopolysaccharide on social behavior in neonatal, adolescent, and adult rats: Implications for autism spectrum disorders. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2014, 39, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Macfabe, D. Autism: Metabolism, mitochondria, and the microbiome. Glob. Adv. Health Med. 2013, 2, 52–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, R.E. Biomarkers of abnormal energy metabolism in children with autism spectrum disorder. N. Am. J. Med. Sci. 2012, 5, 141–147. [Google Scholar] [CrossRef]
- Frye, R.E.; Melnyk, S.; Macfabe, D.F. Unique acyl-carnitine profiles are potential biomarkers for acquired mitochondrial disease in autism spectrum disorder. Transl. Psychiatry 2013, 3, e220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, A.; Meechan, D.W.; Karpinski, B.A.; Paronett, E.M.; Bryan, C.A.; Rutz, H.L.; Radin, E.A.; Lubin, N.; Bonner, E.R.; Popratiloff, A.; et al. Mitochondrial dysfunction leads to cortical under-connectivity and cognitive impairment. Neuron 2019, 102, 1127–1142.e3. [Google Scholar] [CrossRef]
- Lin-Hendel, E.G.; McManus, M.J.; Wallace, D.C.; Anderson, S.A.; Golden, J.A. Differential mitochondrial requirements for radially and non-radially migrating cortical neurons: Implications for mitochondrial disorders. Cell Rep. 2016, 15, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Chacko, B.K.; Kramer, P.A.; Ravi, S.; Benavides, G.A.; Mitchell, T.; Dranka, B.P.; Ferrick, D.; Singal, A.K.; Ballinger, S.W.; Bailey, S.M.; et al. The Bioenergetic Health Index: A new concept in mitochondrial translational research. Clin. Sci. 2014, 127, 367–373. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frye, R.E.; Cakir, J.; Rose, S.; Palmer, R.F.; Austin, C.; Curtin, P.; Arora, M. Mitochondria May Mediate Prenatal Environmental Influences in Autism Spectrum Disorder. J. Pers. Med. 2021, 11, 218. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11030218
Frye RE, Cakir J, Rose S, Palmer RF, Austin C, Curtin P, Arora M. Mitochondria May Mediate Prenatal Environmental Influences in Autism Spectrum Disorder. Journal of Personalized Medicine. 2021; 11(3):218. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11030218
Chicago/Turabian StyleFrye, Richard E., Janet Cakir, Shannon Rose, Raymond F. Palmer, Christine Austin, Paul Curtin, and Manish Arora. 2021. "Mitochondria May Mediate Prenatal Environmental Influences in Autism Spectrum Disorder" Journal of Personalized Medicine 11, no. 3: 218. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11030218