

Molecular and Phenotypic Changes in FLExDUX4 Mice

1
Institute for Biomedical Sciences, The George Washington University, Washington, DC 20037, USA
2
Center for Genetic Medicine Research, Children’s National Hospital, Washington, DC 20010, USA
3
Department of Genomics and Precision Medicine, School of Medicine and Health Science, The George Washington University, Washington, DC 20037, USA
*
Author to whom correspondence should be addressed.
J. Pers. Med. 2023, 13(7), 1040; https://0-doi-org.brum.beds.ac.uk/10.3390/jpm13071040
Submission received: 17 March 2023
/
Revised: 13 June 2023
/
Accepted: 16 June 2023
/
Published: 25 June 2023
(This article belongs to the Special Issue Neuromuscular and Neurodegenerative Diseases: Towards Personalized Medicine, Therapeutics and Improved Mechanistic Understanding)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Facioscapulohumeral muscular dystrophy (FSHD) is caused by the aberrant expression of the double homeobox 4 (DUX4) gene. The FLExDUX4 mouse model carries an inverted human DUX4 transgene which has leaky DUX4 transgene expression at a very low level. No overt muscle pathology was reported before 16 weeks. The purpose of this study is to track and characterize the FLExDUX4 phenotypes for a longer period, up to one year old. In addition, transcriptomic changes in the muscles of 2-month-old mice were investigated using RNA-seq. The results showed that male FLExDUX4 mice developed more severe phenotypes and at a younger age in comparison to the female mice. These include lower body and muscle weight, and muscle weakness measured by grip strength measurements. Muscle pathological changes were observed at older ages, including fibrosis, decreased size of type IIa and IIx myofibers, and the development of aggregates containing TDP-43 in type IIb myofibers. Muscle transcriptomic data identified early molecular changes in biological pathways regulating circadian rhythm and adipogenesis. The study suggests a slow progressive change in molecular and muscle phenotypes in response to the low level of DUX4 expression in the FLExDUX4 mice.
1. Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal dominant disorder caused by the aberrant expression of the double homeobox 4 (DUX4) gene, resulting in progressive muscle loss. Symptoms of FSHD include weakness in the muscles of the face, shoulders, and upper arms, and can include limb and trunk muscles as the disease progresses [1,2,3]. It has a prevalence of 1 in 8333–20,000 people in the world [1,4,5,6,7]. Current therapies for FSHD only address its symptoms, such as shoulder fixation orthotics and physical therapy [2].
FSHD is subclassified into two subtypes, FSHD type 1 and FSHD type 2, which affect approximately 95% and 5% of individuals with FSHD, respectively [1,2]. FSHD type 1 and type 2 are clinically indistinguishable but are classified by different genomic mutations that affect the regulation of epigenetic control over the D4Z4 region. FSHD1 is linked to a contraction of the tandem D4Z4 repeat array on chromosome 4q35 to 1–10 repeats [8,9]. In unaffected individuals, this array contains 11–150 copies of the D4Z4 repeat. FSHD2 is reported to be caused by mutations in the structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1) gene located on chromosome 18, in the DNA-methyltransferase 3 beta gene (DNMT3B) located on chromosome 20, or in ligand-dependent nuclear receptor-interacting factor 1 (LRIF1) on chromosome 1 [6,10,11]. Contraction of the D4Z4 array, haploinsufficiency of SMCHD1, mutations in DNMT3B, or mutations in LRIF1 are reported to be associated with decreased DNA methylation and transcriptionally de-repress the DUX4 gene [6,10,12,13,14,15,16,17,18,19,20]. These genomic mutations cause an indistinguishable clinical presentation because they all epigenetically de-repress the DUX4 gene located in the D4Z4 repeats. Previous studies reported that the development of FSHD requires a combination of two genomic features to develop FSHD. In addition to the genomic mutations mentioned above, the second genomic feature necessary for FSHD pathogenesis is the presence of a functional polyadenylation signal in the pLAM region distal to the D4Z4 array [21,22,23,24,25]. This polyadenylation signal allows for the stabilization of the DUX4 mRNA transcribed from the last D4Z4 repeat for the translation of the DUX4 protein [21,22,23,24,25]. The functional polyadenylation signal, in addition to the genomic mutations listed above, allows for the transcription and translation of the DUX4 mRNA and protein, which causes FSHD.
Previous studies have shown that the DUX4 protein is a transcription factor, which is expressed during early embryonic development or postnatally in the testes, thymus, mesenchymal stem cells, and keratinocytes [26,27,28,29,30,31,32]. The proposed function of DUX4 is that it facilitates early genome activation, also known as the zygotic genome activation, at the two- to four-cell stage of development [26,27,28,33]. In FSHD, the aberrant expression of DUX4 in the muscle causes the expression of germline DUX4 transcriptional targets [26]; the repression of oxidative stress response genes [34,35,36,37]; disruptions in cell cycle and migration [35,38]; disruption of RNA metabolism, splicing, surveillance, and transport [38,39]; and a reduction in myogenic capacity due to the misregulation of MYOD1 and its downstream targets, including structural and contractile components [28,29,33,36,37,40,41,42,43]. The culmination of the effect of DUX4 expression on these pathways results in progressive muscle weakness and loss in individuals with FSHD.
Historically, mouse models for FSHD have had limited experimental use due to either severe phenotypes preventing long-term studies or a lack of detectable DUX4 expression and muscle pathologies [25,44,45,46,47]. The FLExDUX4 model carries an inverted human DUX4 transgene flanked by lox sites, which, when crossed with the ACTA1-MCM, allows for tunable DUX4 expression in the skeletal muscle of mice [48,49,50]. Induction of the DUX4 transgene in the ACTA1-MCM/FLExDUX4 mice led to FSHD-like phenotypes, but required repeated injections of tamoxifen to maintain moderate to severe levels of pathology [48,49]. The single transgenic FLExDUX4 mouse model, while presenting with no overt pathology at a young age, showed leaky expression of DUX4 despite the inverted transgene [48,49]. The absence of pathology compared to the conditional counterparts have left the FLExDUX4 mice relatively under studied. Since patients with FSHD have a wide range of clinical presentations and age of onset, it is important to thoroughly characterize all iterations of the FLExDUX4 model for their pre-clinical relevancy [19,51,52]. In this study, we hypothesized that the long-term expression of a low level of DUX4 will lead to muscle pathologies and functional deficits in older FLExDUX4 mice. To explore this, we investigated phenotypic (e.g., body and muscle weight), functional (e.g., grip strength), pathological (e.g., fiber size and typing), and molecular changes (e.g., transcriptome) in the FLExDUX4 mice at various ages to determine changes during disease progression.
2. Materials and Methods
2.1. IACUC Statement
Animal experiments were approved by the Institutional Animal Care and Use Committee at Children’s National Hospital in Washington, DC. Euthanasia was performed using carbon dioxide asphyxiation, and death was ensured with cervical dislocation. For all experiments, hemizygous B6(Cg)-Gt(ROSA)26Sortm1.1(DUX4*)Plj/J (FLExDUX4) mice were used.
2.2. Genotyping
Genomic DNA was isolated from tail snips using phenol–chloroform extraction. Tail snips were digested in 300 µL digestion buffer (50 mM Tris-HCl pH 7.4, 5 mM EDTA pH 8.0, 0.5% sodium dodecyl sulfate) containing 10 µL proteinase K (20 mg/mL; Ambion, Huntingdon, UK). Tails were incubated overnight at 55 °C with shaking at 300 rpm. Debris was pelleted by centrifugation at 21,000× g for 30 s and the supernatant collected. Phenol–chloroform–isoamyl alcohol (25:24:1) was added to the supernatant in a 1:1 ratio. Samples were inverted by hand for 15 s and centrifuged at 21,000× g for 2 min. The aqueous phase was collected and added to ammonium acetate (150 µL at 7.5 M) and 100% ethanol (750 µL, −20 °C). Samples were centrifuged at 12,000× g at room temperature for 20 min. The pellet was washed twice with 500 µL of chilled 80% ethanol and centrifuged at room temperature at 21,000× g for 5 min. Pellets were air dried for 15 min and re-suspended in 50 µL of water. Sample concentration was determined with a NanoDrop spectrometer and diluted to 100 ng/µL.
PCR amplification of the DUX4 transgene was performed according to published study [48,49], using 20 ng of DNA, 2.74 µL GoTaq Hot Start Polymerase master mix (Promega, Madison, WI, USA), and 400 nM of the Rosa26/FLExDUX4 primers pair or 200 nM of the wild type Rosa26 primers. The reaction volume was brought to 12 µL with nuclease-free water. Primer sequences were Rosa26 (forward): 5′-CAATACCTTTCTGGGAGTTCTCTGCTGC-3′; Rosa26 (reverse): 5′-TGCAGGACAACGCCCACACACC-3′; and Rosa26/FLExDUX4 (reverse): 5′-CTCGTGTAGACAGAGCCTAGACAATTTGTTG-3′. Reactions were performed with a pre-PCR hold at 94 °C for 3 min, then cycled 35 times (94 °C for 20 s, 62 °C for 20 s, and 72 °C for 35 s) and a final extension at 72 °C for 2 min. Gel electrophoresis was performed using a 2% agarose gel using the ZipRuler express DNA ladder to determine band size (ThermoFisher Scientific, Waltham, MA, USA). Wild-type mice had only the 175 bp product produced by the Rosa26 primers. Hemizygous mice had both the 409 bp amplicon of the Rosa26/FLExDUX4 pair and the 175 bp amplicon of the Rosa26 primers [48,49].
2.3. RNA Isolation and RT-qPCR
For muscle, total RNA was isolated from the quadriceps and triceps. Muscles were homogenized in 1 mL of TRIzol, and debris was pelleted out by centrifuging samples at 11,600× g for 5 min. Two hundred microliters of chloroform was added to the supernatant, and the samples were vortexed for 15 s. Samples were incubated at room temperature for 3 min and subsequently centrifuged at 4 °C and 11,600× g for 15 min. The aqueous phase was collected and added to 0.5 mL of isopropyl alcohol. Samples precipitated at room temperature for 10 min and centrifuged at 4 °C and 11,600× g for 10 min. Pellets were washed twice with 1 mL of chilled 75% ethanol and centrifuged at 7500× g for 5 min. Pellets were air dried for 15 min. RNA was purified with the RNeasy Micro kit with DNase digestion (Qiagen, Hilden, Germany).
Complementary DNA (cDNA) was synthesized using the SuperScript IV Reverse Transcriptase kit (ThermoFisher Scientific). Samples were prepared in a 13 µL reaction consisting of 2 µg of total RNA, 1 µL dNTPs (10 mM stock; New England Biolab, Ipswich, MA, USA), 1 µL oligo(dT)12–18 (Life Technologies, Carlsbad, CA, USA), and nuclease-free water and incubated at 65 °C for 5 min. Samples were then chilled on wet ice for 7 min. A solution of 4 µL 5× first strand buffer, 1 µL DTT, 1 µL of SuperScript IV (ThermoFisher Scientific), and 1 µL RNasin (Promega) was added to each reaction and incubated for 25 °C for 5 min, 50 °C for 1 h, and 70 °C for 15 min. Sample cDNA was brought to a volume of 100 µL.
Quantitative PCR was performed in triplicate using the SYBR Green Master Mix (Applied Biosystems, Waltham, MA, USA), 1 µL of cDNA, and 200 µM of forward and reverse primers. The total reaction volume was 20 µL. Reactions were performed with one pre-PCR hold at 50 °C for 2 min, one pre-PCR hold at 95 °C, and 40 cycles of amplification (95 °C for 15 s and 60 °C for 1 min). Relative gene expression was analyzed with the delta delta ct method. The following genes were assayed: DUX4, F-box protein 32 (Fbxo32), and muscle RING-finger protein-1 (Murf1). Primers Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and Hypoxanthine-guanine phosphoribosyltransferase (Hprt1) served as internal controls for Fbxo32, and Murf1. Primers used were Fbxo32/Atrogin1 (forward): 5′-TCAGAGAGGCAGATTCGCAAGC-3′ and (reverse): 5′-GTCAGTGCCCTTCCAGGAGA-3′; Murf1 (forward): 5′-TTGACTTTGGGACAGATGAGG-3′ and (reverse): 5′-AGCGTGTCTCACTCATCTCCTT-3′. Primers for internal controls were Gapdh (forward): 5′-TTGTCAGCAATGCATCCTGC-3′ and (reverse): 5′-CCGTTCAGCTCTGGGATGAC-3′ [53]; Hprt1 (forward): 5′-CGTCGTGATTAGCGATGATG-3′ and (reverse): 5′-TTTTCCAAATCCTCGGCATA-3′ [41].
2.4. Muscle Collection
The gastrocnemius, soleus, tibialis anterior, quadriceps, deltoid, triceps, bicep, masseter, diaphragm, and heart muscles were removed and weighed. Muscles were snap-frozen in isopentane cooled in liquid nitrogen for RNA and protein processing.
2.5. Grip Strength Measurement
Grip strength measurements were performed using a bar and a grid connected to an isometric force transducer (Columbus Instruments, Columbus, OH, USA). Briefly, FLExDUX4 mice were acclimated with five pulls five times each for both forelimb and hindlimb over three days. Grip strength measurements occurred over five days and each mouse performed five pulls for each measurement [54]. Measurements were taken on a predetermined location on the force meter to prevent measurement differences as a result of location on the attached grid. The strongest pull from each day was collected and averaged [54]. Student’s t-test was used to compare the experimental groups to the control groups. Grip strength testing was performed at 5 months, 8 months, and 12 months of age.
2.6. Hematoxylin and Eosin Staining
Snap-frozen muscles were sectioned at 8 µm using a Leica CM1950 cryostat and air-dried overnight at room temperature. Sections were stained with Modified Mayer’s hematoxylin for 30 min. Slides were dipped twice in distilled water and dipped in acid alcohol five times. Slides were rinsed in distilled water for 1 min and stained with eosin for 3 min. Slides were washed in two exchanges of 95% ethanol for 2 min with a final wash in 100% ethanol for 2 min. Slides were then washed in two exchanges of xylene for 2 min and 5 min. Coverslips were mounted with Permount mounting medium (Fisher Scientific). The sections were imaged with a VS120 Olympus scanning microscope.
2.7. Nicotinamide Adenine Dinucleotide Tetrazolium Staining
Frozen quadriceps sections were prepared for NADH-TR staining as described previously [54]. Briefly, 8 µm sections were prepared then incubated in a Tris Buffer (0.05 M, pH 7.6 (Millipore Sigma, Burlington, MA, USA) solution containing nitro-blue tetrazolium (2 mg/mL, Sigma) and NADH (8 mg/5 mL, Sigma) at 37 °C for 30 min. Sections were washed 3 times with deionized water, followed by 3 exchanges each of 30%, 60%, 90%, 60%, and 30% of acetone (1 min each). Slides were rinsed 3 times with deionized water and mounted with aqueous media [54].
2.8. BODIPY Staining
Cryosections (5 µm) were dried at room temperature for 2 h. BODIPY™ 493/503 was used to stain neutral lipids (0.25 µg/mL, ThermoFisher Scientific, Cat#D3922). Hoechst 33342 Solution was used to stain nuclei (0.1 µg/mL). Sections were incubated in solution made of 1× PBS with BODIPY and Hochest for 15 min. Sections were washed five times for 3 min in 1× PBS. The whole sections were imaged with a VS120 Olympus microscope. Images were analyzed with Image J.
2.9. Immunohistochemistry
Frozen quadriceps muscles were sectioned at 5 µm and stained using the Vectastain ABC HRP kit (Peroxidase) for mouse or rabbit antibodies (Vector Laboratories, Newark, CA, USA). Frozen muscle sections were fixed in cold acetone for 10 min and air-dried for 5 min. Sections were then blocked in a solution of 0.3% H2O2 and 0.3 Normal sera in 1× PBS for 5 min. Slides were washed twice with 1× PBS for 5 min. For TDP-43 staining, slides were blocked for 30 min according to the manufacturer’s protocol. Sections were incubated with the TDP-43 primary antibody (1:200, rabbit, Proteintech, Rosemont, IL, USA) overnight at 4 °C. For myosin heavy chain staining, slides were blocked in Vectastain blocking reagent overnight, then incubated with primary antibodies myosin heavy chain type 1 (A4.951), type 2a (2F7), type 2B (10F5), or type 2 × (6H1) (2 µg/mL, mouse, Developmental Studies Hybridoma Bank, The University of Iowa, Iowa City, IA, USA) for 2 h at room temperature [55,56]. DSHB Hybridoma Product A4.951 was deposited by Helen Blau of Stanford University [55,56]. DSHB Hybridoma products 2F7, 10F5, and 6H1 were developed and deposited by Christine Lucas of the University of Sydney [55,56].
After incubation with the primary antibody, all sections were washed 3 times for 15 min in 1× PBS. Sections were incubated with secondary antibodies for 1 h and rinsed 3 times for 15 min in 1× PBS. Slides were developed in DAB reagent for 10 min and washed with deionized water for 5 min. Slides were then washed twice in 1× PBS for 10 min. Slides were mounted with Crystal Mount (Electron Microscopy Sciences).
2.10. Immunofluorescence Staining
Frozen quadriceps were sectioned at a 5 µm thickness and dried at room temperature for 1 h. Slides were then blocked using filtered blocking buffer containing 10% horse serum (Gibco), 1% normal goat serum, and mouse on mouse (Vector Labs) in 1× PBS for 1 h. Primary antibodies were added in antibody buffer (10% horse serum and 1% goat serum in 1× PBS) for 2 h at room temperature. Slides were washed three times for 15 min in 1× PBS. Secondary antibodies were diluted in antibody buffer and incubated on the slides for 1 h at room temperature. Slides were washed in 1× PBS three times for 15 min. Primary antibodies used were Dystrophin (1:100, Abcam, Cambridge, UK) and myosin heavy chain antibodies type 1 (BA-D5), type 2a (SC-71), type 2b (BF-F3), and type 2 × (6H1) (2 µg/mL, Developmental Studies Hybridoma Bank) [57,58]. Clones BA-D5, SC-71, and BF-F3 were deposited to the DSHB by Schiaffino, S. (DSHB Hybridoma Products BA-D5, SC-71, and BF-F3) [57,58].
Secondary antibodies used were DyLight™ 405 AffiniPure Goat Anti-Mouse IgG (1:500, Jackson Immunoresearch Laboratories, Inc., West Grove, PA, USA), Cy™3 AffiniPure Goat Anti-Mouse IgM, µ chain specific (1:500, Jackson Immunoresearch Laboratories, Inc.), Alexa Fluor 647 AffiniPure Goat Anti-Rabbit IgG (H + L) (1:500, Jackson Immunoresearch Laboratories, Inc.), and Alexa Fluor 488 goat anti mouse IgG (1:1000, ThermoFisher Scientific). Slides were washed 3 times in 1× PBS for 15 min and mounted with Fluoromount mounting media (Electron Microscopy Sciences).
2.11. Fiber Size Data Analysis
Quadriceps sections were imaged using a PURNA-Olympus VS-120 scanning microscope, and images were analyzed using Image J (NIH, https://imagej.net/ij/index.html (accessed on 13 June 2023)). Positive fibers were identified after normalizing all sections to the same threshold. The threshold was determined at the point at which fibers with the light blue background stain were eliminated. The minimal Feret’s diameter was determined using the middle section of the quadriceps located between two connective tissue landmarks present in all images (586–799 fibers in each section). For fluorescent staining, images were processed using the ImageJ software, and the minimal Feret’s diameter and fiber area were determined using fiber boundary visualized by dystrophin staining. The fibers measured where located within the same landmarks as the NADH-TR sections. The following numbers of fibers were measured: 256–386 type 2a fibers, 200 type 2b fibers, and 383–539 type 2x fibers in each section. ImageJ calculated the circularity of each fiber using the following formula: circularity = 4pi (area/perimeter2). A value of 1.0 denotes a perfect circle and an elongated polygon as the value approaches 0.0 (https://imagej.nih.gov/ij/plugins/circularity.html (accessed on 13 June 2023)). Five random fields were taken of the whole section. The minimal Feret’s diameter was determined by measuring the fiber boundary visualized by dystrophin staining. A range of 3538–4665 fibers was measured in each section.
All fibers were ranked based upon their size, with the smallest fiber assigned the number one. The rank number was normalized by dividing the rank by the total number of fibers to obtain a value between 0 and 1. Normalized rank was plotted against the fiber diameter or fiber area. The Kolmogorov–Smirnov test was used to determine the significant difference in the fiber size distribution between littermate pairs as previously described [54,59].
2.12. RNA-Sequencing
Total RNA from 2-month-old male FLExDUX4 mice was isolated from triceps muscle using the TRIzol/Qiagen protocol described above (n = 3). Samples were processed by Lexogen GmbH for transcriptome sequencing (Vienna, Austria). Sample RNA was checked for quality using the Nanodrop2000c (ThermoFisher Scientific) and integrity with the Fragment Analyzer high-sensitivity assay (Agilent). The library was sequenced using the QuantSeq 3′mRNA-seq Library Prep kit FWD for Illumina (015UG009V0252) using the standard QuantSeq-FWD protocol. Sequencing was performed on an Illumina NextSeq 500 sequencer with a v2 SR75 High Output kit. Cutadapt version 1.16 was used to remove adapter contaminations, continuous polyA sequences, and continuous polyG sequences at the 3′ end. Reads were aligned to the mouse Mmu_GRCm38.90_ERCC_SIRV genome reference (Genome Reference Consortium) using the STAR tool (spliced transcripts alignment to a reference) version 2.5.3a. Normalization and differential expression analysis was performed with DESeq2 version v1.18.1 with significance determined at adj p < 0.1. Top pathways were determined using Ingenuity Pathway Analysis (IPA).
3. Results
3.1. FLExDUX4 Mice Show Muscle Weakness Measured by Grip Strength Testing
To determine if the FLExDUX4 mice showed muscle weakness, the grip strengths of a cohort of FLExDUX4 mice and their wild-type litter mates (male n = 8–9, female n = 3–4) were measured using an isometric force transducer at 5, 8, and 12 months of age (Figure 1). At all-time points there was a significant difference in the force measurement between the FLExDUX4 and wild-type male (Figure 1A,C) and female mice (Figure 1B,D) (p < 0.05). At 5 months old, male FLExDUX4 mice were an average of 25% weaker in the forelimb (p < 0.001) and 17% weaker in the hindlimb (p < 0.01). At 8 months old, male FLExDUX4 mice were an average of 22% weaker in the forelimb (p < 0.001) and 13% weaker in the hindlimb (p < 0.05). At 12 months old, male FLExDUX4 mice were 21% weaker in the forelimb (p < 0.001) and 17% weaker in the hindlimb (p < 0.05). In female FLExDUX4 mice, the forelimb strength was an average of 18% weaker (p < 0.05), and the hindlimb was an average of 20% weaker at 5 months old (p < 0.05). At 8 months old, female FLExDUX4 mice were an average of 15% weaker in the forelimb (p < 0.05) and 23% weaker in the hindlimb (p < 0.01). At 12 months old, female FLExDUX4 mice were an average of 22% weaker in the forelimb (p < 0.001) and 30% weaker in the hindlimb (p < 0.05).
3.2. FLExDUX4 Mice Display Sex Differences in Body Weight and Muscle Weight
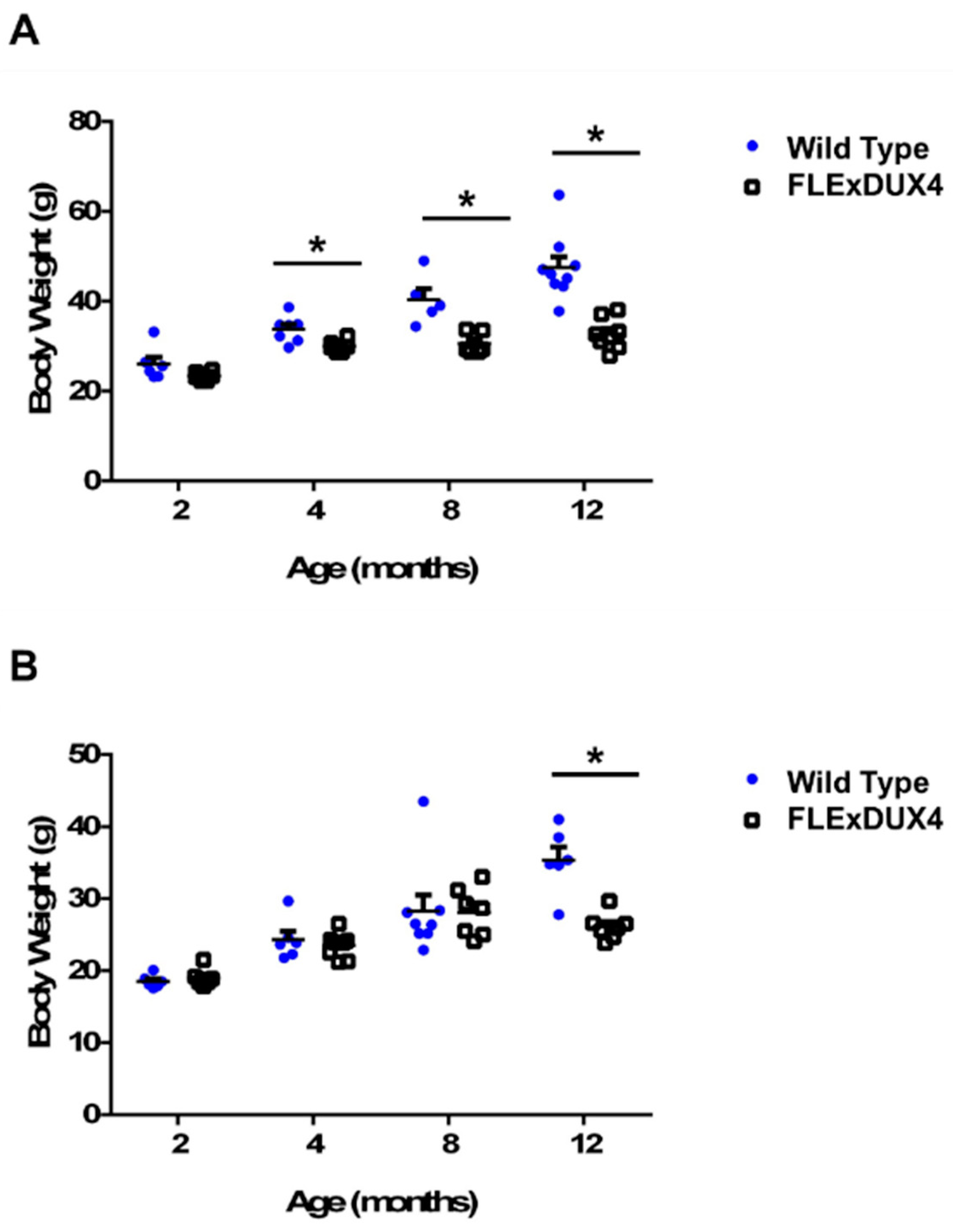
Body weight differences in mice approximately 20 weeks old were previously reported in FLExDUX4 mice [48]; however, muscle weight data were not reported. In this study, we measured the body weight and muscle weight in several cohorts of mice at 2 months, 4 months, 8 months, and 12 months to determine weight changes in the FLExDUX4 mice when they age. The muscles examined include the gastrocnemius, soleus, tibialis anterior, quadriceps, deltoid, triceps, biceps, masseter, diaphragm, and heart. Both male (n = 6–9) and female (n = 6–8) FLExDUX4 mice showed significant differences in body weight (Figure 2) and muscle weight (Supplemental Tables S1 and S2) compared to their wild-type littermates. However, the body weight difference appeared at a much younger age in the male mice (4 months old) compared to the female mice (12 months old). Male FLExDUX4 mice showed body weight differences at 4 months of age and were an average of 3.8 g lighter (11%, p < 0.05). At 8 months old, the FLExDUX4 mice were an average of 9.7 g (24%, p < 0.05) lighter. At 12 months old, male FLExDUX4 mice were an average of 14.7 g (31%, p < 0.05) lighter than their wild-type littermates (Figure 2A). Unlike their male counterparts, female FLExDUX4 mice did not show a significant difference in body weight until 12 months old. At 12 months old, female FLExDUX4 mice were an average of 9.3 g lighter (26%, p < 0.05) than their wild-type littermates (Figure 2B).
The muscle weight data showed that the muscle weight of male FLExDUX4 mice was significantly lower compared to their wild-type littermates at 2 months, 4 months, 8 months, and 12 months old (Supplemental Table S1). In male FLExDUX4 mice, significant differences in the weights of three hindlimb muscles (gastrocnemius, soleus, and quadriceps) were identified at 2 months, 4 months, and 12 months old (p < 0.05). The tibialis anterior muscles showed significant differences at 2 months and 4 months old (p < 0.05). Significant differences in the biceps and triceps of the forearm were observed at 12 months old (p < 0.05). The muscles most frequently affected were the hindlimb muscles: gastrocnemius, soleus, tibialis anterior, and the quadriceps. The deltoid, masseter, heart, and diaphragm were the most frequently spared. In female FLExDUX4 mice, many of the muscles did not show significant differences until 12 months of age (Supplemental Table S2). The muscles that showed significance were the deltoid, masseter, triceps, and quadriceps at 4 months old (p < 0.05). The masseter was the only muscle affected at 8 months of age. At 12 months old, the deltoid, triceps, biceps, gastrocnemius, soleus, tibialis anterior, and the quadriceps were significantly smaller in FLExDUX4 mice (p < 0.05). Additionally, muscle weight in both male and female FLExDUX4 mice and their wild-type littermates increased over time. At 12 months of age, wild-type littermates had higher or similar muscle weights compared to the 8-month-old wild-type mice. However, the FLExDUX4 mice showed a decrease in muscle weight.
3.3. Fiber Size Variations Were Observed in Type IIa and IIx Muscle Fibers in Older FLExDUX4 Mice
Because the male mice showed significant muscle weight differences over time, we further examined muscle pathologies in the male FLExDUX4 mice to determine what contributed to the muscle weight loss. To examine histological changes in FLExDUX4 mice at young and old ages, quadriceps from 2-month-old and 12-month-old male mice were studied. At 2 months old, there was no overt pathology found in the FLExDUX4 mice compared to their wild-type littermates. No significant difference was observed in myofiber size in 2-month-old FLExDUX4 mice. In 12-month-old mice, a deviation in myofiber size (Feret’s diameter) was observed in the smallest 40% of fibers measured, but no difference was observed in the largest fibers measured (Supplemental Figures S1 and S2).
The presence of small myofibers is often reported in diseased muscle. This can be due to regeneration of muscle fibers or muscle fiber atrophy [54]. Analysis of the number of central nucleated fibers did not show a significant difference between the FLExDUX4 and wild-type quadriceps. NADH-TR staining has been used to characterize myofiber types and visualize angular atrophic myofibers [54,60]. Atrophic myofibers are angular-shaped small fibers heavily stained with NADH-TR. To determine if atrophic fibers were present, the 12-month-old quadriceps were stained with NADH-TR. The FLExDUX4 mice presented with smaller NADH-TR positive muscle fibers, and some of them were angular-shaped (Figure 3 and Supplemental Figure S3). Two genes, Atrogin 1 and Murf1, that are commonly involved in muscle atrophy were observed by RT-qPCR, and no significant differences in expression were detected.
Muscle fibers can be subdivided into the oxidative (type I) and glycolytic (type II) myofibers. In mice, a gradient exists between the glycolytic fibers, where the larger type IIb fibers are more glycolytic (light blue with NADH-TR) and smaller type IIa fibers are more oxidative (dark blue with NADH-TR) [60,61,62]. The deviation in fiber sizes suggested that different fiber types were affected in 12-month-old FLExDUX4 mice (Figure 3 and Supplemental Figures S1–S3).
To determine how specific fiber types are affected in the FLExDUX4 mice, quadriceps from 2-month-old and 12-month-old FLExDUX4 mice were co-stained with myosin heavy chain type I, type IIa, and type IIx. Each myofiber was measured to determine fiber size and circularity. Circularity measurements were used in this experiment as a means to quantify myofiber shape. Atrophic myofibers have been shown to be more angular and therefore would have a lower circularity value compared to a healthy myofiber. The data showed that 2-month-old mice did not show significant differences in size or circularity of each muscle fiber type. At 12 months old, the FLExDUX4 mice had significantly smaller type IIa (p < 0.05) and type IIx (p < 0.05) fibers (Figure 4). There was no significant difference in the size of the type IIb fibers. Fiber circularity of the type IIx fibers showed significant differences, indicating that the fibers are more angular-shaped (Figure 5). These data showed that, over time, the oxidative subset of the type II myosin heavy chain fiber types became smaller and atrophic.
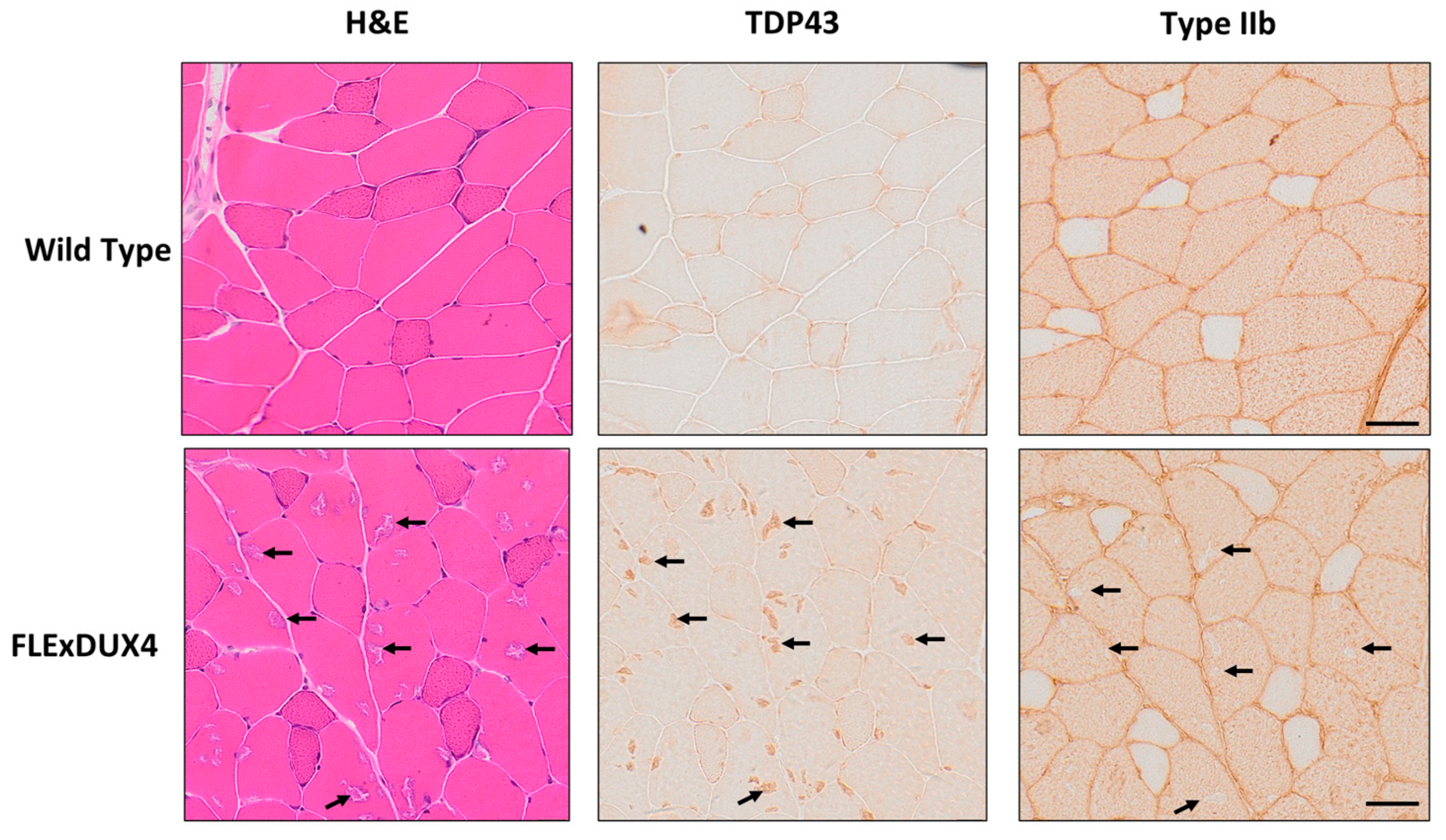
3.4. TDP-43-Positive Aggregates Were Found in the Type IIb Fibers
In addition to the presence of smaller fibers in the 12-month-old FLExDUX4 mice, hematoxylin and eosin-stained sections of the quadriceps muscles appeared to have basophilic aggregates (Figure 6). These aggregates were consistent in appearance to a mouse model that overexpresses TAR DNA-binding protein 43 (TDP-43), and the aggregation of TDP-43 has been observed in FSHD myoblasts in vitro in response to DUX4 expression [63,64,65]. Based on data from the immunochemistry staining of serial sections, the aggregates stained were TDP-43-positive and located specifically in the type IIb fibers.
3.5. RNA Sequencing Revealed Changes in Metabolism and Oxidative Stress Response
To determine molecular changes before the muscle pathology became apparent, we conducted RNA-seq of muscles from 2-month-old male mice. At 2 months of age, there were 573 differentially expressed genes (p < 0.05) in the comparison between the FLExDUX4 vs. wild-type mice.
We used ingenuity pathway analysis (IPA) to determine which genes are enriched in specific functional groups of genes based on the sequencing data. The IPA revealed changes in several pathways involved in growth and muscle remodeling. At two months of age, the top three ranked pathways were Adipogenesis (p < 0.001), circadian rhythm (p < 0.001), and NRF2-mediated oxidative stress response (p < 0.001) (Supplemental Table S3). Canonical circadian rhythm genes, namely Clock, Cry2, and Per2, were shown to be differentially expressed in FLExDUX4 mice [66]. The NRF2 pathway has also been reported to be Clock-controlled and has been shown to have differential expression in FSHD [35,67,68]. In the adipogenesis pathways, Forkhead box protein O1 (Foxo1) and Lipoprotein lipase (Lpl) were both upregulated. The Foxo1 gene was reported to inhibit adipocyte differentiation and initiate muscle atrophy. The activation of Lpl by Foxo1 facilitates a metabolic switch from carbohydrate oxidation to lipid oxidation in muscle [35,67,68,69,70,71,72]. Indeed, BODIPY staining of quadriceps muscle from 4-month-old FLExDUX4 mice showed significant increases in intermuscular fat in the muscle (Supplemental Figure S4). Three metabolic pathways identified based on the IPA (Methylmalonyl, 2-oxobutanoate degradation I, PPARα/RXRα activation) play a role in lipid metabolism and generation of components for the Krebs cycle [73]. The PPARα/RXRα activation in addition to CD27 signaling was reported to play a role in inflammatory pathways [73]. Additional pathways identified include genes in changes in pathways involved in muscle growth (AMPK signaling), and pathways elevated in oxidative stress response (NRF-2 mediated response). These molecular changes were identified in muscles without overt pathologies.
4. Discussion
Most mouse models for FSHD have been limited in experimental use due to either severe phenotypes preventing long-term studies or inconsistent DUX4 expression and pathologies [25,44,45,46,47]. Previously published data on the FLExDUX4 single transgenic animals showed that the FLExDUX4 mice showed body weight differences, had leaky expression of DUX4 mRNA, and did not show overt muscle pathology in 23-week-old female FLExDUX4 mice [48]. A more recent study reported that the FLExDUX4 mice have a mild but significant increase in fibrosis at 6 months old [74]. The goal of this project was to track changes in muscle weight, functional, and muscle pathologies in response to the leaky low-level expression of DUX4 for a long period of time. We found that the FLExDUX4 mice presented with several phenotypes as they aged. Both male and female FLExDUX4 mice had significantly weaker grip strength measurements. Male mice presented with significantly smaller body weight and muscle weight compared to their wild type littermates. In addition, the males developed the phenotypes at a younger age compared to the female FLExDUX4 mice. The RNA-seq data showed that DUX4 expression in myofibers affected gene expression associated with pathways of adipogenesis, circadian rhythm, and NRF2-mediated oxidative stress response. Pathological data showed that prolonged expression of DUX4 leads to TDP-43 positive aggregates in type IIb fibers and a decrease in the size of type IIa and type IIx fibers.
The FLExDUX4 mice showed significant differences in body weight. Male FLExDUX4 mice became significantly smaller once mice reached adulthood at 4 months old which is in concordance with previously reported data by Jones et al. in 20-week-old mice [48]. Different from the previously reported data, we did not observe changes in body weight of FLExDUX4 female mice until 12 months of age [48]. Body weight and muscle weight data suggested that the FLExDUX4 mice showed sex differences. Male FLExDUX4 mice have significantly lower absolute muscle weight starting at 2 months of age and up to 12 months of age with a progressive increase in the number of muscles affected. Few muscles of the female FLExDUX4 mice showed significant weight differences until 12 months old. A previous study suggested that estrogen has a protective effect in DUX4-expressing muscles [75]. Estrogen protection in the FLExDUX4 mice may partially explain the sex differences observed in our experiment. Reproductive senescence in mice occurs between 9 and 12 months of age, which supports the age of onset of the body weight and muscle weight differences in the female FLExDUX4 mice [76]. The data generated from the ACTA1-cre/FLExDUX4 female mice injected with tamoxifen, an estrogen receptor inhibitor used to induce DUX4 expression, also suggested the protective effects of estrogen in the female mice. Female mice were more severely affected compared to their male littermates after DUX4 was induced by tamoxifen injections [49].
The observation of sex differences in the body weight and muscle weight, where males are more severely affected, is similar to the observation in FSHD [41,52,77,78]. The average age of onset is by the second decade of life in males and the third decade of life in females [19,52]. In the FLExDUX4 mice, heart, diaphragm, masseter, and deltoids were mostly spared in the aged mice, which reflects the human FSHD [77,79,80,81]. However, there was no obvious preference of forelimb vs. hindlimb muscles observed in the FLExDUX4 mice. Various mouse models have been generated for the pre-clinical development of therapeutics for FSHD. Another inducible model, iDUX4pA mice, contains an inducible DUX4 gene inserted upstream of the HPRT gene and under the control of the doxycycline-inducible sgTRE promoter [45]. The iDUX4pA model also displays sex differences, but male mice have only been reported to live until 4 months of age, which can limit trial length of a potential therapeutic [45]. Like the FLExDUX4 mice, the iDUX4pA mice have alopecia, smaller body weights, and smaller muscle weights [45,48]. Flow cytometry and transcriptional analysis showed that the iDUX4pA mouse model has increased fibrosis markers in the absence of pathology, as previously observed in the FLExDUX4 model [45,74,82]. Both models have reported leaky DUX4 expression in other tissues [45,48]. Hearing loss and vasculature defects were observed in the iDUX4pA mice [45,82]. Interestingly, decreased body fat was observed in the iDUX4pA model as measured by MRI [45]. We did observe less body fat in the FLExDUX4 mice; however, no quantitative data were collected. The conditional ACTA-MCM/FLExDUX4 mice were also reported to have sex differences where the male mice were more severely affected. Conversely, when the ACTA-MCM/FLExDUX4 mice were administered tamoxifen to induce higher DUX4 expression, the female mice are more severely affected [49]. Chronic induction and expression of DUX4 in female iDUX4pA mice was reported to cause severe phenotypes, including a prominent outward spinal curvature, pronounced muscle atrophy, and muscle weakness at the end of the 6-month study [82]. Pathological changes in muscles of the mice suggest more severe phenotypes, including inflammatory infiltration, fat deposition, and deposition of extracellular matrix [82]. With the transgene induced, the iDUX4pA model is more comparable to the ACTA-MCM/FLExDUX4 mice when the DUX4 is induced by tamoxifen.
Analysis of the grip strength data showed that FLExDUX4 mice were significantly weaker than their wild-type littermates. Previous studies reported no significant difference in time to fatigue or wire hanging assessments [48,49]. These differences may be due to age differences or differences in assays used for accessing muscle strength. The muscles of FLExDUX4 mice do not have overt degeneration/regeneration during the observation period; however, we have previously reported an increase in endomysial fibrosis in the model [74]. Additionally, fibrosis pathways were activated in our current RNA-seq data through IPA at 2 months of age (p < 0.05, ranked 15), suggesting early involvement of the fibrosis pathways. Small increases in fibrosis have been shown to decrease grip strength and increase muscle weakness [83,84,85]. Muscle weakness can also occur due to additional molecular changes in the muscle [54,86,87]. Muscle biopsies from individuals with FSHD were reported to have increased expression of proteins involved in glycolysis and tricarboxylic acid cycle, lipid peroxidation, decreased mitochondrial function, and a decreased response to oxidative stress [41,88]. In our study, we showed that from a young age, genes involved in lipid oxidation and NRF-2 stress response pathways were misregulated. Exposure to reactive oxygen species has been shown to cause decreased contractility in muscle fibers [89]. The combination of fatty acid oxidation and an impaired oxidative stress response may potentially perpetuate oxidative stress that can impact functional output [35,67,68,90,91].
In this study, we identified long-term exposure to low levels of DUX4 expression resulted in fiber-type-specific pathologies as the FLExDUX4 mice age. Basophilic TDP-43 aggregates were observed in type IIb fibers specifically, while the type IIa and IIx fibers decreased in fiber size. The decreased fiber size of type IIa and type IIx muscle fibers in 12-month-old FLExDUX4 muscle suggests that chronic low-level expression of DUX4 negatively affects the fiber maintenance of type IIa and type IIx. Misregulation in circadian rhythm signaling in young FLExDUX4 mice could contribute to impaired muscle maintenance, as MyoD1 is a documented Clock transcriptional target [92]. MyoD-regulated pathways have been reported to be affected in biopsies from individuals with FSHD [37]. Results from the NADH-TR staining showed angular-shaped myofibers with dark blue staining, which suggested these were atrophic fibers [54]; however, RT-qPCR of Atrogin 1 and Murf1 did not show significant differences. It is possible that subtle changes in gene expression were hard to detect when a small proportion of fibers are affected. It can also be that other pathways involved in myofiber atrophy contribute to the observed phenotype. Specific loss of type IIa and IIx fibers in muscles has been reported previously in aging and in FSHD [61,93,94,95,96,97]. Aging studies in 2-year-old murine muscle have shown that there is preferential loss of fiber size in type IIa and IIx fibers, with a 30% observed fiber loss, smaller type 2 fiber sizes, and a sparing of type IIb muscle fibers [93,94,95,96,97]. Expression profiling and proteomics of aging muscles showed increased metabolic- and mitochondrial-induced stress [93,94,97,98]. Recently, proteomic profiling of FSHD myoblasts showed that FSHD myoblasts contain a higher number of mitochondrial proteins in conjunction with reduced mitochondrial protein turnover [99]. This suggested that DUX4 expression impacted the quality of the available mitochondrial proteins, even though the quantity was not reduced [99]. Impairments in quality control mechanisms of the mitochondria have been reported to lead to an excessive generation of reactive oxygen species, disorganization of the myofiber, and malformations in the mitochondria [88,97,100]. In humans, aging mechanisms and FSHD favor the preservation of the oxidative type I and loss of the glycolytic type II [61,93]. Muscle groups affected in FSHD, such as the zygomaticus major, orbicularis orbis, biceps brachii, triceps brachii, and tibialis anterior, contain a large proportion of fast twitch (type 2) fibers and a lower proportion of slow twitch (type 1) fibers [101,102,103,104,105]. Functional deficits and fiber size variation are common pathologies that are shared with the ACTA-MCM/FLExDUX4 model, the AAV-DUX4, and iDUX4pA models [25,45,49,106,107]. However, the aggregation of TDP-43 in type IIb myofibers and preferential decrease in type IIa and IIx fiber sizes are new findings from characterizing the FLExDUX4 mice [48,49,106,107]. To date, TDP-43 has only been shown to aggregate in the nucleus of FSHD myotubes in vitro but not in the cytoplasm [64,65]. Since humans were reported to not have type IIb fibers, this phenomenon may be unique to the mouse model but still worth further investigation [61,93,108,109].
Previous studies have reported the presence of inflammatory infiltrate, fibrosis, and fat deposition in affected patients with FSHD. The FLExDUX4 mice presented with mild but significant increases in fat and fibrosis in response to chronic exposure to low levels of leaky DUX4. However, the low levels of DUX4 expression do not induce severe pathologies, such as muscle necrosis and severe inflammation. When the DUX4 is induced to be expressed at a higher level in the ACTA1-cre/FLExDUX4 mouse model, the mice develop pathological changes, including degeneration/regeneration and inflammation similar to muscle pathologies seen in the affected areas in FSHD [48,49]. Please note that the less affected areas in muscles from individuals with FSHD also do not show overt pathology, which is similar to the observation in the FLExDUX4 mice. Based on these findings, we propose that the FLExDUX4 mice recapitulate the state when the muscles are mildly affected by the low level of DUX4 expression.
In the conditional ACTA-MCM/FLExDUX4 models, continuous induction of the DUX4 transgene is required to maintain what has been coined as moderate levels of pathology because the regenerated muscle fibers do not express equivalently high levels of DUX4 [25,49]. Continuous induction would be required to assess long-term therapeutic efficacy in a pre-clinical trial [106,107]. Of note, the uninduced ACTA-MCM/FLExDUX4 mice do not have overt degeneration and regeneration [49]. However, the DUX4 protein was reported to be detected in uninduced ACTA-MCM/FLExDUX4 mice, which was not detected in the FLExDUX4 mice [48,49]. We show that the FLExDUX4 model can reliably express DUX4 mRNA up to 12 months old, which can be beneficial for testing agents directly targeting the DUX4 transcripts [48,110,111]. This will be an advantage, as consistent DUX4 expression in the model reduces concerns from DUX4 fluctuation due to the degeneration and regeneration of the muscles, such as in the ACTA-MCM/FLExDUX4 model. Further study into the FLExDUX4, uninduced ACTA1-MCM/FLExDUX4, and induced ACTA1-MCM/FLExDUX4 models to determine how different levels of DUX4 affect the pathology of the muscles would help elucidate the disease mechanisms, as would the selection of proper models for specific types of therapeutic approaches.
5. Conclusions
Prolonged expression of a low level of DUX4 led to reductions in muscle and body weights, as well as muscle strength, in the FLExDUX4 mice. Males developed the disease phenotypes at a younger age in comparison to the female mice. Several molecular pathways, including pathways involved in oxidative stress response and fibrosis, were altered in the affected muscles. In addition, prolonged expression of DUX4 lead to preferential decreases in type IIa and IIx myofibers and TDP43-containing aggregates in type IIb myofibers. This study demonstrated that FLExDUX4 mice have measurable phenotypes, some of which reflect human FSHD. The functional, pathological, and molecular changes can potentially be considered to be used as outcome measures when testing agents targeting DUX4.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/jpm13071040/s1, Figure S1: Male FLExDUX4 mice display fiber size differences in small fibers at 12 months old of age; Figure S2: Fiber size distribution of the quadriceps muscle in 12-month-old male FLExDUX4 and wild type littermates using dystrophin stain; Figure S3: Fiber size distribution of the NADH-TR positive fibers in quadriceps in male FLExDUX4 and wild type littermates; Figure S4: BODIPY lipid staining of 4-month-old FLExDUX4 mice and wild type littermates; Table S1: Average muscle weight (mg) that were significantly smaller in male FLExDUX4 mice compared to wild type littermates at different time points; Table S2: Average muscle weight (mg) that were significantly smaller in female FLExDUX4 mice compared to wild type littermates at different time points; Table S3: Canonical pathways that are affected in 2-month-old male FLExDUX4 mice in comparison to wild type littermates.
Author Contributions
K.M. contributed to conceptualization, methodology, data analysis, writing—original draft preparation, and visualization. A.Z. contributed to methodology, visualization, and data analyses. A.J.B. contributed to methodology, performing experiments, and data curation. Y.-W.C. contributed to conceptualization, resources, writing–review and editing, project administration, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially supported by the FSHD society, the FSHD Global Research Foundation, the Muscular Dystrophy Association, and Friends of FSH Research.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board of the Children’s National Hospital for studies involving animals.
Informed Consent Statement
Not applicable.
Data Availability Statement
The RNA-seq data are deposited in NIH GEO database (GSE233832).
Acknowledgments
Myosin heavy chain antibodies were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at the University of Iowa, Department of Biology, Iowa City, IA 52242.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tawil, R.; Van Der Maarel, S.M. Facioscapulohumeral muscular dystrophy. Muscle Nerve 2006, 34, 1–15. [Google Scholar] [CrossRef]
- Statland, J.M.; Tawil, R. Facioscapulohumeral Muscular Dystrophy. Continuum 2016, 22, 1916–1931. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.G.; Carrino, J.A.; Wagner, K.R.; Jacobs, M.A. Whole-body magnetic resonance imaging evaluation of facioscapulohumeral muscular dystrophy. Muscle Nerve 2015, 52, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Deenen, J.C.; Arnts, H.; van der Maarel, S.M.; Padberg, G.W.; Verschuuren, J.J.G.M.; Bakker, E.; Weinreich, S.S.; Verbeek, A.L.; van Engelen, B.G. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 2014, 83, 1056–1059. [Google Scholar] [CrossRef] [Green Version]
- Flanigan, K.M.; Coffeen, C.M.; Sexton, L.; Stauffer, D.; Brunner, S.; Leppert, M.F. Genetic characterization of a large, historically significant Utah kindred with facioscapulohumeral dystrophy. Neuromuscul. Disord. 2001, 11, 525–529. [Google Scholar] [CrossRef]
- Lemmers, R.J.L.F.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.E.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [Green Version]
- Mostacciuolo, M.L.; Pastorello, E.; Vazza, G.; Miorin, M.; Angelini, C.; Tomelleri, G.; Galluzzi, G.; Trevisan, C.P. Facioscapulohumeral muscular dystrophy: Epidemiological and molecular study in a north-east Italian population sample. Clin. Genet. 2009, 75, 550–555. [Google Scholar] [CrossRef]
- van der Maarel, S.M.; Frants, R.R. The D4Z4 repeat-mediated pathogenesis of facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2005, 76, 375–386. [Google Scholar] [CrossRef] [Green Version]
- van Deutekom, J.C.; Wljmenga, C.; Tlenhoven, E.A.; Gruter, A.M. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993, 2, 2037–2042. [Google Scholar] [CrossRef]
- van den Boogaard, M.L.; Lemmers, R.J.L.F.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, K.; Šikrová, D.; Mitsuhashi, S.; Masuda, H.; Sekiguchi, Y.; Sugiyama, A.; Shibuya, K.; Lemmers, R.J.; Goossens, R.; Ogawa, M.; et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology 2020, 94, e2441–e2447. [Google Scholar] [CrossRef]
- Cabianca, D.S.; Casa, V.; Bodega, B.; Xynos, A.; Ginelli, E.; Tanaka, Y.; Gabellini, D. A Long ncRNA Links Copy Number Variation to a Polycomb/Trithorax Epigenetic Switch in FSHD Muscular Dystrophy. Cell 2012, 149, 819–831. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Gearhart, M.D.; Cui, Z.; Bosnakovski, D.; Kim, M.; Schennum, N.; Kyba, M. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016, 44, 5161–5173. [Google Scholar] [CrossRef] [Green Version]
- de Greef, J.C.; Wohlgemuth, M.; Chan, O.A.; Hansson, K.B.; Smeets, D.; Frants, R.R.; Weemaes, C.M.; Padberg, G.W.; van der Maarel, S.M. Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology 2007, 69, 1018–1026. [Google Scholar] [CrossRef]
- Gaillard, M.-C.; Roche, S.; Dion, C.; Tasmadjian, A.; Bouget, G.; Salort-Campana, E.; Vovan, C.; Chaix, C.; Broucqsault, N.; Morere, J.; et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology 2014, 83, 733–742. [Google Scholar] [CrossRef]
- Huichalaf, C.; Micheloni, S.; Ferri, G.; Caccia, R.; Gabellini, D. DNA Methylation Analysis of the Macrosatellite Repeat Associated with FSHD Muscular Dystrophy at Single Nucleotide Level. PLoS ONE 2014, 9, e115278. [Google Scholar] [CrossRef]
- Statland, J.M.; Donlin-Smith, C.M.; Tapscott, S.J.; Lemmers, R.J.; van der Maarel, S.; Tawil, R. Milder phenotype in facioscapulohumeral dystrophy with 7–10 residual D4Z4 repeats. Neurology 2015, 85, 2147–2150. [Google Scholar] [CrossRef]
- van Overveld, P.G.; Lemmers, R.J.; Sandkuijl, L.A.; Enthoven, L.; Winokur, S.T.; Bakels, F.; Padberg, G.W.; van Ommen, G.J.; Frants, R.R.; van der Maarel, S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003, 35, 315–317. [Google Scholar] [CrossRef]
- Zeng, W.; Chen, Y.-Y.; Newkirk, D.A.; Wu, B.; Balog, J.; Kong, X.; Ball, A.R., Jr.; Zanotti, S.; Tawil, R.; Hashimoto, N.; et al. Genetic and Epigenetic Characteristics of FSHD-Associated 4q and 10q D4Z4 that are Distinct from Non-4q/10q D4Z4 Homologs. Hum. Mutat. 2014, 35, 998–1010. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; De Greef, J.C.; Chen, Y.Y.; Chien, R.; Kong, X.; Gregson, H.C.; Winokur, S.T.; Pyle, A.; Robertson, K.D.; Schmiesing, J.A.; et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet. 2009, 5, e1000559. [Google Scholar] [CrossRef] [Green Version]
- Dixit, M.; Ansseau, E.; Tassin, A.; Winokur, S.; Shi, R.; Qian, H.; Sauvage, S.; Mattéotti, C.; van Acker, A.M.; Leo, O.; et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA 2007, 104, 18157–18162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmers, R.J.; De Kievit, P.; Sandkuijl, L.; Padberg, G.W.; Van Ommen, G.-J.B.; Frants, R.R.; van der Maarel, S. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat. Genet. 2002, 32, 235–236. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.F.; Wohlgemuth, M.; Frants, R.R.; Padberg, G.W.; Morava, E.; van der Maarel, S.M. Contractions of D4Z4 on 4qB Subtelomeres Do Not Cause Facioscapulohumeral Muscular Dystrophy. Am. J. Hum. Genet. 2004, 75, 1124–1130. [Google Scholar] [CrossRef] [Green Version]
- Spurlock, G.; Jim, H.-P.; Upadhyaya, M. Confirmation that the specific SSLP microsatellite allele 4qA161 segregates with fascioscapulohumeral muscular dystrophy (FSHD) in a cohort of multiplex and simplex FSHD families. Muscle Nerve 2010, 42, 820–821. [Google Scholar] [CrossRef]
- Wallace, L.M.; Garwick, S.E.; Mei, W.; Belayew, A.; Coppee, F.; Ladner, K.J.; Guttridge, D.; Yang, J.; Harper, S.Q. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann. Neurol. 2011, 69, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 Activates Germline Genes, Retroelements, and Immune Mediators: Implications for Facioscapulohumeral Dystrophy. Dev. Cell 2012, 22, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Padberg, G.W.; van Engelen, B.G. Facioscapulohumeral muscular dystrophy. Curr. Opin. Neurol. 2009, 22, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Hendrickson, P.G.; Doráis, J.A.; Grow, E.J.; Whiddon, J.L.; Lim, J.-W.; Wike, C.L.; Weaver, B.D.; Pflueger, C.; Emery, B.R.; Wilcox, A.L.; et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 2017, 49, 925–934. [Google Scholar] [CrossRef]
- Young, J.M.; Whiddon, J.L.; Yao, Z.; Kasinathan, B.; Snider, L.; Geng, L.N.; Balog, J.; Tawil, R.; van der Maarel, S.M.; Tapscott, S.J. DUX4 Binding to Retroelements Creates Promoters That Are Active in FSHD Muscle and Testis. PLoS Genet. 2013, 9, e1003947. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Chadwick, B.P. Influence of Repressive Histone and DNA Methylation upon D4Z4 Transcription in Non-Myogenic Cells. PLoS ONE 2016, 11, e0160022. [Google Scholar] [CrossRef] [Green Version]
- de la Kethulle de Ryhove, L.; Ansseau, E.; Nachtegael, C.; Pieters, K.; Vanderplanck, C.; Geens, M.; Sermon, K.; Wilton, S.D.; Coppée, F.; Lagneaux, L.; et al. The Role of D4Z4-Encoded Proteins in the Osteogenic Differentiation of Mesenchymal Stromal Cells Isolated from Bone Marrow. Stem Cells Dev. 2015, 24, 2674–2686. [Google Scholar] [CrossRef] [PubMed]
- Gannon, O.; de Long, L.M.; Saunders, N.A. DUX4 Is Derepressed in Late-Differentiating Keratinocytes in Conjunction with Loss of H3K9me3 Epigenetic Repression. J. Investig. Dermatol. 2016, 136, 1299–1302. [Google Scholar] [CrossRef] [Green Version]
- De Iaco, A.; Planet, E.; Coluccio, A.; Verp, S.; Duc, J.; Trono, D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 2017, 49, 941–945. [Google Scholar] [CrossRef]
- Dmitriev, P.; Saada, Y.B.; Dib, C.; Ansseau, E.; Barat, A.; Hamade, A.; Dessen, P.; Robert, T.; Lazar, V.; Louzada, R.A.; et al. DUX4-induced constitutive DNA damage and oxidative stress contribute to aberrant differentiation of myoblasts from FSHD patients. Free. Radic. Biol. Med. 2016, 99, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Harafuji, N.; Belayew, A.; Chen, Y.-W. DUX4 Differentially Regulates Transcriptomes of Human Rhabdomyosarcoma and Mouse C2C12 Cells. PLoS ONE 2013, 8, e64691. [Google Scholar] [CrossRef] [Green Version]
- Winokur, S.T.; Barrett, K.; Martin, J.H.; Forrester, J.R.; Simon, M.; Tawil, R.; Chung, S.-A.; Masny, P.S.; Figlewicz, D.A. Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscul. Disord. 2003, 13, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Winokur, S.T.; Chen, Y.-W.; Masny, P.S.; Martin, J.H.; Ehmsen, J.T.; Tapscott, S.J.; van der Maarel, S.M.; Hayashi, Y.; Flanigan, K.M. Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum. Mol. Genet. 2003, 12, 2895–2907. [Google Scholar] [CrossRef] [Green Version]
- Rickard, A.M.; Petek, L.M.; Miller, D.G. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 2015, 24, 5901–5914. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Snider, L.; Jagannathan, S.; Tawil, R.; van der Maarel, S.M.; Tapscott, S.J.; Bradley, R.K. A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. elife 2015, 4, e04996. [Google Scholar] [CrossRef]
- Barro, M.; Carnac, G.; Flavier, S.; Mercier, J.; Vassetzky, Y.; Laoudj-Chenivesse, D. Myoblasts from affected and non-affected FSHD muscles exhibit morphological differentiation defects. J. Cell. Mol. Med. 2008, 14, 275–289. [Google Scholar] [CrossRef]
- Celegato, B.; Capitanio, D.; Pescatori, M.; Romualdi, C.; Pacchioni, B.; Cagnin, S.; Viganò, A.; Colantoni, L.; Begum, S.; Ricci, E.; et al. Parallel protein and transcript profiles of FSHD patient muscles correlate to the D4Z4 arrangement and reveal a common impairment of slow to fast fibre differentiation and a general deregulation of MyoD-dependent genes. Proteomics 2006, 6, 5303–5321. [Google Scholar] [CrossRef] [Green Version]
- Tassin, A.; Leroy, B.; Laoudj-Chenivesse, D.; Wauters, A.; Vanderplanck, C.; Le Bihan, M.-C.; Coppée, F.; Wattiez, R.; Belayew, A. FSHD Myotubes with Different Phenotypes Exhibit Distinct Proteomes. PLoS ONE 2012, 7, e51865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knopp, P.; Krom, Y.D.; Banerji, C.R.S.; Panamarova, M.; Moyle, L.A.; den Hamer, B.; van der Maarel, S.M.; Zammit, P.S. DUX4 induces a transcriptome more characteristic of a less-differentiated cell state and inhibits myogenesis. J. Cell Sci. 2016, 129, 3816–3831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krom, Y.D.; Thijssen, P.E.; Young, J.M.; den Hamer, B.; Balog, J.; Yao, Z.; Maves, L.; Snider, L.; Knopp, P.; Zammit, P.S.; et al. Intrinsic Epigenetic Regulation of the D4Z4 Macrosatellite Repeat in a Transgenic Mouse Model for FSHD. PLoS Genet. 2013, 9, e1003415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosnakovski, D.; Chan, S.S.K.; Recht, O.O.; Hartweck, L.M.; Gustafson, C.J.; Athman, L.L.; Lowe, D.A.; Kyba, M. Muscle pathology from stochastic low level DUX4 expression in an FSHD mouse model. Nat. Commun. 2017, 8, 550. [Google Scholar] [CrossRef] [Green Version]
- Dandapat, A.; Bosnakovski, D.; Hartweck, L.M.; Arpke, R.W.; Baltgalvis, K.A.; Vang, D.; Baik, J.; Darabi, R.; Perlingeiro, R.C.; Hamra, F.K.; et al. Dominant Lethal Pathologies in Male Mice Engineered to Contain an X-Linked DUX4 Transgene. Cell Rep. 2014, 8, 1484–1496. [Google Scholar] [CrossRef] [Green Version]
- Dandapat, A.; Perrin, B.J.; Cabelka, C.; Razzoli, M.; Ervasti, J.M.; Bartolomucci, A.; Lowe, D.A.; Kyba, M. High Frequency Hearing Loss and Hyperactivity in DUX4 Transgenic Mice. PLoS ONE 2016, 11, e0151467. [Google Scholar] [CrossRef]
- Jones, T.; Jones, P.L. A cre-inducible DUX4 transgenic mouse model for investigating facioscapulohumeral muscular dystrophy. PLoS ONE 2018, 13, e0192657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, T.I.; Chew, G.-L.; Barraza-Flores, P.; Schreier, S.; Ramirez, M.; Wuebbles, R.D.; Burkin, D.J.; Bradley, R.K.; Jones, P.L. Transgenic mice expressing tunable levels of DUX4 develop characteristic facioscapulohumeral muscular dystrophy-like pathophysiology ranging in severity. Skelet. Muscle 2020, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, J.J.; Srikuea, R.; Kirby, T.J.; Peterson, C.A.; Esser, K.A. Inducible Cre transgenic mouse strain for skeletal muscle-specific gene targeting. Skelet. Muscle 2012, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Goselink, R.J.; Mul, K.; van Kernebeek, C.R.; Lemmers, R.J.; van der Maarel, S.M.; Schreuder, T.H.; Erasmus, C.E.; Padberg, G.W.; Statland, J.M.; Voermans, N.C.; et al. Early onset as a marker for disease severity in facioscapulohumeral muscular dystrophy. Neurology 2019, 92, e378–e385. [Google Scholar] [CrossRef]
- Zatz, M.; Marie, S.K.; Cerqueira, A.; Vainzof, M.; Pavanello, R.C.; Passos-Bueno, M.R. The facioscapulohumeral muscular dystrophy (FSHD1) gene affects males more severely and more frequently than females. Am. J. Med. Genet. 1998, 77, 155–161. [Google Scholar] [CrossRef]
- Sasaki, T.; Giltay, R.; Talts, U.; Timpl, R.; Talts, J.F. Expression and Distribution of Laminin α1 and α2 Chains in Embryonic and Adult Mouse Tissues: An Immunochemical Approach. Exp. Cell Res. 2002, 275, 185–199. [Google Scholar] [CrossRef]
- Pandey, S.; Cabotage, J.; Shi, R.; Dixit, M.; Sutherland, M.; Liu, J.; Muger, S.; Harper, S.Q.; Nagaraju, K.; Chen, Y.-W. Conditional over-expression of PITX1 causes skeletal muscle dystrophy in mice. Biol. Open 2012, 1, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Lucas, C.A.; Kang, L.H.; Hoh, J.F. Monospecific Antibodies against the Three Mammalian Fast Limb Myosin Heavy Chains. Biochem. Biophys. Res. Commun. 2000, 272, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.; Silberstein, L.; Hays, A.P.; Blau, H.M. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 1988, 52, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Azzarello, G.; Sartore, S.; Saggin, L.; Gorza, L.; D’Andrea, E.; Chieco-Bianchi, L.; Schiaffino, S. Myosin isoform expression in rat rhabdomyosarcoma induced by Moloney murine sarcoma virus. J. Cancer Res. Clin. Oncol. 1987, 113, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Gorza, L.; Sartore, S.; Saggin, L.; Ausoni, S.; Vianello, M.; Gundersen, K.; Lømo, T. Three myosin heavy chain isoforms in type 2 skeletal muscle fibres. J. Muscle Res. Cell Motil. 1989, 10, 197–205. [Google Scholar] [CrossRef]
- Briguet, A.; Courdier-Fruh, I.; Foster, M.; Meier, T.; Magyar, J.P. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul. Disord. 2004, 14, 675–682. [Google Scholar] [CrossRef]
- Suga, T.; Kimura, E.; Morioka, Y.; Ikawa, M.; Li, S.; Uchino, K.; Uchida, Y.; Yamashita, S.; Maeda, Y.; Chamberlain, J.S.; et al. Muscle Fiber Type-Predominant Promoter Activity in Lentiviral-Mediated Transgenic Mouse. PLoS ONE 2011, 6, e16908. [Google Scholar] [CrossRef] [Green Version]
- Talbot, J.; Maves, L. Skeletal muscle fiber type: Using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 518–534. [Google Scholar] [CrossRef] [Green Version]
- Nowak, L.; Reyes, P.F. Muscle Biopsy: A Diagnostic Tool in Muscle Diseases. J. Histotechnol. 2008, 31, 101–108. [Google Scholar] [CrossRef]
- Tawara, N.; Yamashita, S.; Kawakami, K.; Kurashige, T.; Zhang, Z.; Tasaki, M.; Yamamoto, Y.; Nishikami, T.; Doki, T.; Zhang, X.; et al. Muscle-dominant wild-type TDP-43 expression induces myopathological changes featuring tubular aggregates and TDP-43-positive inclusions. Exp. Neurol. 2018, 309, 169–180. [Google Scholar] [CrossRef]
- Homma, S.; Beermann, M.L.; Boyce, F.M.; Miller, J.B. Expression of FSHD-related DUX4-FL alters proteostasis and induces TDP-43 aggregation. Ann. Clin. Transl. Neurol. 2015, 2, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Homma, S.; Beermann, M.L.; Yu, B.; Boyce, F.M.; Miller, J.B. Nuclear bodies reorganize during myogenesis in vitro and are differentially disrupted by expression of FSHD-associated DUX4. Skelet. Muscle 2016, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Langmesser, S.; Tallone, T.; Bordon, A.; Rusconi, S.; Albrecht, U. Interaction of circadian clock proteins PER2 and CRY with BMAL1 and CLOCK. BMC Mol. Biol. 2008, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Tsumagari, K.; Chang, S.-C.; Lacey, M.; Baribault, C.; Chittur, S.V.; Sowden, J.; Tawil, R.; Crawford, G.E.; Ehrlich, M. Gene expression during normal and FSHD myogenesis. BMC Med. Genom. 2011, 4, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wible, R.S.; Ramanathan, C.; Sutter, C.H.; Olesen, K.M.; Kensler, T.W.; Liu, A.C.; Sutter, T.R. NRF2 regulates core and stabilizing circadian clock loops, coupling redox and timekeeping in Mus musculus. elife 2018, 7, e31656. [Google Scholar] [CrossRef]
- Cheng, Z.; White, M.F. Targeting Forkhead Box O1 from the Concept to Metabolic Diseases: Lessons from Mouse Models. Antioxid. Redox Signal. 2011, 14, 649–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, D.N.; Heuvel, A.P.J.V.D.; Birnbaum, M.J. The role of FoxO in the regulation of metabolism. Oncogene 2008, 27, 2320–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Tang, Y.; Zhao, Y.; Zhao, J.; Zhang, L.; Wei, W.; Chen, J. MiR-144-3p Targets FoxO1 to Reduce Its Regulation of Adiponectin and Promote Adipogenesis. Front. Genet. 2020, 11, 603144. [Google Scholar] [CrossRef]
- McCarthy, J.J.; Esser, K.A. Anabolic and catabolic pathways regulating skeletal muscle mass. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARalpha in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [Green Version]
- Bittel, A.J.; Sreetama, S.C.; Bittel, D.C.; Horn, A.; Novak, J.S.; Yokota, T.; Zhang, A.; Maruyama, R.; Lim, K.R.Q.; Jaiswal, J.K.; et al. Membrane Repair Deficit in Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 5575. [Google Scholar] [CrossRef] [PubMed]
- Teveroni, E.; Pellegrino, M.; Sacconi, S.; Calandra, P.; Cascino, I.; Farioli-Vecchioli, S.; Puma, A.; Garibaldi, M.; Morosetti, R.; Tasca, G.; et al. Estrogens enhance myoblast differentiation in facioscapulohumeral muscular dystrophy by antagonizing DUX4 activity. J. Clin. Investig. 2017, 127, 1531–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinton, R.D. Minireview: Translational Animal Models of Human Menopause: Challenges and Emerging Opportunities. Endocrinology 2012, 153, 3571–3578. [Google Scholar] [CrossRef]
- Ricci, G.; Scionti, I.; Sera, F.; Govi, M.; D’amico, R.; Frambolli, I.; Mele, F.; Filosto, M.; Vercelli, L.; Ruggiero, L.; et al. Large scale genotype–phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain 2013, 136, 3408–3417. [Google Scholar] [CrossRef] [Green Version]
- Tonini, M.; Passos-Bueno, M.; Cerqueira, A.; Matioli, S.; Pavanello, R.; Zatz, M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul. Disord. 2004, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Padberg, G.; Lunt, P.; Koch, M.; Fardeau, M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 1991, 1, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Tawil, R.; Figlewicz, D.A.; Griggs, R.C.; Weiffenbach, B.; The FSH Consortium. Facioscapulohumeral dystrophy: A distinct regional myopathy with a novel molecular pathogenesis. Ann. Neurol. 1998, 43, 279–282. [Google Scholar] [CrossRef]
- van der Maarel, S.M.; Frants, R.R.; Padberg, G.W. Facioscapulohumeral muscular dystrophy. Biochim. Biophys. Acta 2007, 1772, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Bosnakovski, D.; Shams, A.S.; Yuan, C.; Da Silva, M.T.; Ener, E.T.; Baumann, C.W.; Lindsay, A.J.; Verma, M.; Asakura, A.; Lowe, D.A.; et al. Transcriptional and cytopathological hallmarks of FSHD in chronic DUX4-expressing mice. J. Clin. Investig. 2020, 130, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- Abdelmagid, S.M.; Barr, A.E.; Rico, M.; Amin, M.; Litvin, J.; Popoff, S.N.; Safadi, F.F.; Barbe, M.F. Performance of Repetitive Tasks Induces Decreased Grip Strength and Increased Fibrogenic Proteins in Skeletal Muscle: Role of Force and Inflammation. PLoS ONE 2012, 7, e38359. [Google Scholar] [CrossRef] [Green Version]
- Mann, C.J.; Perdiguero, E.; Kharraz, Y.; Aguilar, S.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 2011, 1, 21. [Google Scholar] [CrossRef] [Green Version]
- Piñol-Jurado, P.; Suárez-Calvet, X.; Fernández-Simón, E.; Gallardo, E.; de la Oliva, N.; Martínez-Muriana, A.; Gómez-Gálvez, P.; Escudero, L.M.; Pérez-Peiró, M.; Wollin, L.; et al. Nintedanib decreases muscle fibrosis and improves muscle function in a murine model of dystrophinopathy. Cell Death Dis. 2018, 9, 776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aartsma-Rus, A.; van Putten, M. Assessing functional performance in the mdx mouse model. J. Vis. Exp. 2014, 85, e51303. [Google Scholar] [CrossRef] [Green Version]
- Pastoret, C.; Sebille, A. mdx mice show progressive weakness and muscle deterioration with age. J. Neurol. Sci. 1995, 129, 97–105. [Google Scholar] [CrossRef]
- Turki, A.; Hayot, M.; Carnac, G.; Pillard, F.; Passerieux, E.; Bommart, S.; de Mauverger, E.R.; Hugon, G.; Pincemail, J.; Pietri, S.; et al. Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free. Radic. Biol. Med. 2012, 53, 1068–1079. [Google Scholar] [CrossRef]
- Prochniewicz, E.; Lowe, D.A.; Spakowicz, D.; Higgins, L.; O’Conor, K.; Thompson, L.V.; Ferrington, D.; Thomas, D.D. Functional, structural, and chemical changes in myosin associated with hydrogen peroxide treatment of skeletal muscle fibers. Am. J. Physiol. Physiol. 2008, 294, C613–C626. [Google Scholar] [CrossRef] [Green Version]
- Burri, L.; Thoresen, G.H.; Berge, R.K. The Role of PPARalpha Activation in Liver and Muscle. PPAR Res. 2010, 2010, 542359. [Google Scholar] [CrossRef] [Green Version]
- Wilking, M.; Ndiaye, M.; Mukhtar, H.; Ahmad, N.; Haddadi, M.; Jahromi, S.R.; Nongthomba, U.; Shivanandappa, T.; Ramesh, S.; Mahasneh, A.A.; et al. Circadian Rhythm Connections to Oxidative Stress: Implications for Human Health. Antioxid. Redox Signal. 2013, 19, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, J.L.; Zhang, X.; McCarthy, J.J.; McDearmon, E.L.; Hornberger, T.A.; Russell, B.; Campbell, K.S.; Arbogast, S.; Reid, M.B.; Walker, J.R.; et al. CLOCK and BMAL1 regulate MyoD and are necessary for maintenance of skeletal muscle phenotype and function. Proc. Natl. Acad. Sci. USA 2010, 107, 19090–19095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murgia, M.; Toniolo, L.; Nagaraj, N.; Ciciliot, S.; Vindigni, V.; Schiaffino, S.; Reggiani, C.; Mann, M. Single Muscle Fiber Proteomics Reveals Fiber-Type-Specific Features of Human Muscle Aging. Cell Rep. 2017, 19, 2396–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goljanek-Whysall, K.; Soriano-Arroquia, A.; McCormick, R.; Chinda, C.; McDonagh, B. miR-181a regulates p62/SQSTM1, parkin, and protein DJ-1 promoting mitochondrial dynamics in skeletal muscle aging. Aging Cell 2020, 19, e13140. [Google Scholar] [CrossRef] [Green Version]
- Kirkendall, D.T.; Garrett, W.E. The Effects of Aging and Training on Skeletal Muscle. Am. J. Sports Med. 1998, 26, 598–602. [Google Scholar] [CrossRef]
- Evans, W.J.; Lexell, J. Human aging, muscle mass, and fiber type composition. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1995, 50, 11–16. [Google Scholar] [CrossRef]
- Lin, I.-H.; Chang, J.-L.; Hua, K.; Huang, W.-C.; Hsu, M.-T.; Chen, Y.-F. Skeletal muscle in aged mice reveals extensive transformation of muscle gene expression. BMC Genet. 2018, 19, 55. [Google Scholar] [CrossRef]
- Huang, D.-D.; Yan, X.-L.; Fan, S.-D.; Chen, X.-Y.; Yan, J.-Y.; Dong, Q.-T.; Chen, W.-Z.; Liu, N.-X.; Chen, X.-L.; Yu, Z. Nrf2 deficiency promotes the increasing trend of autophagy during aging in skeletal muscle: A potential mechanism for the development of sarcopenia. Aging 2020, 12, 5977–5991. [Google Scholar] [CrossRef]
- Nishimura, Y.; Bittel, A.J.; Stead, C.A.; Chen, Y.W.; Burniston, J.G. Facioscapulohumeral muscular dystrophy is associated with altered myoblast proteome dynamics. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lei, H.; Kazlauskas, A. A reactive oxygen species-mediated, self-perpetuating loop persistently activates platelet-derived growth factor receptor alpha. Mol. Cell Biol. 2014, 34, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Banerji, C.R.S.; Zammit, P.S. Pathomechanisms and biomarkers in facioscapulohumeral muscular dystrophy: Roles of DUX4 and PAX7. EMBO Mol. Med. 2021, 13, e13695. [Google Scholar] [CrossRef] [PubMed]
- Burrows, A.M.; Parr, L.A.; Durham, E.L.; Matthews, L.C.; Smith, T.D. Human Faces Are Slower than Chimpanzee Faces. PLoS ONE 2014, 9, e110523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elder, G.C.; Bradbury, K.; Roberts, R. Variability of fiber type distributions within human muscles. J. Appl. Physiol. 1982, 53, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Henriksson-Larsén, K.B.; Lexell, J.; Sjöström, M. Distribution of different fibre types in human skeletal muscles. I. Method for the preparation and analysis of cross-sections of whole tibialis anterior. Histochem. J. 1983, 15, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Loonen, T.G.J.; Horlings, C.G.C.; Vincenten, S.C.C.; Beurskens, C.H.G.; Knuijt, S.; Padberg, G.W.A.M.; Statland, J.M.; Voermans, N.C.; Maal, T.J.J.; van Engelen, B.G.M.; et al. Characterizing the face in facioscapulohumeral muscular dystrophy. J. Neurol. 2021, 268, 1342–1350. [Google Scholar] [CrossRef]
- Lu-Nguyen, N.; Dickson, G.; Malerba, A.; Popplewell, L. Long-Term Systemic Treatment of a Mouse Model Displaying Chronic FSHD-like Pathology with Antisense Therapeutics That Inhibit DUX4 Expression. Biomedicines 2022, 10, 1623. [Google Scholar] [CrossRef]
- Lu-Nguyen, N.; Malerba, A.; Herath, S.; Dickson, G.; Popplewell, L. Systemic antisense therapeutics inhibiting DUX4 expression ameliorates FSHD-like pathology in an FSHD mouse model. Hum. Mol. Genet. 2021, 30, 1398–1412. [Google Scholar] [CrossRef]
- Murach, K.A.; Dungan, C.M.; Kosmac, K.; Voigt, T.B.; Tourville, T.W.; Miller, M.S.; Bamman, M.M.; Peterson, C.A.; Toth, M.J. Fiber typing human skeletal muscle with fluorescent immunohistochemistry. J. Appl. Physiol. 2019, 127, 1632–1639. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber Types in Mammalian Skeletal Muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.R.Q.; Bittel, A.; Maruyama, R.; Echigoya, Y.; Nguyen, Q.; Huang, Y.; Dzierlega, K.; Zhang, A.; Chen, Y.-W.; Yokota, T. DUX4 Transcript Knockdown with Antisense 2′-O-Methoxyethyl Gapmers for the Treatment of Facioscapulohumeral Muscular Dystrophy. Mol. Ther. 2021, 29, 848–858. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Maruyama, R.; Echigoya, Y.; Nguyen, Q.; Zhang, A.; Khawaja, H.; Chandra, S.S.; Jones, T.; Jones, P.; Chen, Y.-W.; et al. Inhibition of DUX4 expression with antisense LNA gapmers as a therapy for facioscapulohumeral muscular dystrophy. Proc. Natl. Acad. Sci. USA 2020, 117, 16509–16515. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
FLExDUX4 mice display muscle weakness from 5 months to 12 months of age. Forelimb (A) and hindlimb (C) grip strength measurements of male wild-type (blue) and FLExDUX4 (white) mice (Wild-type n = 9; FLExDUX4 n = 8). Forelimb (B) and hindlimb (D) grip strength measurements of female wild-type and FLExDUX4 mice (Wild-type n = 3; FLExDUX4 n = 4). Error bars mark ± the standard error of the mean. * p < 0.05, ** p < 0.01, *** p < 0.001.
Figure 1.
FLExDUX4 mice display muscle weakness from 5 months to 12 months of age. Forelimb (A) and hindlimb (C) grip strength measurements of male wild-type (blue) and FLExDUX4 (white) mice (Wild-type n = 9; FLExDUX4 n = 8). Forelimb (B) and hindlimb (D) grip strength measurements of female wild-type and FLExDUX4 mice (Wild-type n = 3; FLExDUX4 n = 4). Error bars mark ± the standard error of the mean. * p < 0.05, ** p < 0.01, *** p < 0.001.

Figure 2.
Male FLExDUX4 mice showed body weight differences. (A) Male FLExDUX4 mice showed body weight differences as early as 4 months of age. (B) Female FLExDUX4 mice did not show body weight differences until 12 months of age. * p < 0.05. Error bars are ± the standard error of the mean.
Figure 2.
Male FLExDUX4 mice showed body weight differences. (A) Male FLExDUX4 mice showed body weight differences as early as 4 months of age. (B) Female FLExDUX4 mice did not show body weight differences until 12 months of age. * p < 0.05. Error bars are ± the standard error of the mean.

Figure 3.
Small and angular-shaped NADH-TR-positive myofibers in quadriceps muscle of 12-month-old male FLExDUX4 mice. (A) Cross-section of quadriceps stained with NADH-TR in wild-type and FLExDUX4 mice. White arrows depict examples of small angular myofibers. (B) S-curve depicting the fiber sizes of NADH-TR-positive fibers of the FLExDUX4 mice and their wild-type littermates, ranked by size. The scale bar marks a 100 µm distance. A total of 586–799 fibers were measured from the same area of the quadriceps muscles in each section (n = 3).
Figure 3.
Small and angular-shaped NADH-TR-positive myofibers in quadriceps muscle of 12-month-old male FLExDUX4 mice. (A) Cross-section of quadriceps stained with NADH-TR in wild-type and FLExDUX4 mice. White arrows depict examples of small angular myofibers. (B) S-curve depicting the fiber sizes of NADH-TR-positive fibers of the FLExDUX4 mice and their wild-type littermates, ranked by size. The scale bar marks a 100 µm distance. A total of 586–799 fibers were measured from the same area of the quadriceps muscles in each section (n = 3).

Figure 4.
FLExDUX4 mice had smaller type IIa and type IIx fibers at 12 months of age. (A) Type IIa (green) and type IIx (red) fibers were measured. Sarcolemma was stained by dystrophin staining (magenta). Scale bar measures 50 µm. (B) Type IIa (left) and type IIx (right) are significantly smaller in muscles of the FLExDUX4 mice. The average fiber size of all measured fibers from each mouse (n = 3). Error bars mark the standard error. * p < 0.05. Error bars are ± the standard error of the mean.
Figure 4.
FLExDUX4 mice had smaller type IIa and type IIx fibers at 12 months of age. (A) Type IIa (green) and type IIx (red) fibers were measured. Sarcolemma was stained by dystrophin staining (magenta). Scale bar measures 50 µm. (B) Type IIa (left) and type IIx (right) are significantly smaller in muscles of the FLExDUX4 mice. The average fiber size of all measured fibers from each mouse (n = 3). Error bars mark the standard error. * p < 0.05. Error bars are ± the standard error of the mean.

Figure 5.
The circularity is reduced in type IIx fibers in FLExDUX4 mice at 12 months of age. Error bars are ± the standard error of the mean. * p < 0.05.
Figure 5.
The circularity is reduced in type IIx fibers in FLExDUX4 mice at 12 months of age. Error bars are ± the standard error of the mean. * p < 0.05.

Figure 6.
TDP-43-positive aggregates are in type IIb myofibers in quadriceps of 12-month-old male FLExDUX4 mice. Serial sections of 12-month-old FLExDUX4 male quadriceps were taken. Basophilic aggregates in sections stained with hematoxylin and eosin (H&E) were observed in the quadriceps of the FLExDUX4 mice. The TDP-43-positive fibers are also stained positive for the myosin heavy chain type IIb. Black arrows denote examples of aggregates present in the same myofiber. Scale bar measures 50 µm.
Figure 6.
TDP-43-positive aggregates are in type IIb myofibers in quadriceps of 12-month-old male FLExDUX4 mice. Serial sections of 12-month-old FLExDUX4 male quadriceps were taken. Basophilic aggregates in sections stained with hematoxylin and eosin (H&E) were observed in the quadriceps of the FLExDUX4 mice. The TDP-43-positive fibers are also stained positive for the myosin heavy chain type IIb. Black arrows denote examples of aggregates present in the same myofiber. Scale bar measures 50 µm.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Murphy, K.; Zhang, A.; Bittel, A.J.; Chen, Y.-W. Molecular and Phenotypic Changes in FLExDUX4 Mice. J. Pers. Med. 2023, 13, 1040. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm13071040
AMA Style
Murphy K, Zhang A, Bittel AJ, Chen Y-W. Molecular and Phenotypic Changes in FLExDUX4 Mice. Journal of Personalized Medicine. 2023; 13(7):1040. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm13071040
Chicago/Turabian StyleMurphy, Kelly, Aiping Zhang, Adam J. Bittel, and Yi-Wen Chen. 2023. "Molecular and Phenotypic Changes in FLExDUX4 Mice" Journal of Personalized Medicine 13, no. 7: 1040. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm13071040
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.