
β-lactam Resistance in Pseudomonas aeruginosa: Current Status, Future Prospects



Department of Biochemistry, University of Otago, P.O. Box 56, Dunedin 9054, New Zealand
*
Author to whom correspondence should be addressed.
Pathogens 2021, 10(12), 1638; https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10121638
Submission received: 2 November 2021
/
Revised: 6 December 2021
/
Accepted: 16 December 2021
/
Published: 18 December 2021
(This article belongs to the Section Bacterial Pathogens)
Abstract
:Pseudomonas aeruginosa is a major opportunistic pathogen, causing a wide range of acute and chronic infections. β-lactam antibiotics including penicillins, carbapenems, monobactams, and cephalosporins play a key role in the treatment of P. aeruginosa infections. However, a significant number of isolates of these bacteria are resistant to β-lactams, complicating treatment of infections and leading to worse outcomes for patients. In this review, we summarize studies demonstrating the health and economic impacts associated with β-lactam-resistant P. aeruginosa. We then describe how β-lactams bind to and inhibit P. aeruginosa penicillin-binding proteins that are required for synthesis and remodelling of peptidoglycan. Resistance to β-lactams is multifactorial and can involve changes to a key target protein, penicillin-binding protein 3, that is essential for cell division; reduced uptake or increased efflux of β-lactams; degradation of β-lactam antibiotics by increased expression or altered substrate specificity of an AmpC β-lactamase, or by the acquisition of β-lactamases through horizontal gene transfer; and changes to biofilm formation and metabolism. The current understanding of these mechanisms is discussed. Lastly, important knowledge gaps are identified, and possible strategies for enhancing the effectiveness of β-lactam antibiotics in treating P. aeruginosa infections are considered.
1. Introduction
Pseudomonas aeruginosa is a Gram-negative bacillus that is found in many environments including water and soil, and in association with animals [1]. It is also a major opportunistic pathogen, being one of the most frequent causes of acute infections in hospitalised patients and in patients with predisposing conditions such as severe burns, catheterisation, or neutropenia, causing septicaemia, urinary tract infections, and bacteraemia [2,3,4,5]. P. aeruginosa is a primary cause of hospital- and ventilator-acquired pneumonia [6,7,8]. It also causes severe eye infections and chronic infections in patients with cystic fibrosis or chronic obstructive pulmonary disease [9,10,11]. Infections with P. aeruginosa are often associated with higher mortality and morbidity than those with other pathogens [12,13].
Examples of P. aeruginosa infections that have been intensively studied include chronic infections in people with CF and acute infections in burns patients. Chronic infections in the lungs of people with CF can last for many years and are the leading cause of morbidity and reduced life expectancy in these individuals [14]. The eradication of P. aeruginosa infections in individuals with CF is associated with a better long-term outcome [15,16]. P. aeruginosa infections in CF are also strongly associated with mortality in childhood [17]. Acute P. aeruginosa infections are the leading cause of death in burn victims [18,19]. Treatment of burn wounds becomes increasingly difficult when P. aeruginosa infection occurs [20]. In a study of over 5000 patients over twenty years, 55% of burn victim mortality was due to multidrug-resistant P. aeruginosa [18]. Other acute P. aeruginosa infections, in particular bloodstream infections, also have very high mortality rates [21,22,23,24,25].
P. aeruginosa infections can be acquired from most aquatic and damp environments, with these bacteria being commonly isolated from showers, sinks, drains, and even liquid soap [26,27]. For example, in one between 9.7% and 68.1% of tap water samples from intensive care units were contaminated with P. aeruginosa [27]. Wash hand basins and other moist areas are a source of hospital outbreaks of P. aeruginosa infections [28,29]. Outbreaks have also been linked to a P. aeruginosa-contaminated hand soap dispenser [30] and bottled water supplied to an intensive care unit [31]. Analysis of P. aeruginosa isolated from newly infected CF patients found that for 13 of 25 patients, the P. aeruginosa genotype matched that of isolates obtained from a sink in their house, while one of the isolates matched to the patient’s nebuliser used for antibiotic inhalation, identifying these environments as possible sources of infection [32].
Infections by multidrug-resistant (MDR) isolates (bacteria resistant to one antibiotic from three or more antibiotic classes [33]; Table 1) are particularly problematic. Acute infection with antibiotic-resistant P. aeruginosa results in thousands of deaths worldwide each year [5,34,35]. In a meta-analysis of 23 studies of over 10,000 P. aeruginosa infections, mortality was 34% in patients with antibiotic-resistant P. aeruginosa compared with 22% in those infected with antibiotic-susceptible P. aeruginosa [5]. Similar findings resulted from a separate meta-analysis, with hospital patients who had non-MDR P. aeruginosa infections having a mortality rate of 24.8% (of 2388 cases) and patients with MDR infections having a mortality rate of 44.6% (of 813 cases) [36]. A factor contributing towards mortality of P. aeruginosa infections is the time between the onset of infection and treatment. Treatment within 24 h had a mortality rate of 27.7% compared to a mortality rate of 43.4% if effective treatment was delayed for 24 h [23]. Initial treatment with antibiotics to which the bacteria were resistant was associated with a mortality rate of 40.6%, emphasising the importance of early treatment with antibiotics that are effective against the infecting P. aeruginosa [23]. Isolation of infecting bacteria followed by antibiotic susceptibility testing is used to determine which antibiotics are likely to be efficacious. However, the timeframe involved in this process (about two days) can delay effective treatment. For some bacterial species, genomic sequencing and analysis provides a more rapid method to identify effective antibiotics [37] but this approach is not yet available for P. aeruginosa.
The frequency of P. aeruginosa infections, the clinical challenges and poor outcomes associated with these infections, and in particular the proportion of P. aeruginosa isolates that are resistant to antibiotics have resulted in these bacteria being classified as one of a group of six pathogens (the ESKAPE pathogens) that are the most problematic to treat [38,39]. P. aeruginosa that have become resistant to the carbapenem class of antibiotics are classified by the World Health Organisation as one of the three “Priority 1: Critical” groups of bacteria for which new treatment strategies are most critically needed [40].
2. Antibiotics Used against P. aeruginosa
Oral and intravenous delivery of antibiotics can be used to treat a wide range of P. aeruginosa infections including septicemia, lung infections, and bone infections, while inhalation of specific antibiotics is also used for the treatment of lung infections in individuals with CF or other forms of lung disease [11,41,42,43]. There are a variety of antibiotic classes used to treat P. aeruginosa (Table 1), each having different targets within the bacterial cell.
β-lactams, the topic of this review, inhibit the synthesis of peptidoglycan, a key component of the cell envelope [54]. This inhibits the ability of bacteria to replicate and divide while also reducing the integrity of the cell wall leading to cellular lysis [54]. Aminoglycosides and fluoroquinolones inhibit protein and DNA synthesis, respectively, and polymixins disrupt the bacterial cell membrane. Antibiotics from different classes can be used in combination to increase the likelihood of effectiveness when resistance phenotype is not known and to suppress the emergence of resistance [3].
3. The Problem of P. aeruginosa Antibiotic Resistance
The low outer membrane permeability of P. aeruginosa, coupled to the presence of efflux pumps and other genetic features and its ability to form biofilms, provides it with intrinsic resistance to moderate concentrations of antibiotics including β-lactams [55,56]. Biofilms involve the attachment of cells to a surface by adhesins and encasing the bacterial cells in an extracellular matrix [57]. Biofilms contribute to antibiotic resistance by being difficult for antibiotics to penetrate, because the biofilm lifestyle alters the metabolism of the bacteria, and because biofilms can contain dormant antibiotic-insensitive persister cells [56,57,58,59,60]. Nonetheless, P. aeruginosa isolated from the general environment are generally susceptible to antibiotics including β-lactams [61]. However, isolates from clinical settings are frequently resistant to antibiotics, and a high proportion of P. aeruginosa infections are by antibiotic-resistant bacteria [62,63]. For example, in one study of hospitalised patients, 37% of 826 P. aeruginosa isolates from a CF unit, and 49% of 224 isolates from an intensive care unit were antibiotic-resistant [64]. These isolates were resistant to a wide variety of antibiotics, including gentamicin (58%), carbapenems (55%), and colistin (6%) [65]. In a study of over 1000 P. aeruginosa infections in neonatal and paediatric patients, over 10% of isolates were resistant to carbapenems (imipenem, meropenem, doripenem), and approximately 15% to cephalosporins (cefepime and ceftazidime) [66]. Large case studies and meta-analyses have found that people infected with carbapenem-resistant P. aeruginosa often [67,68,69] though not always [70], have a significantly higher risk of death than people infected with carbapenem susceptible isolates.
The US Centers for Disease Control and Prevention (CDC) has estimated that there were 32,600 hospitalizations and 2700 deaths from multidrug-resistant P. aeruginosa infections in the USA in 2019 [34]. In Europe, there were on average approximately 72,000 infections with antibiotic-resistant P. aeruginosa and 4155 attributable deaths per million people in the period 2007–2015 [71], with the numbers of such infections increasing by about three-fold over the time period. The median cost per hospitalization and treatment of patients with antibiotic-resistant P. aeruginosa infections was US$99,672, while for patients with susceptible P. aeruginosa infections the median cost was US$69,502 [5]. Contributing factors of the increased financial burden of antibiotic-resistant P. aeruginosa infections are the increased price of drug regimens and increased days of mechanical ventilation (15 versus 11 days) and increased hospitalization length [5,72]. The total healthcare costs associated with infections by antibiotic-resistant P. aeruginosa in the USA were estimated at US$767 million in 2017 [34].
P. aeruginosa is capable of developing resistance to all classes of antibiotics through chromosomal mutations [73]. This is well studied in chronic CF infections, where patients are commonly infected with an antibiotic-sensitive P. aeruginosa isolate that develops resistance over the course of prolonged infection and treatment [74,75]. Likelihood of infecting P. aeruginosa being β-lactam resistant increases with age in CF patients, supporting a model whereby antibiotic resistance develops over the course of treatment [76]. Eradication of P. aeruginosa infections becomes increasingly difficult as antibiotic resistance occurs, resulting in worsening patient conditions [15,16].
Severe acute burn wounds can be directly infected with antibiotic-resistant P. aeruginosa [77]. Outbreaks of MDR and extensive drug-resistant (XDR; resistant to all but one or two classes of antibiotics) strains are a large issue in burn care units as treatment becomes increasingly difficult [78,79]. A 2016 outbreak of an XDR P. aeruginosa infected 10 patients within a burn unit resulting in the death of two patients from septic shock [78]. Although the development of antibiotic resistance is not commonly observed in burn wound infections with sensitive P. aeruginosa strains (because of shorter infection times), a case study has shown that a sensitive P. aeruginosa strain became resistant within 14 days from the beginning of treatment [13]. Analysis of wastewater from a burns hospital found that out of 100 P. aeruginosa isolates 66% were MDR [64], illustrating how antibiotic-resistant isolates can be spread into the general environment.
4. Penicillin-Binding Proteins and Peptidoglycan Synthesis
Penicillin-binding proteins (PBPs) are, as their name suggests, the key targets of β-lactam antibiotics. They are involved in the synthesis of the layer of peptidoglycan that forms part of the cell envelope and provides structural integrity to P. aeruginosa cells [80]. The peptidoglycan layer consists of linear β(1–4)-Linked disaccharides of N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM) strands containing on average about 24 NAG-NAM units that are cross-linked together by amide bonds between peptide side chains [81,82]. This structure resists internal cellular pressures while also giving cells their inherent shape [82]. The peptide side chains are synthesised as pentapeptide precursors, with cross-linking between diaminopimelic acid and D-alanine residues bridging different strands of NAG-NAM polymers [83] (Figure 1). Cross-links are formed in a process involving the removal of a terminal D-alanyl residue. In mature peptidoglycan, approximately 50% of the peptide units are cross-linked. In uncross-linked peptides, approximately 40% of all D-alanine have been removed by LD-carboxypeptidases that remove the D-alanine at position four of the peptide sidechain, preventing cross-linking [83].
PBPs are involved in the later stages of peptidoglycan synthesis and remodelling of peptidoglycan during cell growth and division [84,85]. P. aeruginosa has eight PBPs, numbered 1a, 1b, 2, 3, 3a, 4, 5, and 7 in order of decreasing molecular mass. The PBPs fall into two categories, high molecular mass (HMM) PBPs (1a, 1b, 2, 3, and 3a) and lower molecular mass (LMM) PBPs (4, 5, and 7) [86]. PBPs play a role in cellular division and controlling cellular morphology through the incorporation of NAG-NAM units into growing peptidoglycan chains via a glycosyltransferase activity, cross-linking of different NAG-NAM units through their peptide side chains, and modifying peptidoglycan peptide chains (Figure 1) [87,88]. There is functional redundancy of PBPs, with only PBP3 being essential for growth [79]. The transpeptidase domains of all PBPs contain three amino acid sequence motifs Ser-XX-Lys (catalytic serine), Ser-X-Gln, and Lys-Ser-Gly-Thr [89], which all play a role in DD-transpeptidase activity (cross-linking of NAG-NAM chains), DD-carboxypeptidase activity (cleaving terminal D-Ala from peptide chains), and DD-endopeptidase activity (cleaving the peptide cross-link made by DD-transpeptidase activity) [87,90]. HMM PBPs 1a and 1b have both glycosyltransferase and transpeptidase domains, while PBPs 2, 3, and 3a only have transpeptidase domains [86,91,92]. Glycosyltransferase domains catalyse the extension of NAG-NAM polymers by incorporation of NAG-NAM units and DD-transpeptidase domains catalyse peptide cross-linking. The DD-transpeptidase domains bind to the pentapeptide precursor and catalyse the formation of a cross-link between the D-alanine residue at the fourth position of a pentapeptide side chain and a diaminopimelate residue of a tri-, tetra-, or penta-peptide of an adjacent peptide sidechain, on a different NAG-NAM strand (Figure 1) [84]. During the formation of this cross-link, the terminal D-Ala of the pentapeptide is removed [84].
LMM PBPs in P. aeruginosa have DD-carboxypeptidase activity [93,94]. Deletions of genes encoding LMM PBPs increased the presence of pentapeptides in peptidoglycan which indicates the lack of DD-carboxypeptidase activity [93]. PBP5 is the main DD-carboxypeptidase, followed by PBP4 and, lastly, PBP7 [93]. The LMM PBP PBP4 also regulates ampC (β-lactamase) expression [95]. Although deletion of all three LMM PBPs (4, 5, and 7) cause a significant increase in pentapeptide levels, there was no significant effect on cell morphology [93].
As well as being required for peptidoglycan synthesis, PBPs play a role in peptidoglycan recycling. Recycling occurs as part of the process of peptidoglycan turnover and remodelling that is required for cell growth and division. Recycling of peptidoglycan components released during hydrolysis by lytic transglycosylases and carboxypeptidases reduces the metabolic burden of peptidoglycan synthesis [96], as described below. LMM PBPs with endopeptidase activity form an important part of this process [96]. PBP4 has both a DD-endopeptidase and DD-carboxypeptidase activity [97,98].
5. β-lactam Antibiotics: Inhibitors of PBPs
Antibiotics in the β-lactam class have bactericidal activity against a broad spectrum of bacteria [99]. The bactericidal properties of β-lactams are due to their ability to inhibit the transpeptidase and DD-carboxypeptidase activities of PBPs [54]. β-lactams are structural mimics of the terminal D-alanine-D-alanyl residues of peptidoglycan pentapeptide precursors which PBPs bind to perform their catalytic functions (Figure 2) [100,101]. Once the β-lactam has entered the active site of a PBP, it covalently binds to the catalytic serine in the Ser-XX-Lys motif permanently, inactivating the PBP [102,103]. The inhibition of PBPs can result in filamentation and can also reduce the structural integrity of the cell wall resulting in lysis of the bacteria [104,105,106].
β-lactam antibiotics all have a core structure of a four-membered lactam ring which is closed by an amide bond (Figure 2) [108]. β-lactams are assigned into subclasses on the basis of the nature of the chemical groups attached to this core structure, with subclasses containing multiple members [100]. The differences in side chains affect many characteristics of the β-lactams, including the affinity towards different penicillin-binding proteins PBPs, ability to cross cell envelopes, their chemical stability, and resistance to degradation by β-lactamases [100].
The affinities of anti-Pseudomonal β-lactams for different PBPs are shown in Table 2. Several β-lactams have a high affinity for a number of different PBPs. Notably, however, all of those that have a low minimum inhibitory concentration (MIC) and are used in clinical practice have a high affinity for PBP3, the only essential PBP in P. aeruginosa. Conversely, cephalexin that is not clinically effective has a low affinity towards PBP3 and is not effective at killing P. aeruginosa (high MIC). Faropenem has a high affinity for PBP3 but nonetheless is inefficient at killing P. aeruginosa demonstrating that affinity for PBP3 is not by itself sufficient to ensure effectiveness. This high MIC for faropenem is attributed to a combination of intrinsic resistance mechanisms [109].
As well as causing cell lysis and filamentation, in E. coli, inhibition of PBPs can lead to unnecessary recycling of the cell wall, depleting cellular resources and contributing to cellular death [104]. This process depends on the product of the slt gene, a transglycosylase [104]. The slt gene product of P. aeruginosa appears to have an equivalent function to its E. coli homologue [113,114,115] and may contribute to cellular resource depletion in P. aeruginosa in the presence of β-lactams.
The mode of antibiotic delivery and the use of combinations of different antibiotics have been found to be important in the treatment of P. aeruginosa infections. Continuous infusion of meropenem, aztreonam, and ceftazidime can eradicate MDR P. aeruginosa infections [116,117]. This finding is supported by a meta-study which found that continuous infusion of meropenem had a higher success rate than intermittent dosages for MDR P. aeruginosa [118]. Recent studies have indicated that dual antibiotic regimens improve the survival of patients with MDR P. aeruginosa. The combination of a carbapenem antibiotic with colistin increased the eradication of P. aeruginosa infections [42]. Dual β-lactam therapy also has been shown to have enhanced efficacy in killing P. aeruginosa [119], with an example being ceftazidime-avibactam paired with meropenem [120].
6. Mechanisms of β-lactam Resistance
Antibiotic-resistant isolates of P. aeruginosa arise through genetic changes in antibiotic-susceptible bacteria (acquired resistance). Acquired resistance can occur through mutations affecting a wide range of cellular functions. The primary mechanisms for the development of β-lactam resistance through mutation include alterations to the PBP3 target protein, decreased antibiotic uptake, increased export, and degradation of antibiotic molecules (Figure 3) [121]. In addition, horizontal gene transfer can lead to the acquisition of antibiotic-degrading enzymes (β-lactamases) from other bacteria [122]. Metabolic changes and increased biofilm production may also play a role in resistance [123]. Resistance mechanisms are described in detail below.
β-lactam resistance can arise from genetic changes that reduce antibiotic uptake through porins, increase degradation of β-lactams, alter the PBP3 target protein, or increase antibiotic efflux. Resistance often involves a combination of these mechanisms. CM, cytoplasmic membrane; P, periplasm; OM, outer membrane.
The availability of methods for relatively inexpensive whole-genome sequencing has greatly accelerated the discovery of genetic changes that contribute to resistance. A number of studies have used experimental evolution to develop β-lactam-resistant P. aeruginosa from sensitive strains, followed by whole-genome sequencing to identify the mutations causing resistance [124,125,126,127]. Whole-genome sequencing of isolates from chronically-infected patients has shown that genes acquiring mutations during resistance development in vitro also acquire mutations that are likely to contribute to resistance during infection [128]. Whole-genome sequencing of clinical isolates has also enhanced understanding of the contributions of horizontally acquired genes to β-lactam resistance. Collectively, these studies allow a good understanding of the mechanisms of β-lactam resistance in P. aeruginosa. Different resistance mechanisms are discussed in detail below.
7. Target-Site Modification: Changes to PBP3
Mutational changes to PBPs that are associated with resistance act by reducing the affinities of PBPs for β-lactams [129,130,131,132]. PBP3 is encoded by the ftsI gene. The essential nature of PBP3 in P. aeruginosa was shown through the conditional expression of the ftsI gene using an inducible promoter [86]. Inhibition of ftsI expression led to long filaments of cells, indicating a defect in cell division. As the only essential PBP in P. aeruginosa, PBP3 is the primary target for β-lactams [86]. Experimentally-evolved mutants of P. aeruginosa resistant to β-lactams frequently have mutations in ftsI [124,126,132], and PBP3 sequence variants are also common in isolates from patients that have reduced β-lactam susceptibility [133,134,135].
The structure of PBP3 bound to different β-lactams has been determined [102]. PBP3 is comprised of a short cytoplasmic N-terminal domain, a transmembrane helix, a domain predicted to play a role in protein-protein interactions, and a transpeptidase domain [136,137] (Figure 4). The active site of PBP3 contains the protein sequence motifs Ser-XX-Lys (residues 294–297), Ser-x-Asn (residues 349–351), and Lys-Ser-Gly-Thr (residues 484–487) that are present in all PBPs [136,137]. β-lactams bind to the catalytic serine (S294) of the Ser-XX-Lys motif [102]. Ceftazidime, aztreonam, meropenem, and imipenem all bind at the active site but cause different conformational changes [102]. β-lactams that are effective anti-Pseudomonas agents, including aztreonam, meropenem, imipenem, doripenem, ceftazidime, and ceftolozane, all have a high affinity for PBP3 [42,43] (Table 2) emphasising that PBP3 is a key β-lactam target.
Variants of PBP3 likely to contribute to β-lactam resistance in clinical isolates were identified by comparing the PBP3 sequence of antibiotic-resistant clinical isolates with those of antibiotic naive strains (environmental or pre-treatment) [126,128,135,138,139,140,141,142,143,144,145,146,147]. The most frequent PBP3 variants that are likely to affect β-lactam activity are amino acid substitutions that, as might be expected, are centred around the active side and, in particular, are near the catalytic serine of the Ser-XX-Lys motif (Figure 4, Supplementary Table S1). Variants in experimentally-evolved resistant bacteria also cluster around the active site and are often at the same residues as those in clinical isolates [124,125,126,127,148]. Variants around the active sites of PBPs also contribute to β-lactam resistance in other species [131,149,150], and some cases have been shown to reduce the affinity of β-lactams for the PBP [150,151]. It seems likely that sequence variants around the active sites in PBPs cause slight confirmational changes that reduce the ability of β-lactams to bind and the active site and react with the catalytic serine [129,132]. Consistent with this prediction, the PBP3 variant F533L reduces the affinity of PBP3 for meropenem [132]. It is currently not known whether, or how, PBP3 variants away from the catalytic domain, such as the frequently observed G63C/D variants (Figure 4, Supplementary Table S2), contribute to β-lactam resistance.
As different β-lactams bind slightly differently to the PBP3 active site, mutations around the active site are likely to have different effects with different β-lactams. There is some evidence to support this prediction. Analysis of isolates from a CF patent with PBP3 variants V465G or A244T showed that both variants were associated with aztreonam and cefsulodin resistance, whereas only the V465G variant was associated with ceftazidime and piperacillin resistance [140]. In a separate study, the PBP3 variant R504C was associated with ceftazidime and cefsulodin resistance, whereas the variant P527S was associated with resistance to aztreonam, cefepime, ceftazidime, and cefsulodin [134]. Biochemical assays are limited, but in one study the sequence variant A539T reduced affinity for meropenem and ceftazidime, and sequence variant F533L selectively reduced affinity for meropenem but not ceftazidime [132]. Collectively these findings support the notion that different PBP3 sequence variants can have different impacts on different antibiotics, but more work is needed to fully understand the relationships between PBP3 sequence variants and β-lactam resistance.
8. Reduced Uptake of β-lactams: The Role of Porins
Variants in the oprD gene are commonly found in isolates of P. aeruginosa that are resistant to carbapenems [76,152,153,154,155,156,157,158]. The oprD gene encodes the OprD porin that plays a role in the uptake of small basic amino acids and small peptides. OprD also mediates uptake of carbapenems other than faropenem [153,159]. Basic amino acids act as competitive inhibitors towards the uptake of carbapenems, and so the growth environment influences carbapenem susceptibility [160]. Disruption of the oprD gene via point mutations, frameshifts, premature stop codons, or large deletions all contribute to carbapenem resistance [153,155,156,157]. These mutations reduce or abolish the uptake of carbapenems through OprD [152,157,158]. In addition to contributing to carbapenem resistance, loss of oprD increases the ability of P. aeruginosa to colonize mucosal environments, and increases resistance towards acidic environments in a mouse model of infection [161].
Mutations to OprD do not affect susceptibility to other classes of β-lactams, indicating that this porin is not involved in their uptake [159,162,163]. How antibiotics in these classes access the periplasm of P. aeruginosa is not fully understood [162]. Most porins (other than OprD) do not mediate antibiotic uptake [162]. However, loss of porin OpdP confers a reduced susceptibility to meropenem and, in an OprD-lacking strain, to imipenem and doripenem [163]. Heterologous expression assays confirmed that OpdD can contribute to susceptibility to carbapenems presumably by providing a channel into the periplasm [163]. Loss of OprF may cause a slight increase in piperacillin resistance [162], and loss of OpdH reduced susceptibility to ceftazidime but not to other cephalosporins [164], suggesting that these porins may also play a role in the entry of β-lactams into P. aeruginosa. Diffusion through the lipid layer of the outer membrane likely also plays a role in the uptake of β-lactams [165].
9. P. aeruginosa Efflux Systems
Most isolates of P. aeruginosa contain 12 Resistance-Nodulation-Division efflux system pumps, which play a role in virulence, stress response, and both intrinsic and acquired antibiotic resistance [109,166,167,168]. Each of these efflux pumps is comprised of a protein that spans the cytoplasmic membrane, an outer membrane protein, and a periplasmic component that links the two [169]. Efflux pumps form channels from the cytoplasm to the outside of the cell that export a wide range of substrates, in a process driven by proton motive force [169]. As well as compounds in the cytoplasm, chemicals present in the periplasm can be exported. Different pumps export different compounds, although how substrate selection occurs is not well understood. Overexpression studies have shown that efflux pumps MexAB-OprM, MexXY-OprM, and MexCD-OprJ are the most important in the context of β-lactam resistance [170,171,172,173,174], although each pump can export a wide range of antibiotics (Table 3). These efflux systems are clinically relevant as they are overexpressed in many antibiotic-resistant clinical isolates [76,138,152,156,166,175,176,177,178,179]. For example, in one multicentre study, 39 of 80 CF isolates overproduced at least one of these efflux systems, with 65 having increased expression of MexXY-OprM, 36 of MexAB-OprM, and two of MexCD-OprJ [76].
The MexAB-OprM efflux pump plays a role in the intrinsic resistance of P. aeruginosa to a wide range of β-lactams, with deletion of the mexABoprM operon increasing susceptibility to faropenem, sulopenem, ritipenem, temocillin, and ticarcillin [109,167]. Increased mexABoprM expression (acquired resistance) is often observed in carbapenem-resistant clinical isolates–for example, 16 of 23 isolates and 28 of 32 isolates in two separate studies [152,175]–and contributes to resistance to a wide range of β-lactams including meropenem, ceftazidime, aztreonam, ticarcillin and carbenicillin [185,186].
Overexpression of the mexABoprM genes occurs because of mutations affecting the repressor proteins MexR, NalC, or NalD [179]. Deletion of the mexR gene leads to the highest levels of mexABoprM expression, followed by deletion of nalD and nalC [186,187]. MexR, the primary regulator of the mexABoprM operon, plays a role in sensing oxidative stress. MexR binds to the promoter of the mexAoprM operon repressing expression, but under conditions of oxidative stress, MexR disassociates increasing expression of the efflux pump [188,189]. Mutations in the DNA-binding domain of MexR inhibit its ability to bind to the mexABoprM promoter, increasing expression [190]. Overexpression of mexR significantly increases susceptibility to aztreonam consistent with its role in the repression of mexABoprM and the involvement of this efflux pump in aztreonam resistance [190]. NalD is a second repressor of the mexABoprM operon, acting similarly to mexR, and mutations in nalD cause increased expression of the mexABoprM genes and are associated with β-lactam resistance [186,187]. Mutations in nalC lead to overexpression of gene PA3719, which in turn leads to mexABoprM overexpression [191].
Increased mexXYoprM expression also contributes to resistance to multiple β-lactams (Table 3). Expression of the mexXY operon is regulated by the repressor MexZ that binds to the promoter of the mexXY operon [192]. Disruption of protein synthesis causes increased synthesis of the AmrZ protein that interacts with MexZ, dislodging it from the promoter of the mexXY operon and inducing expression [171]. MexXY uses OprM from the mexABoprM as the outer membrane component [193]. Mutations in mexZ are the most common cause of mexXY overexpression in clinical isolates [194,195].
MexCDOprJ is capable of exporting a wide variety of antibiotics (Table 3). The mexCDoprJ operon is regulated by the repressor NfxB [73,196], and expression is induced by membrane-damaging agents [197]. Mutations in nfxB can cause mexCDoprJ overexpression, although this is infrequently observed in clinical isolates [76,198,199]. Mutations in mexD can lead to increased resistance to cephalosporin-β-lactamase inhibitor combinations (ceftolozane-tazobactam and ceftazidime-avibactam) through altered substrate specificity of MexCDOprJ [200].
Increased expression of the efflux pump mexEF-oprN is associated with imipenem resistance [201], although MexEF-OprN does not export β-lactams [202]. Overexpression of the mexEF-oprN genes occurs because of mutations affecting the repressor protein NfxC and MexS that influence the expression of the mexABoprM operon and the oprD gene [76,183,184,203]. Increased expression of mexEF-OprN is, therefore, likely to be a consequence of mutations that affect antibiotic susceptibility, rather than a direct contributor to β-lactam resistance.
It should be noted that overexpression of efflux pumps can also increase antibiotic susceptibility, with overexpression of both MexEF-OprN and MexCD-OprJ making P. aeruginosa more susceptible to imipenem, ticarcillin, aztreonam, and aminoglycosides [172,204]. Therefore, the benefits of overexpressing efflux pumps are likely dependent on the environment, with different antibiotics selecting for or against the expression of different efflux pumps.
10. Degradation of β-lactams by β-lactamases
β-lactamases are enzymes that cleave open the β-lactam ring of β-lactam antibiotics through hydrolysis, inactivating the antibiotic [205]. They are categorised into four classes (A to D) based on their amino-acid sequence similarity [205,206]. Within each class, enzymes are further categorised into families based on the protein sequence. Families are named on the basis of substrate β-lactam, or geographic location where they were first identified. Enzymes in classes A, C, and D have a catalytic serine for substrate hydrolysis. Class B enzymes are metallo-β-lactamases that catalyse the hydrolysis of β-lactam rings in reaction mechanisms involving a metal ion, most commonly a zinc ion [206,207,208]. Due to their different mechanism of action metallo-β-lactamases have likely had different evolutionary origins from the other classes of β-lactamases [207]. Metallo-β-lactamases have a broad activity spectrum degrading all β-lactams except monobactams [209]. The presence of Metallo-β-lactamases is significantly associated with carbapenem resistance [210]. β-lactamases that are capable of degrading carbapenems (carbapenemases) are especially problematic because of the critical role of carbapenems in managing P. aeruginosa infections [211]. Carbapenemase-producing isolates are often a cause of severe infections and are becoming more frequently detected in hospitals [67,68,69,212,213,214,215,216]. Almost all carbapenemases are class A, B, or D β-lactamases as class C β-lactamases have only low activity against carbapenems. For a detailed review of carbapenemases, see [217].
The effectiveness of β-lactamases is reduced by enzyme inhibitors and these are often co-administered with β-lactam antibiotics, increasing antibiotic efficacy. β-lactamase inhibitors used in treating P. aeruginosa infections include clavulanate, tazobactam, avibactam, and vaborbactam for Class A β-lactamases, relebactam for Class A and Class C enzymes, and avibactam for class C and a limited number of class D β-lactamases [217,218,219,220,221]. Class B enzymes can be inhibited by metal ion chelators, but currently, these are not in clinical use as β-lactamase inhibitors [217,222].
11. β-lactamases Encoded by the Core Genome
Like many other Gram-negative bacteria, P. aeruginosa has a chromosomally encoded Class C β-lactamase, AmpC [73,205]. Class C β-lactamases have high activity against penicillins and cephalosporins [206,223]. AmpC contributes to intrinsic resistance to many penicillins, including faropenem, ritipenem, and sulopenem, as shown by increased susceptibility to these antibiotics when the ampC gene is deleted [109]. Antibiotic-resistant P. aeruginosa clinical isolates often have high levels of ampC expression, reducing susceptibility to ceftazidime, cefepime, aztreonam, and piperacillin, although having little or no effect on susceptibility to carbapenems [224,225]. Expression of the ampC gene is regulated through a complex signalling pathway. Increased expression of ampC can occur through activation of this pathway by the presence of β-lactams (Figure 5), or by mutations that alter the pathway.
The expression of ampC is controlled by the transcriptional regulator AmpR (Figure 5A) [226]. During peptidoglycan synthesis and recycling, the peptidoglycan precursor UDP-NAM pentapeptide is formed and binds to AmpR. The D-Ala-D-Ala of the pentapeptide plays a primary role in interacting with AmpR [227]. The resulting complex binds to the divergent ampC-ampR promoter and inhibits transcription of ampC [95,228]. In the presence of β-lactams, there are increased amounts of NAG-NAM pentapeptide units formed following hydrolysis of mature peptidoglycan (Figure 5B). These are imported into the cytoplasm through the AmpG permease and they, as well as NAM penta- and tri-peptides generated by removal of the NAG moiety, bind to AmpR. The binding of these molecules causes AmpR to activate the expression of ampC [95,228].
Peptidoglycan fragments that have been imported into the cytoplasm are processed for recycling by NagZ, AmpD, and other enzymes [95,226,228,229,230] (Figure 5). Inhibition of PBPs increases the intracellular concentrations of NAG-NAM pentapeptide, NAM pentapeptide, and NAM tripeptide [95]. Excess pentapeptides arising from the action of β-lactams are thought to saturate AmpD, raising the intracellular concentration of the AmpR-activating compounds [95,228]. The cytoplasmic concentration of NAG-NAM pentapeptide is also influenced by LMM PBPs, in particular the PBP4 protein [95]. Inhibition of PBP4 by mutation or β-lactams is a major inducer of ampC expression [93]. Inhibition of PBP4 does not significantly increase NAG-NAM pentapeptide levels in the periplasm [83]. Instead, inhibition of PBP4 is thought to increase peptidoglycan recycling [95,97] resulting in an increase in the intracellular concentration of NAG-NAM pentapeptide.
Mutations in ampR contribute to β-lactam resistance [231,232]. These mutations inhibit the UDP-NAM pentapeptide from binding to AmpR leading to constitutive expression of ampC at high levels [97,233,234]. Mutations in dacB that encodes PBP4 are also found commonly in β-lactam resistant clinical isolates and lead to increased ampC expression [178,235]. PBP4 inhibition also activates the CreBC signalling [236], which is a global regulator of bacterial fitness, biofilm development, and ampC expression. It is not yet known how PBP4 inhibition activates CreBC signalling or how CreBC signalling regulates ampC expression [236].
Mutations in ampD are also often found in clinical isolates, increasing β-lactam resistance [97,230,234,237,238]. AmpD plays a role in recycling peptidoglycan by cleaving NAG-NAM from the peptide side chains that are imported from the periplasm into the cytoplasm [229,230]. Disabling or impairing the function of AmpD through mutation results in increased quantities of partially recycled peptidoglycan building up especially the NAG-NAM tripeptide [95].
As well as reduced β-lactam susceptibility through increased expression of the ampC gene, the catalytic activity of AmpC towards many penicillins and most cephalosporins can be increased by mutations altering the enzyme (Figure 6) [238,239,240]. Additionally, ampC mutations can reduce the affinity of inhibitors such as avibactam and tazobactam for AmpC [239,240,241,242]. For a full review on ampC point mutations see [240].
Two other β-lactamases are encoded in the core genome of P. aeruginosa. One is a class A β-lactamase PIB-1 (PA5542) capable of degrading imipenem [243], and the second is a class D OXA-50 like β-lactamase (PA5514/poxB) capable of degrading carbapenems [244,245]. Inactivation of PIB-1 increases the susceptibility of P. aeruginosa to carbapenems, and overexpression of PoxB reduces susceptibility to meropenem [243,244,245]. The contributions of these enzymes to antibiotic resistance in clinical settings, if any, has not yet been studied.
The structure of AmpC is shown with avibactam (blue) bound to the catalytic serine (pink) at the active site. Deletions that contribute to β-lactam resistance [240] are shown in black. The locations of amino acid variants that contribute to β-lactam resistance [240] are shown in red with the side chains displayed. Sequence variants P180L and F147L reduce susceptibility to ceftazidime and ceftolozane-tazobactam. Variants V239A, G242R, E247K, E247G, and Y249H reduce susceptibility to ticarcillin, ceftazidime, ceftolozane-tazobactam, piperacillin-tazobactam and cefepime, and except for E247K aztreonam. Variants L320P, N373I, and ΔT316-ΔQ321reduce susceptibility to a wide range of cephalosporins. Variants that reduce the effectiveness of the AmpC inhibitor tazobactam are G183V, E247K (shown in red), and the deletion ΔG229–ΔE247 [242]. Variant N347Y (cyan) reduces the effectiveness of the inhibitor avibactam [241]. Amino acid residues are numbered relative to the start codon. The image is based on the crystal structure 4HEF_1 [246].
12. β-lactamases Acquired by Horizontal Gene Transfer
Many clinical isolates of P. aeruginosa have additional β-lactamases that have been acquired by horizontal gene transfer [209,210,247,248,249]. Enzymes in classes, A, B, and D can all be acquired in this way. The most prevalent horizontally acquired class A β-lactamases in P. aeruginosa include enzymes in the SHV, TEM, KPC, and GES families [250,251]. For example, a study in 88 isolates of P. aeruginosa from patients in Germany found that 44% had a SHV, 23% had a TEM, 14% had a KPC, and 2% had a GES [250]. A novel class A carbapenemase, GPC-1, has also been recently discovered in P. aeruginosa [252].
Amongst class B Metallo-β-lactamases, which include carbapenemases, two of the most prevalent horizontally acquired enzymes are VIM and IMP [210,250,253,254]. Of 207 isolates of P. aeruginosa from patients in China, 55% had a class B β-lactamase, of which 32% were a VIM and 29% an IMP with the remainder being in the SIM, NDM SPM, and GIM families [255]. In the previously mentioned study of isolates from German patients, IMP had a prevalence of 16%, and VIM was present in 6% of isolates [250]. The difference in prevalence between the two studies may be due to factors such as a differing distribution of β-lactamases around the globe and different treatment protocols.
Transferable Class C β-lactamases are relatively rare in species such as P. aeruginosa that have chromosomally encoded ampC [256,257]. Transferable Class C β-lactamases can have activity against penicillins, cephalosporins, and monobactams [256]. These enzymes are thought to have originated from chromosomally-encoded enzymes that have been transferred to mobile elements [257]. Class C β-lactamases present in P. aeruginosa as a result of horizontal gene transfer include FOX-4 in a high-risk strain of P. aeruginosa (ST308) [258] and CMY-2 that was found in a variety of clinical isolates [259].
Class D β-lactamases that are increasingly problematic in P. aeruginosa include the OXA family of β-lactams, named for their high activity against oxacillin [209,249]. Class D β-lactamases have a broad activity spectrum capable of degrading all β-lactams [260]. Many Class D OXA β-lactamases, such as the OXA-10-like β-lactamase that confers resistance to ceftazidime, were first discovered in P. aeruginosa [249]. In one study, of 1173 P. aeruginosa isolates from patients, 15.4% had OXA β-lactamases [261]. In a separate study, of 75 β-lactamase-producing isolates, OXA-1 was the most common β-lactamase being found in 37.3% of isolates, followed by OXA-4 (in 32%), GES-1 (in 16%), and VEB-1 (in 13.3%) [262]. OXA-1 and OXA-4 were both present in 18.7% of isolates. In a further study of 184 carbapenem-resistant P. aeruginosa isolates, the OXA-type carbapenemases present were OXA-23 in 6.5% of isolates, OXA-40 in 0.5% and OXA-58 in 0.5% [263].
The acquisition of carbapenemases by P. aeruginosa has contributed towards a significant proportion of overall carbapenemase resistance [264]. In a study of 232 carbapenem-resistant P. aeruginosa isolates, 71 isolates had carbapenemases that had likely been acquired through HGT [265]. In P. aeruginosa acquisition of β-lactamase genes by horizontal transfer occurs via plasmids or through integrative and conjugative elements (ICEs) that integrate into the chromosome of recipient cells following transfer. For example, the gene encoding an OXA-198 enzyme in isolates of P. aeruginosa from Belgium was carried on an IncP-type plasmid [266]. IMP was the first transferable Metallo-β-lactamase found in P. aeruginosa (in 1991) and was on a conjugative plasmid [247]. In a separate study, 11 VIM β-lactamases and one IMP β-lactamase from P. aeruginosa isolates were found on self-mobilizing plasmids [248]. Conversely, the gene encoding a class A GES-6 enzyme was present on an ICE element in a P. aeruginosa [267], and bioinformatic analysis shows that β-lactamases are commonly located on ICEs in P. aeruginosa [268]. Multiple antibiotic-modifying genes can be present on a single mobile genetic element [269,270,271]. β-lactamases are often present on integrons that facilitate their capture by mobile genetic elements [270,271,272]. This can result in the presence of multiple β-lactamase genes, such as carbapenemase- and cephalosporinase-encoding genes, on a single mobile element conferring resistance to a wide range of β-lactams [261,269,270,271]. Compounding the problem, integrons can also carry genes conferring resistance to other antibiotic classes such as aminoglycosides, so that horizontal gene transfer can result in multidrug-resistant bacteria [273,274].
Horizontal gene transfer can occur between as well as within species, providing the potential for P. aeruginosa to acquire β-lactamases from unrelated bacteria such as the Enterobacteriaceae. KPCs first discovered in Klebsiella pneumoniae are now found in P. aeruginosa and many other gram-negative bacteria such as Enterobacter spp., E. coli, Proteus mirabilis, and Salmonella spp. [122]. Conversely, VIMs were first discovered in P. aeruginosa and are now found widely spread amongst gram-negative bacteria [122].
Carbapenemase-producing bacteria have been found in hospital drainage systems (wastewater) and sewage systems and in the general wastewater downstream of hospital treatment systems [216,275,276,277,278]. This indicates that hospitals may be a large source of the dissemination of carbapenemase-producing isolates into the general environment, providing potential for inter-species gene transfer. However, carbapenemases have also been found in many other environments. Carbapenemase-producing bacteria have been found in most aquatic environments including rivers, the sea, well water, sewage, and drinking water [279,280]. Non-aquatic environments also harbour carbapenemases. In one study, 4 out of 856 bacterial isolates from samples of vegetables had carbapenemases-encoding genes [281]. The widespread presence of carbapenemase-encoding genes in the general environment increases the risk of acquisition of carbapenemase resistance genes by P. aeruginosa.
13. Lifestyle and Metabolism: Other Contributors to Resistance
The primary mechanisms of β-lactam resistance are outlined above, but mutational changes affecting other pathways can also influence susceptibility to β-lactams. Bacteria near the centre of a biofilm have low metabolism and growth because of low oxygen and nutrient levels, factors that contribute towards β-lactam resistance as β-lactams only kill actively growing cells [57]. Mutations in genes that regulate biofilm production are observed in many clinical isolates of P. aeruginosa from the lungs of patients with CF [282,283,284] and may reduce the susceptibility of the bacteria to β-lactams. For example, mutations in wspF were present in 68% of clinical isolates that have increased production of extracellular matrix [285]. wspF belongs to the Wsp signalling complex, a major regulator of biofilm formation [286]. The loss of wspF via mutations is predicted to leave the Wsp signalling complex in an active conformation leading to a signalling cascade upregulating genes responsible for adhesion and biofilm formation [286]. Similarly, point mutation and deletions of rpoS increase adhesion and biofilm production [287,288]. In experimental evolution studies, rpoS was commonly mutated in P. aeruginosa grown as biofilms and then exposed to imipenem [287]. Mutations in rpoS have also been found in many clinical isolates [287].
Experimental evolution studies have shown that mutations in a wide variety of other genes are also associated with an increased ability to tolerate β-lactams [55]. Many of these mutations affect lipopolysaccharide (LPS) synthesis (e.g., wapR/galU) [289] or alter other aspects of metabolism (e.g., aroB/acyl-CoA thiolase) [124,290] or membrane composition. Mutations in the galU gene that plays a role in the synthesis of lipopolysaccharide occur in CF clinical isolates of P. aeruginosa [135] and increase ceftazidime and meropenem tolerance [74]. How these mutations contribute towards β-lactam resistance is not well studied. It may be that altered LPS synthesis reduces the permeability of the outer membrane for β-lactams. There is some evidence that mutations affecting LPS synthesis can reduce bacterial fitness in vitro [291] but whether they affect fitness in vivo is not known.
Mutations that lead to enhanced activity of AlgU, a regulator of alginate biosynthesis, are predicted to cause a metabolic burden, and mutations in rpoN which nitrogen metabolism, reducing the growth rate in many clinical isolates [292]. Slower growth rates in P. aeruginosa are associated with antibiotic resistance [292]. Mutations in genes encoding enzymes of metabolism, such as triosephospate isomerase (central carbon metabolism), N-acetylglutamate synthase (arginine metabolism), and 3-dehydroquinate synthase (synthesis of aromatic amino acids) also increase tolerance to β-lactams [55,124,289]. Whether these mutations act by reducing growth rate by altering antibiotic susceptibility because of reduced cellular respiration [293] is not yet clear.
During chronic infection, P. aeruginosa can also evolve morphotypes termed small colony variants (SCVs) because of their appearance during growth on agar plates [294]. SCVs have an increased ability to resist antibiotics. SCVs arise from changes to signalling pathways, including the Wsp signalling pathway described above, and also to changes in metabolic pathways [295]. However, the relationship between metabolic changes, the SCV phenotype, and the increased ability to tolerate β-lactams is not yet fully understood.
14. Conclusions and Prospects for the Future
β-lactam antibiotics are a key tool in the treatment of P. aeruginosa infections and will remain so for the foreseeable future. However, the occurrence of resistant isolates of this major and problematic pathogen, complicating treatment, is a major concern. Clinical isolates that have acquired carbapenemases are a serious problem as carbapenems are one of the last lines of defence against P. aeruginosa [211]. The importance of β-lactams in treating P. aeruginosa infections, and the widespread occurrence of antibiotic-resistant bacteria, has led to a large amount of research into the mechanism of β-lactam action and bacterial resistance. Consequently, we have a good understanding of how β-lactams act and on resistance mechanisms, including mutations that reduce carbapenem uptake or upregulate efflux pumps, target site mutations that alter PBP3 or increase AmpC expression and activity, and acquisition of antibiotic modifying enzymes through HGT.
Nonetheless, there are still some important knowledge gaps. Sequence variants in PBP3 are an important contributor to resistance, but the effects of different variants on affinity for, and effectiveness of, different antibiotics are not yet fully understood. A better understanding of the effects of PBP3 variants on β-lactam affinity will be important in the design of new β-lactams, as well as helping to understand cross-resistance between currently used antibiotics. Some other resistance mechanisms are class-specific. For example, mutations altering OprD contribute to carbapenem resistance but not cephalosporin resistance, whereas mutations affecting PBP4 increase resistance to cephalosporins but not carbapenems. β-lactamases acquired by horizontal gene transfer also exhibit specificity for different classes of β-lactams, as well as different susceptibilities to β-lactam inhibitors. Refining our understanding of class-specific resistance mechanisms has the potential to allow fine-tuning of β-lactam use in clinical practice.
More broadly, while the primary contributors to β-lactam resistance are relatively well understood, the relationship between genotype (genome sequence) and phenotype is not yet fully clear. How do different combinations of resistance mechanisms affect β-lactam susceptibility, what is the contribution of intrinsic resistance and to what extent does it vary between isolates? It is especially difficult to determine if variants in gene promoters and regulatory regions or non-coding RNAs have an effect. Fully understanding the relationship between genotype and phenotype has the potential to allow the development of genome-based tools for rapid prediction of antibiotic susceptibility [296,297], as has been done for other bacterial species [37,298,299,300]. More rapid treatment with antibiotics would reduce the mortality associated with P. aeruginosa infections.
How can the occurrence of P. aeruginosa resistant to β-lactams be minimised? One approach that has already been implemented in a variety of settings is antimicrobial stewardship–limiting β-lactam use to cases where there will be a clear benefit [301]. Initial studies applying this approach to P. aeruginosa are encouraging, with the rate of antibiotic resistance (including β-lactam resistance) being lowered once stewardship programmes were implemented [302,303].
Additional approaches to minimise the emergence of resistant bacteria are to use combinations of antibiotics or to avoid prolonged exposure to an antibiotic in chronic infections by alternating between different antibiotic classes–“antibiotic cycling” [304]. The first of these relies on the principle that if an infecting bacterium has become resistant to one antibiotic, it will still be susceptible to a second. As well as antibiotic combinations, phage therapy in parallel with antibiotic treatment is undergoing extensive testing in clinical trials and results are encouraging [305,306,307,308,309]. Powdered phage cocktails are being specifically designed for the treatment of respiratory infections [310]. Peptides with anti-microbial activity are also being actively explored as potential tools in overcoming antibiotic-resistant bacteria [311] and peptide–β-lactam combinations may present another approach to preventing the emergence of resistance.
The “antibiotic cycling” approach is based in part on the concept that mutations that confer antibiotic resistance may reduce the fitness of the bacteria in the absence of antibiotics, allowing resistant mutants to be outcompeted by antibiotic-susceptible bacteria. Further research on these approaches can be expected to provide clear principles on the most effective use of β-lactams for treatment while minimising the emergence of resistant bacteria. More broadly, there is limited research on the circumstances in which P. aeruginosa acquires β-lactamase genes by HGT. However, as in the clinic, the presence of antibiotics in the general environment will promote antibiotic resistance. Minimising the discharge of β-lactams from clinical settings such as hospitals into wastewater will reduce the selection of antibiotic-resistant bacteria.
An alternative approach to circumventing resistance would be to develop new β-lactams that are unaffected by resistance mechanisms such as β-lactamases, along with new β-lactamase inhibitors. Examples of new β-lactams in development include the carbapenem benapenem [312,313] and a monobactam BOS-228 [312,314]. Combinations of β-lactams with a number of new β-lactamase inhibitors are undergoing clinical trials [315,316]. Diazabicyclooctane-derived β-lactamase inhibitors can inhibit class A and C β-lactamases but have limited activity against class D β-lactamases, whereas enmetazobactam and LN-1-255 show activity against class A, C, and D β-lactamases [316]. Three boronic acid-derived β-lactamases inhibitors VNRX-5236, taniborbactam, and xeruborbactam are also undergoing clinical trials [316]. Of particular significance, taniborbactam and xeruborbactam are pan-β-lactamase inhibitors that can inhibit all classes of β-lactamases, including metallo-β-lactamases which are not targeted by other inhibitors. A novel metallo-β-lactamase inhibitor ANT431 is also at a pre-clinical stage [317,318]. A number of other compounds are also under development, although how many will find their way into the clinic, and whether they will be subject to the same resistance mechanisms as existing compounds, is not yet clear [319,320].
Rational development of new anti-Pseudomonas antibiotics is greatly enhanced by an understanding of the mechanisms of action of, and resistance to, existing antibiotics [320]. Carbapenems can enter P. aeruginosa via OprD, but other antibiotics are thought to diffuse through the lipid bilayer of the outer membrane. A better understanding of how β-lactams access the periplasm may enable more effective uptake. One approach that has been explored is to conjugate β-lactams to siderophores, low molecular weight compounds imported into the periplasm by P. aeruginosa to enable the acquisition of iron a key nutrient [321,322]. A newly approved cephalosporin-siderophore, cefiderocol (FDA 2019 and European Union 2020), is showing promising results against P. aeruginosa [323,324] shown to be active against greater than 95% of P. aeruginosa isolates [325] Resistance towards cefiderocol can arise through mutations in AmpC and in TonB-dependent transporters required for entry of the antibiotic into bacterial cells [237,326,327] and the extent to which resistance becomes a clinical problem remains to be determined. Encapsulating β-lactams into liposomes or loading them into nanoparticles for targeted antibiotic release may also increase the local concentration of antibiotics, enhancing entry into the bacterial cells [315,328,329].
As well as β-lactamases, other P. aeruginosa proteins contribute to resistance and are potential targets for antibiotics. For example, inhibitors of AmpR would be expected to increase the susceptibility of P. aeruginosa to AmpC-susceptible β-lactams, and inhibition of the MexABOprM efflux pump would also increase susceptibility. Phe-Arg-β-naphthylamide (PAβN) is a broad-spectrum efflux inhibitor that reduces MICs towards antibiotics in vitro [330,331]. PAβN is toxic to humans but its effectiveness in vitro demonstrates the potential of efflux pump inhibitors as targets for anti-Pseudomonal therapy.
In conclusion, β-lactams will be key members of the anti-Pseudomonas armamentarium for many years to come. Future research on their modes of action and or how to overcome the threat of resistant bacteria will be essential to maintain and maximise their usage.
Supplementary Materials
The following are available online at www.mdpi.com/article/10.3390/pathogens10121638/s1, Table S1: P. aeruginosa PBP3 variants within the transpeptidase domain, Table S2: P. aeruginosa PBP3 variants outside the transpeptidase domain.
Author Contributions
Conceptualization, K.A.G. and I.L.L.; Investigation and review of the literature, K.A.G. and I.L.L.; Writing—original draft preparation, K.A.G. and I.L.L.; Writing, review and editing, K.A.G. and I.L.L. Funding acquisition, I.L.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by grants from the New Zealand Health Research Council (17/372 and 20/812). Karl A. Glen is supported by a Postgraduate Scholarship from the University of Otago.
Institutional Review Board Statement
Not applicable as this study did not involve humans or animals.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable as no data was generated for this review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Crone, S.; Vives-Flórez, M.; Kvich, L.; Saunders, A.M.; Malone, M.; Nicolaisen, M.H.; Martínez-García, E.; Rojas-Acosta, C.; Catalina Gomez-Puerto, M.; Calum, H. The environmental occurrence of Pseudomonas aeruginosa. APMIS 2020, 128, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, M.; Vena, A.; Croxatto, A.; Righi, E.; Guery, B. How to manage Pseudomonas aeruginosa infections. Drugs Context 2018, 7, 212527. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, M.; Vena, A.; Russo, A.; Croxatto, A.; Calandra, T.; Guery, B. Rational approach in the management of Pseudomonas aeruginosa infections. Curr. Opin. Infect. Dis. 2018, 31, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Sievert, D.M.; Ricks, P.; Edwards, J.R.; Schneider, A.; Patel, J.; Srinivasan, A.; Kallen, A.; Limbago, B.; Fridkin, S. Antimicrobial-resistant pathogens associated with healthcare-associated infections summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect. Control Hosp. Epidemiol. 2013, 34, 1–14. [Google Scholar] [CrossRef]
- Nathwani, D.; Raman, G.; Sulham, K.; Gavaghan, M.; Menon, V. Clinical and economic consequences of hospital-acquired resistant and multidrug-resistant Pseudomonas aeruginosa infections: A systematic review and meta-analysis. Antimicrob. Resist. Infect. Control 2014, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Luyt, C.-E.; Hékimian, G.; Koulenti, D.; Chastre, J. Microbial cause of ICU-acquired pneumonia: Hospital-acquired pneumonia versus ventilator-associated pneumonia. Curr. Opin. Crit. Care 2018, 24, 332–338. [Google Scholar] [CrossRef]
- Sarda, C.; Fazal, F.; Rello, J. Management of ventilator-associated pneumonia (VAP) caused by resistant gram-negative bacteria: Which is the best strategy to treat? Expert Rev. Respir. Med. 2019, 13, 787–798. [Google Scholar] [CrossRef]
- Raineri, E.; Porcella, L.; Acquarolo, A.; Crema, L.; Albertario, F.; Candiani, A. Ventilator-Associated Pneumonia Caused by Pseudomonas aeruginosa in Intensive Care Unit: Epidemiology and Risk Factors. J. Med. Microbiol. Diagn. 2014, 3, 1. [Google Scholar] [CrossRef]
- Hilliam, Y.; Kaye, S.; Winstanley, C. Pseudomonas aeruginosa and microbial keratitis. J. Med. Microbiol. 2020, 69, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langan, K.M.; Kotsimbos, T.; Peleg, A.Y. Managing Pseudomonas aeruginosa respiratory infections in cystic fibrosis. Curr. Opin. Infect. Dis. 2015, 28, 547–556. [Google Scholar] [CrossRef]
- Garcia-Clemente, M.; de la Rosa, D.; Máiz, L.; Girón, R.; Blanco, M.; Olveira, C.; Canton, R.; Martinez-García, M.A. Impact of Pseudomonas aeruginosa Infection on Patients with Chronic Inflammatory Airway Diseases. J. Clin. Med. 2020, 9, 3800. [Google Scholar] [CrossRef]
- Gransden, W.R.; Leibovici, L.; Eykyn, S.J.; Pitlik, S.D.; Samra, Z.; Konisberger, H.; Drucker, M.; Phillips, I. Risk factors and a clinical index for diagnosis of Pseudomonas aeruginosa bacteremia. Clin. Microbiol. Infect. 1995, 1, 119–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tredget, E.E.; Shankowsky, H.A.; Rennie, R.; Burrell, R.E.; Logsetty, S. Pseudomonas infections in the thermally injured patient. Burns 2004, 30, 3–26. [Google Scholar] [CrossRef]
- Puchelle, E.; Bajolet, O.; Abély, M. Airway mucus in cystic fibrosis. Paediatr. Respir. Rev. 2002, 3, 115–119. [Google Scholar] [CrossRef]
- Mayer-Hamblett, N.; Kloster, M.; Rosenfeld, M.; Gibson, R.L.; Retsch-Bogart, G.Z.; Emerson, J.; Thompson, V.; Ramsey, B.W. Impact of sustained eradication of new Pseudomonas aeruginosa infection on long-term outcomes in cystic fibrosis. Clin. Infect. Dis. 2015, 61, 707–715. [Google Scholar] [CrossRef]
- Taccetti, G.; Campana, S.; Festini, F.; Mascherini, M.; Döring, G. Early eradication therapy against Pseudomonas aeruginosa in cystic fibrosis patients. Eur. Respir. J. 2005, 26, 458–461. [Google Scholar] [CrossRef] [Green Version]
- Zolin, A.; Bossi, A.; Cirilli, N.; Kashirskaya, N.; Padoan, R. Cystic Fibrosis Mortality in Childhood. Data from European Cystic Fibrosis Society Patient Registry. Int. J. Environ. Res. Public Health 2018, 15, 2020. [Google Scholar] [CrossRef] [Green Version]
- Williams, F.N.; Herndon, D.N.; Hawkins, H.K.; Lee, J.O.; Cox, R.A.; Kulp, G.A.; Finnerty, C.C.; Chinkes, D.L.; Jeschke, M.G. The leading causes of death after burn injury in a single pediatric burn center. Crit. Care 2009, 13, R183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norbury, W.; Herndon, D.N.; Tanksley, J.; Jeschke, M.G.; Finnerty, C.C. Infection in Burns. Surg. Infect. 2016, 17, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Church, D.; Elsayed, S.; Reid, O.; Winston, B.; Lindsay, R. Burn wound infections. Clin. Microbiol. Rev. 2006, 19, 403–434. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.E.; D’Agata, E.M.C.; Paterson, D.L.; Clarke, L.; Qureshi, Z.A.; Potoski, B.A.; Peleg, A.Y. Pseudomonas aeruginosa bacteremia over a 10-year period: Multidrug resistance and outcomes in transplant recipients. Transpl. Infect. Dis. 2009, 11, 227–234. [Google Scholar] [CrossRef]
- Choi, Y.; Paik, J.H.; Kim, J.H.; Han, S.B.; Durey, A. Clinical predictors of Pseudomonas aeruginosa bacteremia in emergency department. Emerg. Med. Int. 2018, 2018, 7581036. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.I.; Kim, S.H.; Kim, H.B.; Park, S.W.; Choe, Y.J.; Oh, M.D.; Kim, E.C.; Choe, K.W. Pseudomonas aeruginosa bacteremia: Risk factors for mortality and influence of delayed receipt of effective antimicrobial therapy on clinical outcome. Clin. Infect. Dis. 2003, 37, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, E.-J.; Kang, C.-I.; Ha, Y.E.; Kang, S.-J.; Park, S.Y.; Chung, D.R.; Peck, K.R.; Lee, N.Y.; Song, J.-H. Risk factors for mortality in patients with Pseudomonas aeruginosa bacteremia: Clinical impact of antimicrobial resistance on outcome. Microb. Drug Resist. 2011, 17, 305–312. [Google Scholar] [CrossRef]
- Lachiewicz, A.M.; Hauck, C.G.; Weber, D.J.; Cairns, B.A.; van Duin, D. Bacterial infections after burn injuries: Impact of multidrug resistance. Clin. Infect. Dis. 2017, 65, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Bédard, E.; Prévost, M.; Déziel, E. Pseudomonas aeruginosa in premise plumbing of large buildings. MicrobiologyOpen 2016, 5, 937–956. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, M.; Lepper, P.M.; Haller, M. Ecology of Pseudomonas aeruginosa in the intensive care unit and the evolving role of water outlets as a reservoir of the organism. Am. J. Infect. Control 2005, 33, S41–S49. [Google Scholar] [CrossRef]
- Parcell, B.J.; Oravcova, K.; Pinheiro, M.; Holden, M.T.G.; Phillips, G.; Turton, J.F.; Gillespie, S.H. Pseudomonas aeruginosa intensive care unit outbreak: Winnowing of transmissions with molecular and genomic typing. J. Hosp. Infect. 2018, 98, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Quick, J.; Cumley, N.; Wearn, C.M.; Niebel, M.; Constantinidou, C.; Thomas, C.M.; Pallen, M.J.; Moiemen, N.S.; Bamford, A.; Oppenheim, B.; et al. Seeking the source of Pseudomonas aeruginosa infections in a recently opened hospital: An observational study using whole-genome sequencing. BMJ Open 2014, 4, e006278. [Google Scholar] [CrossRef] [Green Version]
- Lanini, S.; D’Arezzo, S.; Puro, V.; Martini, L.; Imperi, F.; Piselli, P.; Montanaro, M.; Paoletti, S.; Visca, P.; Ippolito, G. Molecular epidemiology of a Pseudomonas aeruginosa hospital outbreak driven by a contaminated disinfectant-soap dispenser. PLoS ONE 2011, 6, e17064. [Google Scholar] [CrossRef]
- Eckmanns, T.; Oppert, M.; Martin, M.; Amorosa, R.; Zuschneid, I.; Frei, U.; Ruden, H.; Weist, K. An outbreak of hospital-acquired Pseudomonas aeruginosa infection caused by contaminated bottled water in intensive care units. Clin. Microbiol. Infect. 2008, 14, 454–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schelstraete, P.; Van daele, S.; De Boeck, K.; Proesmans, M.; Lebecque, P.; Leclercq-Foucart, J.; Malfroot, A.; Vaneechoutte, M.; De Baets, F. Pseudomonas aeruginosa in the home environment of newly infected cystic fibrosis patients. Eur. Respir. J. 2008, 31, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDC. Antibiotic Resistance Threats in the United States, 2019; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019. [Google Scholar]
- Hirsch, E.B.; Tam, V.H. Impact of multidrug-resistant Pseudomonas aeruginosa infection on patient outcomes. Expert Rev. Pharm. Outcomes Res. 2010, 10, 441–451. [Google Scholar]
- Matos, E.C.O.d.; Andriolo, R.B.; Rodrigues, Y.C.; Lima, P.D.L.d.; Carneiro, I.C.d.R.S.; Lima, K.V.B. Mortality in patients with multidrug-resistant Pseudomonas aeruginosa infections: A meta-analysis. Rev. Soc. Bras. Med. Trop. 2018, 51, 415–420. [Google Scholar] [CrossRef] [Green Version]
- Bradley, P.; Gordon, N.C.; Walker, T.M.; Dunn, L.; Heys, S.; Huang, B.; Earle, S.; Pankhurst, L.J.; Anson, L.; De Cesare, M. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun. 2015, 6, 10063. [Google Scholar] [CrossRef] [Green Version]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- De Oliveira, D.M.; Forde, B.M.; Kidd, T.J.; Harris, P.N.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Taconelli, E.; Magrini, N. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Elborn, J.S.; Vataire, A.-L.; Fukushima, A.; Aballea, S.; Khemiri, A.; Moore, C.; Medic, G.; Hemels, M.E.H. Comparison of inhaled antibiotics for the treatment of chronic Pseudomonas aeruginosa lung infection in patients with cystic fibrosis: Systematic literature review and network meta-analysis. Clin. Ther. 2016, 38, 2204–2226. [Google Scholar] [CrossRef] [Green Version]
- Horcajada, J.P.; Montero, M.; Oliver, A.; Sorlí, L.; Luque, S.; Gómez-Zorrilla, S.; Benito, N.; Grau, S. Epidemiology and treatment of multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa infections. Clin. Microbiol. Rev. 2019, 32, e00031-19. [Google Scholar] [CrossRef]
- Talwalkar, J.S.; Murray, T.S. The approach to Pseudomonas aeruginosa in cystic fibrosis. Clin. Chest Med. 2016, 37, 69–81. [Google Scholar] [CrossRef]
- Castle, S.S. Ticarcillin. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Castle, S.S. Piperacillin. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Scholar, E. Aztreonam. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Hansen, C.; Skov, M. Evidence for the efficacy of aztreonam for inhalation solution in the management of Pseudomonas aeruginosa in patients with cystic fibrosis. Ther. Adv. Respir. Dis. 2015, 9, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Taccetti, G.; Francalanci, M.; Pizzamiglio, G.; Messore, B.; Carnovale, V.; Cimino, G.; Cipolli, M. Cystic Fibrosis: Recent Insights into Inhaled Antibiotic Treatment and Future Perspectives. Antibiotics 2021, 10, 338. [Google Scholar] [CrossRef]
- Blanco-Aparicio, M.; Saleta Canosa, J.L.; Valiño López, P.; Martín Egaña, M.T.; Vidal Garcia, I.; Montero Martínez, C. Eradication of Pseudomonas aeruginosa with inhaled colistin in adults with non-cystic fibrosis bronchiectasis. Chronic Respir. Dis. 2019, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- King, D.E.; Malone, R.; Lilley, S.H. New classification and update on the quinolone antibiotics. Am. Fam. Physician 2000, 61, 2741–2748. [Google Scholar]
- Smith, S.; Rowbotham, N.J.; Charbek, E. Inhaled antibiotics for pulmonary exacerbations in cystic fibrosis. Cochrane Database Syst. Rev. 2018, 10, CD008319. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Cascella, M. Beta Lactam Antibiotics. In StatPearls; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Campanella, T.A.; Gallagher, J.C. A clinical review and critical evaluation of Imipenem-Relebactam: Evidence to date. Infect. Drug Resist. 2020, 13, 4297. [Google Scholar] [CrossRef]
- Kong, K.-F.; Schneper, L.; Mathee, K. β-lactam antibiotics: From antibiosis to resistance and bacteriology. APMIS 2010, 118, 1–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Ortega, C.; Wiegand, I.; Olivares, J.; Hancock, R.E.; Martinez, J.L. Genetic determinants involved in the susceptibility of Pseudomonas aeruginosa to β-lactam antibiotics. Antimicrob. Agents Chemother. 2010, 54, 4159–4167. [Google Scholar] [CrossRef] [Green Version]
- Breidenstein, E.B.M.; de la Fuente-Núñez, C.; Hancock, R.E.W. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef]
- Ciofu, O.; Tolker-Nielsen, T. Tolerance and Resistance of Pseudomonas aeruginosa Biofilms to Antimicrobial Agents-How P. aeruginosa Can Escape Antibiotics. Front. Microbiol. 2019, 10, 913. [Google Scholar] [CrossRef] [Green Version]
- Povolotsky, T.L.; Keren-Paz, A.; Kolodkin-Gal, I. Metabolic Microenvironments Drive Microbial Differentiation and Antibiotic Resistance. Trends Genet. 2021, 37, 4–8. [Google Scholar] [CrossRef]
- Soares, A.; Alexandre, K.; Etienne, M. Tolerance and Persistence of Pseudomonas aeruginosa in Biofilms Exposed to Antibiotics: Molecular Mechanisms, Antibiotic Strategies and Therapeutic Perspectives. Front. Microbiol. 2020, 11, 2057. [Google Scholar] [CrossRef] [PubMed]
- Hengzhuang, W.; Ciofu, O.; Yang, L.; Wu, H.; Song, Z.; Oliver, A.; Hoiby, N. High β-lactamase levels change the pharmacodynamics of β-lactam antibiotics in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2013, 57, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, K.A.; Wardell, S.J.T.; Patrick, W.M.; Brockway, B.; Reid, D.W.; Winstanley, C.; Bell, S.C.; Lamont, I.L. Genomic and phenotypic comparison of environmental and patient-derived isolates of Pseudomonas aeruginosa suggest that antimicrobial resistance is rare within the environment. J. Med. Microbiol. 2019, 68, 1591–1595. [Google Scholar] [CrossRef]
- Nordmann, P.; Poirel, L. Epidemiology and Diagnostics of Carbapenem Resistance in Gram-negative Bacteria. Clin. Infect. Dis. 2019, 69, S521–S528. [Google Scholar] [CrossRef] [Green Version]
- Shortridge, D.; Gales, A.C.; Streit, J.M.; Huband, M.D.; Tsakris, A.; Jones, R.N. Geographic and Temporal Patterns of Antimicrobial Resistance in Pseudomonas aeruginosa Over 20 Years From the SENTRY Antimicrobial Surveillance Program, 1997–2016. Open Forum Infect. Dis. 2019, 6, S63–S68. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimpour, M.; Nikokar, I.; Ghasemi, Y.; Sedigh Ebrahim-Saraie, H.; Araghian, A.; Farahbakhsh, M.; Ghassabi, F. Antibiotic resistance and frequency of class 1 integrons among Pseudomonas aeruginosa isolates obtained from wastewaters of a burn center in Northern Iran. Ann. Ig. 2018, 30, 112–119. [Google Scholar] [CrossRef]
- Feretzakis, G.; Loupelis, E.; Sakagianni, A.; Skarmoutsou, N.; Michelidou, S.; Velentza, A.; Martsoukou, M.; Valakis, K.; Petropoulou, S.; Koutalas, E. A 2-year single-centre audit on antibiotic resistance of Pseudomonas aeruginosa, Acinetobacter baumannii and Klebsiella pneumoniae strains from an intensive care unit and other wards in a general public hospital in Greece. Antibiotics 2019, 8, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, J.G.; Weiner, L.M.; Milstone, A.M.; Saiman, L.; Magill, S.S.; See, I. Pathogen distribution and antimicrobial resistance among pediatric healthcare-associated infections reported to the National Healthcare Safety Network, 2011–2014. Infect. Control Hosp. Epidemiol. 2018, 39, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, X.-L.; Huang, A.-W.; Liu, S.-L.; Liu, W.-J.; Zhang, N.; Lu, X.-Z. Mortality attributable to carbapenem-resistant Pseudomonas aeruginosa bacteremia: A meta-analysis of cohort studies. Emerg. Microbes Infect. 2016, 5, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Li, X.; Li, W.; Du, X.; He, J.-Q.; Tao, C.; Feng, Y. Influence of carbapenem resistance on mortality of patients with Pseudomonas aeruginosa infection: A meta-analysis. Sci. Rep. 2015, 5, 11715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkhair, A.; Al-Muharrmi, Z.; Al’Adawi, B.; Al Busaidi, I.; Taher, H.B.; Al-Siyabi, T.; Al Amin, M.; Hassan, K.S. Prevalence and 30-day all-cause mortality of carbapenem-and colistin-resistant bacteraemia caused by Acinetobacter baumannii, Pseudomonas aeruginosa, and Klebsiella pneumoniae: Description of a decade-long trend. Int. J. Infect. Dis. 2019, 85, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.Y.; Lauderdale, T.L.; Wang, J.T.; Chang, S.C. Carbapenem-resistant Pseudomonas aeruginosa in Taiwan: Prevalence, risk factors, and impact on outcome of infections. J. Microbiol. Immunol. Infect. 2016, 49, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Kaier, K.; Heister, T.; Gotting, T.; Wolkewitz, M.; Mutters, N.T. Measuring the in-hospital costs of Pseudomonas aeruginosa pneumonia: Methodology and results from a German teaching hospital. BMC Infect. Dis. 2019, 19, 1028. [Google Scholar] [CrossRef] [PubMed]
- Lister Philip, D.; Wolter Daniel, J.; Hanson Nancy, D. Antibacterial-Resistant Pseudomonas aeruginosa: Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef] [Green Version]
- López-Causapé, C.; Cabot, G.; del Barrio-Tofiño, E.; Oliver, A. The Versatile Mutational Resistome of Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 685. [Google Scholar] [CrossRef]
- López-Causapé, C.; Rojo-Molinero, E.; Macià, M.D.; Oliver, A. The problems of antibiotic resistance in cystic fibrosis and solutions. Expert Rev. Respir. Med. 2015, 9, 73–88. [Google Scholar] [CrossRef]
- Llanes, C.; Pourcel, C.; Richardot, C.; Plesiat, P.; Fichant, G.; Cavallo, J.D.; Merens, A.; Group, G.S. Diversity of β-lactam resistance mechanisms in cystic fibrosis isolates of Pseudomonas aeruginosa: A French multicentre study. J. Antimicrob. Chemother. 2013, 68, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- Pitten, F.A.; Panzig, B.; Schröder, G.; Tietze, K.; Kramer, A. Transmission of a multiresistant Pseudomonas aeruginosa strain at a German University Hospital. J. Hosp. Infect. 2001, 47, 125–130. [Google Scholar] [CrossRef]
- Aguilera-Sáez, J.; Andreu-Solà, V.; Larrosa Escartín, N.; Rodríguez Garrido, V.; Armadans Gil, L.; Sánchez García, J.M.; Campins, M.; Baena Caparrós, J.; Barret, J.P. Extensively drug-resistant Pseudomonas aeruginosa outbreak in a burn unit: Management and solutions. Ann. Burn. Fire Disasters 2019, 32, 47–55. [Google Scholar]
- Douglas, M.W.; Mulholland, K.; Denyer, V.; Gottlieb, T. Multi-drug resistant Pseudomonas aeruginosa outbreak in a burns unit—An infection control study. Burns 2001, 27, 131–135. [Google Scholar] [CrossRef]
- Vollmer, W.; Holtje, J.V. The architecture of the murein (peptidoglycan) in gram-negative bacteria: Vertical scaffold or horizontal layer(s)? J. Bacteriol. 2004, 186, 5978–5987. [Google Scholar] [CrossRef] [Green Version]
- Torrens, G.; Escobar-Salom, M.; Pol-Pol, E.; Camps-Munar, C.; Cabot, G.; López-Causapé, C.; Rojo-Molinero, E.; Oliver, A.; Juan, C. Comparative analysis of peptidoglycans from Pseudomonas aeruginosa isolates recovered from chronic and acute infections. Front. Microbiol. 2019, 10, 1868. [Google Scholar] [CrossRef]
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [Green Version]
- Akira Yanai, K.K. Teruhiko Beppu and Kei Arima. Peptidoglycan of Pseudomonas aeruginosa. Agric. Biol. Chem. 1975, 40, 1505–1508. [Google Scholar]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef] [Green Version]
- Heywood, A.; Lamont, I.L. Cell envelope proteases and peptidases of Pseudomonas aeruginosa: Multiple roles, multiple mechanisms. FEMS Microbiol. Rev. 2020, 44, 857–873. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Y.-M.; Davies, C. Penicillin-binding protein 3 is essential for growth of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e01651-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffin, C.; Ghuysen, J.M. Biochemistry and comparative genomics of SxxK superfamily acyltransferases offer a clue to the mycobacterial paradox: Presence of penicillin-susceptible target proteins versus lack of efficiency of penicillin as therapeutic agent. Microbiol. Mol. Biol. Rev. 2002, 66, 702–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Heijenoort, J.; Gutmann, L. Correlation between the structure of the bacterial peptidoglycan monomer unit, the specificity of transpeptidation, and susceptibility to β-lactams. Proc. Natl. Acad. Sci. USA 2000, 97, 5028–5030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffin, C.; Ghuysen, J.M. Multimodular penicillin-binding proteins: An enigmatic family of orthologs and paralogs. Microbiol. Mol. Biol. Rev. 1998, 62, 1079–1093. [Google Scholar] [CrossRef] [Green Version]
- Tomberg, J.; Temple, B.; Fedarovich, A.; Davies, C.; Nicholas, R.A. A highly conserved interaction involving the middle residue of the SXN active-site motif is crucial for function of class B penicillin-binding proteins: Mutational and computational analysis of PBP 2 from N. gonorrhoeae. Biochemistry 2012, 51, 2775–2784. [Google Scholar] [CrossRef] [Green Version]
- Macheboeuf, P.; Contreras-Martel, C.; Job, V.; Dideberg, O.; Dessen, A. Penicillin Binding Proteins: Key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol. Rev. 2006, 30, 673–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvage, E.; Terrak, M. Glycosyltransferases and transpeptidases/penicillin-binding proteins: Valuable targets for new antibacterials. Antibiotics 2016, 5, 12. [Google Scholar] [CrossRef]
- Ropy, A.; Cabot, G.; Sánchez-Diener, I.; Aguilera, C.; Moya, B.; Ayala, J.A.; Oliver, A. Role of Pseudomonas aeruginosa low-molecular-mass penicillin-binding proteins in AmpC expression, β-lactam resistance, and peptidoglycan structure. Antimicrob. Agents Chemother. 2015, 59, 3925–3934. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.S.; Chowdhury, C.; Nelson, D.E. Physiological functions of D-alanine carboxypeptidases in Escherichia coli. Trends Microbiol. 2008, 16, 309–317. [Google Scholar] [CrossRef]
- Torrens, G.; Hernandez, S.B.; Ayala, J.A.; Moya, B.; Juan, C.; Cava, F.; Oliver, A. Regulation of AmpC-driven β-lactam resistance in Pseudomonas aeruginosa: Different pathways, different signaling. mSystems 2019, 4, e00524-19. [Google Scholar] [CrossRef] [Green Version]
- Vollmer, W.; Joris, B.; Charlier, P.; Foster, S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 2008, 32, 259–286. [Google Scholar] [CrossRef] [Green Version]
- Aguilera Rossi, C.G.; Gómez-Puertas, P.; Ayala Serrano, J.A. In vivo functional and molecular characterization of the Penicillin-Binding Protein 4 (DacB) of Pseudomonas aeruginosa. BMC Microbiol. 2016, 16, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Hesek, D.; Blázquez, B.; Lastochkin, E.; Boggess, B.; Fisher, J.F.; Mobashery, S. Catalytic spectrum of the penicillin-binding protein 4 of Pseudomonas aeruginosa, a nexus for the induction of β-lactam antibiotic resistance. J. Am. Chem. Soc. 2015, 137, 190–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, J.; Oesch, G.; Kuster, S.P. Bacteriostatic versus bactericidal antibiotics for patients with serious bacterial infections: Systematic review and meta-analysis. J. Antimicrob. Chemother. 2014, 70, 382–395. [Google Scholar] [CrossRef] [Green Version]
- Dumancas, G.G.; Hikkaduwa Koralege, R.S.; Mojica, E.R.E.; Murdianti, B.S.; Pham, P.J. Penicillins. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 768–772. [Google Scholar] [CrossRef]
- Zeng, X.; Lin, J. β-lactamase induction and cell wall metabolism in gram-negative bacteria. Front. Microbiol. 2013, 4, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Zaniewski, R.P.; Marr, E.S.; Lacey, B.M.; Tomaras, A.P.; Evdokimov, A.; Miller, J.R.; Shanmugasundaram, V. Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2010, 107, 22002–22007. [Google Scholar] [CrossRef] [Green Version]
- Beadle, B.M.; Nicholas, R.A.; Shoichet, B.K. Interaction energies between β-lactam antibiotics and E. coli penicillin-binding protein 5 by reversible thermal denaturation. Protein Sci. 2001, 10, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Uehara, T.; Bernhardt, T.G. β-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 2014, 159, 1300–1311. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Kahne, D.; Kishony, R. Distinct single-cell morphological dynamics under β-lactam antibiotics. Mol. Cell 2012, 48, 705–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horii, T.; Ichiyama, S.; Ohta, M.; Kobayashi, M. Relationship between morphological changes and endotoxin release induced by carbapenems in Pseudomonas aeruginosa. J. Med. Microbiol. 1999, 48, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Das, S.; Dawson, N.L.; Dobrijevic, D.; Ward, J.; Orengo, C. Novel computational protocols for functionally classifying and characterising serine β-lactamases. PLoS Comput. Biol. 2016, 12, e1004926. [Google Scholar] [CrossRef] [Green Version]
- Liras, P.; Martín, J.F. β-lactam Antibiotics. In Encyclopedia of Microbiology, 3rd ed.; Schaechter, M., Ed.; Academic Press: Oxford, UK, 2009; pp. 274–289. [Google Scholar] [CrossRef]
- Okamoto, K.; Gotoh, N.; Nishino, T. Pseudomonas aeruginosa reveals high intrinsic resistance to penem antibiotics: Penem resistance mechanisms and their interplay. Antimicrob. Agents Chemother. 2001, 45, 1964–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, T.A.; Shang, W.; Bush, K.; Flamm, R.K. Affinity of doripenem and comparators to penicillin-binding proteins in Escherichia coli and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2008, 52, 1510–1512. [Google Scholar] [CrossRef] [Green Version]
- Davies, T.A.; Page, M.G.; Shang, W.; Andrew, T.; Kania, M.; Bush, K. Binding of ceftobiprole and comparators to the penicillin-binding proteins of Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, and Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2007, 51, 2621–2624. [Google Scholar] [CrossRef] [Green Version]
- Fontana, R.; Cornaglia, G.; Ligozzi, M.; Mazzariol, A. The final goal: Penicillin-binding proteins and the target of cephalosporins. Clin. Microbiol. Infect. 2000, 6 (Suppl. S3), 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dik, D.A.; Madukoma, C.S.; Tomoshige, S.; Kim, C.; Lastochkin, E.; Boggess, W.C.; Fisher, J.F.; Shrout, J.D.; Mobashery, S. Slt, MltD, and MltG of Pseudomonas aeruginosa as targets of Bulgecin A in potentiation of β-lactam antibiotics. ACS Chem. Biol. 2019, 14, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Lamers, R.P.; Nguyen, U.T.; Nguyen, Y.; Buensuceso, R.N.C.; Burrows, L.L. Loss of membrane-bound lytic transglycosylases increases outer membrane permeability and β-lactam sensitivity in Pseudomonas aeruginosa. MicrobiologyOpen 2015, 4, 879–895. [Google Scholar] [CrossRef]
- Lee, M.; Batuecas, M.T.; Tomoshige, S.; Domínguez-Gil, T.; Mahasenan, K.V.; Dik, D.A.; Hesek, D.; Millán, C.; Usón, I.; Lastochkin, E. Exolytic and endolytic turnover of peptidoglycan by lytic transglycosylase Slt of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2018, 115, 4393–4398. [Google Scholar] [CrossRef] [Green Version]
- Domenig, C.; Traunmüller, F.; Kozek, S.; Wisser, W.; Klepetko, W.; Steininger, R.; Spiss, C.; Thalhammer, F. Continuous β-lactam antibiotic therapy in a double-lung transplanted patient with a multidrug-resistant Pseudomonas aeruginosa infection. Transplantation 2001, 71, 744–745. [Google Scholar] [CrossRef]
- Moriyama, B.; Henning, S.A.; Childs, R.; Holland, S.M.; Anderson, V.L.; Morris, J.C.; Wilson, W.H.; Drusano, G.L.; Walsh, T.J. High-dose continuous infusion β-lactam antibiotics for the treatment of resistant Pseudomonas aeruginosa infections in immunocompromised patients. Ann. Pharmacother. 2010, 44, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Pang, X.; Wu, X.; Shan, C.; Jiang, S. Clinical outcomes of prolonged infusion (extended infusion or continuous infusion) versus intermittent bolus of meropenem in severe infection: A meta-analysis. PLoS ONE 2018, 13, e0201667. [Google Scholar] [CrossRef]
- Siriyong, T.; Murray, R.M.; Bidgood, L.E.; Young, S.A.; Wright, F.; Parcell, B.J.; Voravuthikunchai, S.P.; Coote, P.J. Dual β-lactam combination therapy for multi-drug resistant Pseudomonas aeruginosa infection: Enhanced efficacy in vivo and comparison with monotherapies of penicillin-binding protein inhibition. Sci. Rep. 2019, 9, 9098. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, S.; Singh, N.B.; Kebriaei, R.; Rice, S.A.; Stamper, K.C.; Castanheira, M.; Rybak, M.J. Evaluation of the synergy of ceftazidime-avibactam in combination with meropenem, amikacin, aztreonam, colistin, or fosfomycin against well-characterized multidrug-resistant Klebsiella pneumoniae and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e00779-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4, 4.2.15. [Google Scholar] [CrossRef] [Green Version]
- Karballaei Mirzahosseini, H.; Hadadi-Fishani, M.; Morshedi, K.; Khaledi, A. Meta-analysis of biofilm formation, antibiotic resistance pattern, and biofilm-related genes in Pseudomonas aeruginosa isolated from clinical samples. Microb. Drug Resist. 2020, 26, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Wardell, S.J.T.; Rehman, A.; Martin, L.W.; Winstanley, C.; Patrick, W.M.; Lamont, I.L. A large-scale whole-genome comparison shows that experimental evolution in response to antibiotics predicts changes in naturally evolved clinical Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e01619-19. [Google Scholar] [CrossRef]
- Yen, P.; Papin, J.A. History of antibiotic adaptation influences microbial evolutionary dynamics during subsequent treatment. PLoS Biol. 2017, 15, e2001586. [Google Scholar] [CrossRef] [Green Version]
- Jorth, P.; McLean, K.; Ratjen, A.; Secor, P.R.; Bautista, G.E.; Ravishankar, S.; Rezayat, A.; Garudathri, J.; Harrison, J.J.; Harwood, R.A. Evolved aztreonam resistance is multifactorial and can produce hypervirulence in Pseudomonas aeruginosa. mBio 2017, 8, e00517-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz-García, F.; Hernando-Amado, S.; Martínez, J.L. Mutation-Driven Evolution of Pseudomonas aeruginosa in the Presence of either Ceftazidime or Ceftazidime-Avibactam. Antimicrob. Agents Chemother. 2018, 62, e01379-18. [Google Scholar] [CrossRef] [Green Version]
- Marvig, R.L.; Sommer, L.M.; Molin, S.; Johansen, H.K. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 2015, 47, 57–64. [Google Scholar] [CrossRef]
- Sumita, Y.; Fakasawa, M. Potent activity of meropenem against Escherichia coli arising from its simultaneous binding to penicillin-binding proteins 2 and 3. J. Antimicrob. Chemother. 1995, 36, 53–64. [Google Scholar] [CrossRef]
- Kocaoglu, O.; Carlson, E.E. Profiling of β-Lactam selectivity for penicillin-binding proteins in Escherichia coli strain DC2. Antimicrob. Agents Chemother. 2015, 59, 2785–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diawara, I.; Nayme, K.; Katfy, K.; Barguigua, A.; Kettani-Halabi, M.; Belabbes, H.; Timinouni, M.; Zerouali, K.; Elmdaghri, N. Analysis of amino acid motif of penicillin-binding proteins 1a, 2b, and 2x in invasive Streptococcus pneumoniae nonsusceptible to penicillin isolated from pediatric patients in Casablanca, Morocco. BMC Res. Notes 2018, 11, 632. [Google Scholar] [CrossRef]
- Cabot, G.; Zamorano, L.; Moya, B.; Juan, C.; Navas, A.; Blazquez, J.; Oliver, A. Evolution of Pseudomonas aeruginosa antimicrobial resistance and fitness under low and high mutation rates. Antimicrob. Agents Chemother. 2016, 60, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanheira, M.; Doyle, T.B.; Smith, C.J.; Mendes, R.E.; Sader, H.S. Combination of MexAB-OprM overexpression and mutations in efflux regulators, PBPs and chaperone proteins is responsible for ceftazidime/avibactam resistance in Pseudomonas aeruginosa clinical isolates from US hospitals. J. Antimicrob. Chemother. 2019, 74, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.T.; Sinha, U.; Zhang, Y.; Wang, P.W.; Donaldson, S.L.; Coburn, B.; Waters, V.J.; Yau, Y.C.W.; Tullis, D.E.; Guttman, D.S.; et al. Penicillin-binding protein 3 is a common adaptive target among Pseudomonas aeruginosa isolates from adult cystic fibrosis patients treated with β-lactams. Int. J. Antimicrob. Agents 2019, 53, 620–628. [Google Scholar] [CrossRef]
- López-Causapé, C.; Sommer, L.M.; Cabot, G.; Rubio, R.; Ocampo-Sosa, A.A.; Johansen, H.K.; Figuerola, J.; Cantón, R.; Kidd, T.J.; Molin, S. Evolution of the Pseudomonas aeruginosa mutational resistome in an international cystic fibrosis clone. Sci. Rep. 2017, 7, 5555. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Nettleship, J.E.; Males, A.; Stuart, D.I.; Owens, R.J. Crystal structures of penicillin-binding protein 3 in complexes with azlocillin and cefoperazone in both acylated and deacylated forms. FEBS Lett. 2016, 590, 288–297. [Google Scholar] [CrossRef] [Green Version]
- Sainsbury, S.; Bird, L.; Rao, V.; Shepherd, S.M.; Stuart, D.I.; Hunter, W.N.; Owens, R.J.; Ren, J. Crystal structures of penicillin-binding protein 3 from Pseudomonas aeruginosa: Comparison of native and antibiotic-bound forms. J. Mol. Biol. 2011, 405, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- del Barrio-Tofiño, E.; López-Causapé, C.; Cabot, G.; Rivera, A.; Benito, N.; Segura, C.; Montero, M.M.; Sorlí, L.; Tubau, F.; Gómez-Zorrilla, S. Genomics and susceptibility profiles of extensively drug-resistant Pseudomonas aeruginosa isolates from Spain. Antimicrob. Agents Chemother. 2017, 61, e01589-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabot, G.; Lopez-Causape, C.; Ocampo-Sosa, A.A.; Sommer, L.M.; Dominguez, M.A.; Zamorano, L.; Juan, C.; Tubau, F.; Rodriguez, C.; Moya, B.; et al. Deciphering the resistome of the widespread Pseudomonas aeruginosa sequence type 175 international high-risk clone through whole-genome sequencing. Antimicrob. Agents Chemother. 2016, 60, 7415–7423. [Google Scholar] [CrossRef] [Green Version]
- Diaz Caballero, J.; Clark, S.T.; Coburn, B.; Zhang, Y.; Wang, P.W.; Donaldson, S.L.; Tullis, D.E.; Yau, Y.C.; Waters, V.J.; Hwang, D.M.; et al. Selective sweeps and parallel pathoadaptation drive Pseudomonas aeruginosa evolution in the cystic fibrosis lung. mBio 2015, 6, e00981-15. [Google Scholar] [CrossRef] [Green Version]
- Jorth, P.; Staudinger, B.J.; Wu, X.; Hisert, K.B.; Hayden, H.; Garudathri, J.; Harding, C.L.; Radey, M.C.; Rezayat, A.; Bautista, G.; et al. Regional isolation drives bacterial diversification within cystic fibrosis lungs. Cell Host Microbe 2015, 18, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Markussen, T.; Marvig, R.L.; Gómez-Lozano, M.; Aanæs, K.; Burleigh, A.E.; Høiby, N.; Johansen, H.K.; Molin, S.; Jelsbak, L. Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. mBio 2014, 5, e01592-14. [Google Scholar] [CrossRef] [Green Version]
- Marvig, R.L.; Johansen, H.K.; Molin, S.; Jelsbak, L. Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 2013, 9, e1003741. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Jelsbak, L.; Marvig, R.L.; Damkiaer, S.; Workman, C.T.; Rau, M.H.; Hansen, S.K.; Folkesson, A.; Johansen, H.K.; Ciofu, O.; et al. Evolutionary dynamics of bacteria in a human host environment. Proc. Natl. Acad. Sci. USA 2011, 108, 7481–7486. [Google Scholar] [CrossRef] [Green Version]
- Chotirmall, S.H.; Sherrard, L.J.; Tai, A.S.; Wee, B.A.; Ramsay, K.A.; Kidd, T.J.; Ben Zakour, N.L.; Whiley, D.M.; Beatson, S.A.; Bell, S.C. Within-host whole genome analysis of an antibiotic resistant Pseudomonas aeruginosa strain sub-type in cystic fibrosis. PLoS ONE 2017, 12, e0172179. [Google Scholar] [CrossRef] [Green Version]
- Colque, C.A.; Albarracin Orio, A.G.; Feliziani, S.; Marvig, R.L.; Tobares, A.R.; Johansen, H.K.; Molin, S.; Smania, A.M. Hypermutator Pseudomonas aeruginosa exploits multiple genetic pathways to develop multidrug resistance during long-term infections in the airways of cystic fibrosis patients. Antimicrob. Agents Chemother. 2020, 64, e02142-19. [Google Scholar] [CrossRef]
- McLean, K.; Lee, D.; Holmes, E.A.; Penewit, K.; Waalkes, A.; Ren, M.; Lee, S.A.; Gasper, J.; Manoil, C.; Salipante, S.J. Genomic analysis identifies novel Pseudomonas aeruginosa resistance genes under selection during inhaled aztreonam therapy in vivo. Antimicrob. Agents Chemother. 2019, 63, e00866-19. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Jonker, M.J.; Moustakas, I.; Brul, S.; Ter Kuile, B.H. Dynamics of Mutations during Development of Resistance by Pseudomonas aeruginosa against Five Antibiotics. Antimicrob. Agents Chemother. 2016, 60, 4229–4236. [Google Scholar] [CrossRef] [Green Version]
- Thegerström, J.; Matuschek, E.; Su, Y.-C.; Riesbeck, K.; Resman, F. A novel PBP3 substitution in Haemophilus influenzae confers reduced aminopenicillin susceptibility. BMC Microbiol. 2018, 18, 1–7. [Google Scholar] [CrossRef]
- Behmard, E.; Ahmadi, A.; Najafi, A. Influence of the T to S mutation at the STMK motif on antibiotic resistance of penicillin binding protein 1A: A comprehensive computational study. J. Mol. Graph. Model. 2019, 87, 185–191. [Google Scholar] [CrossRef]
- Nagai, K.; Davies, T.A.; Jacobs, M.R.; Appelbaum, P.C. Effects of amino acid alterations in penicillin-binding proteins (PBPs) 1a, 2b, and 2x on PBP affinities of penicillin, ampicillin, amoxicillin, cefditoren, cefuroxime, cefprozil, and cefaclor in 18 clinical isolates of penicillin-susceptible, -intermediate, and -resistant pneumococci. Antimicrob. Agents Chemother. 2002, 46, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Giske, C.G.; Buaro, L.; Sundsfjord, A.; Wretlind, B. Alterations of porin, pumps, and penicillin-binding proteins in carbapenem resistant clinical isolates of Pseudomonas aeruginosa. Microb. Drug Resist. 2008, 14, 23–30. [Google Scholar] [CrossRef]
- Li, H.; Luo, Y.-F.; Williams, B.J.; Blackwell, T.S.; Xie, C.-M. Structure and function of OprD protein in Pseudomonas aeruginosa: From antibiotic resistance to novel therapies. Int. J. Med. Microbiol. 2012, 302, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, D.; Talukdar, A.D.; Choudhury, M.D.; Maurya, A.P.; Chanda, D.D.; Chakravorty, A.; Bhattacharjee, A. Carbapenem nonsusceptibility with modified OprD in clinical isolates of Pseudomonas aeruginosa from India. Indian J. Med. Microbiol. 2017, 35, 137–139. [Google Scholar] [CrossRef]
- Shu, J.C.; Kuo, A.J.; Su, L.H.; Liu, T.P.; Lee, M.H.; Su, I.N.; Wu, T.L. Development of carbapenem resistance in Pseudomonas aeruginosa is associated with OprD polymorphisms, particularly the amino acid substitution at codon 170. J. Antimicrob. Chemother. 2017, 72, 2489–2495. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-Y.; Ko, K.S. OprD mutations and inactivation, expression of efflux pumps and AmpC, and metallo-β-lactamases in carbapenem-resistant Pseudomonas aeruginosa isolates from South Korea. Int. J. Antimicrob. Agents 2012, 40, 168–172. [Google Scholar] [CrossRef]
- Kos, V.N.; McLaughlin, R.E.; Gardner, H.A. Identification of unique in-frame deletions in OprD among clinical isolates of Pseudomonas aeruginosa. Pathog. Dis. 2016, 74, ftw031. [Google Scholar] [CrossRef] [Green Version]
- Ochs, M.M.; McCusker, M.P.; Bains, M.; Hancock, R.E.W. Negative regulation of the Pseudomonas aeruginosa outer membrane porin OprD selective for imipenem and basic amino acids. Antimicrob. Agents Chemother. 1999, 43, 1085–1090. [Google Scholar] [CrossRef] [Green Version]
- Iyer, R.; Sylvester, M.A.; Velez-Vega, C.; Tommasi, R.; Durand-Reville, T.F.; Miller, A.A. Whole-cell-based assay to evaluate structure permeation relationships for carbapenem passage through the Pseudomonas aeruginosa porin OprD. ACS Infect. Dis. 2017, 3, 310–319. [Google Scholar] [CrossRef]
- Muramatsu, H.; Horii, T.; Morita, M.; Hashimoto, H.; Kanno, T.; Maekawa, M. Effect of basic amino acids on susceptibility to carbapenems in clinical Pseudomonas aeruginosa isolates. Int. J. Med. Microbiol. 2003, 293, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Skurnik, D.; Roux, D.; Cattoir, V.; Danilchanka, O.; Lu, X.; Yoder-Himes, D.R.; Han, K.; Guillard, T.; Jiang, D.; Gaultier, C.; et al. Enhanced in vivo fitness of carbapenem-resistant oprD mutants of Pseudomonas aeruginosa revealed through high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 20747–20752. [Google Scholar] [CrossRef] [Green Version]
- Ude, J.; Tripathi, V.; Buyck, J.M.; Söderholm, S.; Cunrath, O.; Fanous, J.; Claudi, B.; Egli, A.; Schleberger, C.; Hiller, S.; et al. Outer membrane permeability: Antimicrobials and diverse nutrients bypass porins in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2021, 118, e2107644118. [Google Scholar] [CrossRef]
- Isabella, V.M.; Campbell, A.J.; Manchester, J.; Sylvester, M.; Nayar, A.S.; Ferguson, K.E.; Tommasi, R.; Miller, A.A. Toward the rational design of carbapenem uptake in Pseudomonas aeruginosa. Chem. Biol. 2015, 22, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Tamber, S.; Maier, E.; Benz, R.; Hancock Robert, E.W. Characterization of OpdH, a Pseudomonas aeruginosa Porin Involved in the Uptake of Tricarboxylates. J. Bacteriol. 2007, 189, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Samanta, S.; Bodrenko, I.; Acosta-Gutiérrez, S.; D’Agostino, T.; Pathania, M.; Ghai, I.; Schleberger, C.; Bumann, D.; Wagner, R.; Winterhalter, M.; et al. Getting drugs through small pores: Exploiting the porins pathway in Pseudomonas aeruginosa. ACS Infect. Dis. 2018, 4, 1519–1528. [Google Scholar] [CrossRef] [Green Version]
- Chalhoub, H.; Pletzer, D.; Weingart, H.; Braun, Y.; Tunney, M.M.; Elborn, J.S.; Rodriguez-Villalobos, H.; Plésiat, P.; Kahl, B.C.; Denis, O. Mechanisms of intrinsic resistance and acquired susceptibility of Pseudomonas aeruginosa isolated from cystic fibrosis patients to temocillin, a revived antibiotic. Sci. Rep. 2017, 7, 40208. [Google Scholar] [CrossRef]
- Buyck, J.M.; Guénard, S.; Plésiat, P.; Tulkens, P.M.; Van Bambeke, F. Role of MexAB-OprM in intrinsic resistance of Pseudomonas aeruginosa to temocillin and impact on the susceptibility of strains isolated from patients suffering from cystic fibrosis. J. Antimicrob. Chemother. 2012, 67, 771–775. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Espada, R.; Shahrour, H.; Pitts, B.; Stewart, P.S.; Sánchez-Gómez, S.; Martínez-de-Tejada, G. A permeability-increasing drug synergizes with bacterial efflux pump inhibitors and restores susceptibility to antibiotics in multi-drug resistant Pseudomonas aeruginosa strains. Sci. Rep. 2019, 9, 3452. [Google Scholar] [CrossRef] [Green Version]
- Fernando, D.M.; Kumar, A. Resistance-nodulation-division multidrug efflux pumps in gram-negative bacteria: Role in virulence. Antibiotics 2013, 2, 163–181. [Google Scholar] [CrossRef] [Green Version]
- Köhler, T.; Michea-Hamzehpour, M.; Plesiat, P.; Kahr, A.L.; Pechere, J.C. Differential selection of multidrug efflux systems by quinolones in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1997, 41, 2540–2543. [Google Scholar] [CrossRef] [Green Version]
- Guénard, S.; Muller, C.; Monlezun, L.; Benas, P.; Broutin, I.; Jeannot, K.; Plésiat, P. Multiple mutations lead to MexXY-OprM-dependent aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llanes, C.; Köhler, T.; Patry, I.; Dehecq, B.; van Delden, C.; Plésiat, P. Role of the MexEF-OprN efflux system in low-level resistance of Pseudomonas aeruginosa to ciprofloxacin. Antimicrob. Agents Chemother. 2011, 55, 5676–5684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-Z.; Zhang, L.; Poole, K. Interplay between the MexA-MexB-OprM multidrug efflux system and the outer membrane barrier in the multiple antibiotic resistance of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2000, 45, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Masuda, N.; Sakagawa, E.; Ohya, S.; Gotoh, N.; Tsujimoto, H.; Nishino, T. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3322–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayoub Moubareck, C.; Hammoudi Halat, D.; Akkawi, C.; Nabi, A.; AlSharhan, M.A.; AlDeesi, Z.O.; Peters, C.C.; Celiloglu, H.; Karam Sarkis, D. Role of outer membrane permeability, efflux mechanism, and carbapenemases in carbapenem-nonsusceptible Pseudomonas aeruginosa from Dubai hospitals: Results of the first cross-sectional survey. Int. J. Infect. Dis. 2019, 84, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, D.; Das Talukdar, A.; Dutta Choudhury, M.; Maurya, A.P.; Paul, D.; Dhar Chanda, D.; Chakravorty, A.; Bhattacharjee, A. Transcriptional analysis of MexAB-OprM efflux pumps system of Pseudomonas aeruginosa and its role in carbapenem resistance in a tertiary referral hospital in India. PLoS ONE 2015, 10, e0133842. [Google Scholar] [CrossRef] [PubMed]
- Köhler, T.; Michéa-Hamzehpour, M.; Henze, U.; Gotoh, N.; Kocjancic Curty, L.; Pechère, J.-C. Characterization of MexE–MexF–OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 1997, 23, 345–354. [Google Scholar] [CrossRef]
- Moyá, B.; Beceiro, A.; Cabot, G.; Juan, C.; Zamorano, L.; Alberti, S.; Oliver, A. Pan-β-lactam resistance development in Pseudomonas aeruginosa clinical strains: Molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob. Agents Chemother. 2012, 56, 4771–4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Xu, Y.; Wang, Z.; Fang, Y.; Shen, J. Overexpression of MexAB-OprM efflux pump in carbapenem-resistant Pseudomonas aeruginosa. Arch. Microbiol. 2016, 198, 565–571. [Google Scholar] [CrossRef]
- Aires, J.R.; Köhler, T.; Nikaido, H.; Plésiat, P. Involvement of an active efflux system in the natural resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 1999, 43, 2624–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, Y.; Tomida, J.; Kawamura, Y. MexXY multidrug efflux system of Pseudomonas aeruginosa. Front. Microbiol. 2012, 3, 408. [Google Scholar] [CrossRef] [Green Version]
- Köhler, T.; Michea-Hamzehpour, M.; Epp, S.F.; Pechere, J.C. Carbapenem activities against Pseudomonas aeruginosa: Respective contributions of OprD and efflux systems. Antimicrob. Agents Chemother. 1999, 43, 424–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobel Mara, L.; Neshat, S.; Poole, K. Mutations in PA2491 (mexS) promote MexT-dependent mexEF-oprN expression and multidrug resistance in a clinical Strain of Pseudomonas aeruginosa. J. Bacteriol. 2005, 187, 1246–1253. [Google Scholar] [CrossRef] [Green Version]
- Köhler, T.; Epp, S.F.; Curty, L.K.; Pechère, J.C. Characterization of MexT, the regulator of the MexE-MexF-OprN multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 6300–6305. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Yoneyama, H.; Nakae, T. nalB-type mutations causing the overexpression of the MexAB-OprM efflux pump are located in the mexR gene of the Pseudomonas aeruginosa chromosome. FEMS Microbiol. Lett. 1999, 179, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Sobel, M.L.; Hocquet, D.; Cao, L.; Plesiat, P.; Poole, K. Mutations in PA3574 (nalD) lead to increased MexAB-OprM expression and multidrug resistance in laboratory and clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2005, 49, 1782–1786. [Google Scholar] [CrossRef] [Green Version]
- Morita, Y.; Cao, L.; Gould, V.C.; Avison, M.B.; Poole, K. nalD encodes a second repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 8649–8654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Yi, C.; Zhang, J.; Zhang, W.; Ge, Z.; Yang, C.G.; He, C. Structural insight into the oxidation-sensing mechanism of the antibiotic resistance of regulator MexR. EMBO Rep. 2010, 11, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.; Adewoye, L.; Poole, K. MexR repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa identification of MexR binding sites in the mexA-mexR intergenic region. J. Bacteriol. 2001, 183, 807–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Akama, H.; Yoshihara, E.; Nakae, T. Mutations affecting DNA-binding activity of the MexR repressor of mexR-mexA-mexB-oprM operon expression. J. Bacteriol. 2003, 185, 6195–6198. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Srikumar, R.; Poole, K. MexAB-OprM hyperexpression in NalC-type multidrug-resistant Pseudomonas aeruginosa: Identification and characterization of the nalC gene encoding a repressor of PA3720-PA3719. Mol. Microbiol. 2004, 53, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Eda, S.; Gotoh, N.; Yoshihara, E.; Nakae, T. MexZ-mediated regulation of mexXY multidrug efflux pump expression in Pseudomonas aeruginosa by binding on the mexZ-mexX intergenic DNA. FEMS Microbiol. Lett. 2004, 238, 23–28. [Google Scholar] [CrossRef]
- Masuda, N.; Sakagawa, E.; Ohya, S.; Gotoh, N.; Tsujimoto, H.; Nishino, T. Contribution of the MexX-MexY-oprM efflux system to intrinsic resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 2242–2246. [Google Scholar] [CrossRef] [Green Version]
- Frimodt-Møller, J.; Rossi, E.; Haagensen, J.A.J.; Falcone, M.; Molin, S.; Johansen, H.K. Mutations causing low level antibiotic resistance ensure bacterial survival in antibiotic-treated hosts. Sci. Rep. 2018, 8, 12512. [Google Scholar] [CrossRef] [Green Version]
- Seupt, A.; Schniederjans, M.; Tomasch, J.; Haussler, S. Expression of the MexXY aminoglycoside efflux pump and presence of an aminoglycoside-modifying enzyme in clinical Pseudomonas aeruginosa isolates are highly correlated. Antimicrob. Agents Chemother. 2020, 65, e01166-20. [Google Scholar] [CrossRef]
- Gotoh, N.; Tsujimoto, H.; Tsuda, M.; Okamoto, K.; Nomura, A.; Wada, T.; Nakahashi, M.; Nishino, T. Characterization of the MexC-MexD-OprJ multidrug efflux system in ΔmexA-mexB-oprM mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1998, 42, 1938–1943. [Google Scholar] [CrossRef] [Green Version]
- Fraud, S.; Campigotto, A.J.; Chen, Z.; Poole, K. MexCD-OprJ multidrug efflux system of Pseudomonas aeruginosa: Involvement in chlorhexidine resistance and induction by membrane-damaging agents dependent upon the AlgU stress response sigma factor. Antimicrob. Agents Chemother. 2008, 52, 4478–4482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purssell, A.; Poole, K. Functional characterization of the NfxB repressor of the mexCD–oprJ multidrug efflux operon of Pseudomonas aeruginosa. Microbiology 2013, 159, 2058–2073. [Google Scholar] [CrossRef] [Green Version]
- Higgins, P.G.; Fluit, A.C.; Milatovic, D.; Verhoef, J.; Schmitz, F.J. Mutations in GyrA, ParC, MexR and NfxB in clinical isolates of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2003, 21, 409–413. [Google Scholar] [CrossRef]
- Gomis-Font, M.A.; Pitart, C.; Del Barrio-Tofiño, E.; Zboromyrska, Y.; Cortes-Lara, S.; Mulet, X.; Marco, F.; Vila, J.; López-Causapé, C.; Oliver, A. Emergence of resistance to novel cephalosporin-β-lactamase inhibitor combinations through the modification of the Pseudomonas aeruginosa MexCD-OprJ efflux pump. Antimicrob. Agents Chemother. 2021, 65, e0008921. [Google Scholar] [CrossRef]
- Köhler, T.; van Delden, C.; Curty, L.K.; Hamzehpour, M.M.; Pechere, J.C. Overexpression of the MexEF-OprN multidrug efflux system affects cell-to-cell signaling in Pseudomonas aeruginosa. J. Bacteriol. 2001, 183, 5213–5222. [Google Scholar] [CrossRef] [Green Version]
- Maseda, H.; Yoneyama, H.; Nakae, T. Assignment of the substrate-selective subunits of the MexEF-OprN multidrug efflux pump of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 658–664. [Google Scholar] [CrossRef] [Green Version]
- Maseda, H.; Uwate, M.; Nakae, T. Transcriptional regulation of the mexEF-oprN multidrug efflux pump operon by MexT and an unidentified repressor in nfxC-type mutant of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2010, 311, 36–43. [Google Scholar] [CrossRef]
- Jeannot, K.; Elsen, S.; Köhler, T.; Attree, I.; Delden, C.v.; Plésiat, P. Resistance and virulence of Pseudomonas aeruginosa clinical strains overproducing the MexCD-OprJ efflux pump. Antimicrob. Agents Chemother. 2008, 52, 2455–2462. [Google Scholar] [CrossRef] [Green Version]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-lactamases and β-lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Boyd, S.E.; Livermore, D.M.; Hooper, D.C.; Hope, W.W. Metallo-β-lactamases: Structure, function, epidemiology, treatment options, and the development pipeline. Antimicrob. Agents Chemother. 2020, 64, e00397-20. [Google Scholar] [CrossRef] [PubMed]
- Palzkill, T. Metallo-β-lactamase structure and function. Ann. N. Y. Acad. Sci. 2013, 1277, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Queenan, A.M.; Bush, K. Carbapenemases: The versatile β-lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458. [Google Scholar] [CrossRef] [Green Version]
- Adam, M.A.; Elhag, W.I. Prevalence of metallo-β-lactamase acquired genes among carbapenems susceptible and resistant Gram-negative clinical isolates using multiplex PCR, Khartoum hospitals, Khartoum Sudan. BMC Infect. Dis. 2018, 18, 668. [Google Scholar] [CrossRef]
- Meletis, G. Carbapenem resistance: Overview of the problem and future perspectives. Ther. Adv. Infect. Dis. 2016, 3, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintos-Pascual, I.; Cantero-Caballero, M.; Munez Rubio, E.; Sanchez-Romero, I.; Asensio-Vegas, A.; Ramos-Martinez, A. Epidemiology and clinical of infections and colonizations caused by Enterobacterales producing carbapenemases in a tertiary hospital. Rev. Española Quimioter. 2020, 33, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Betancur, J.C.; Appel, T.M.; Esparza, G.; Gales, A.C.; Levy-Hara, G.; Cornistein, W.; Vega, S.; Nunez, D.; Cuellar, L.; Bavestrello, L.; et al. Update on the epidemiology of carbapenemases in Latin America and the Caribbean. Expert Rev. Anti-Infect. Ther. 2021, 19, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Fuster, B.; Tormo, N.; Salvador, C.; Gimeno, C. Detection of two simultaneous outbreaks of Klebsiella pneumoniae coproducing OXA-48 and NDM-1 carbapenemases in a tertiary-care hospital in Valencia, Spain. New Microbes New Infect. 2020, 34, 100660. [Google Scholar] [CrossRef] [PubMed]
- Kollenda, H.; Frickmann, H.; Ben Helal, R.; Wiemer, D.F.; Naija, H.; El Asli, M.S.; Egold, M.; Bugert, J.J.; Handrick, S.; Wolfel, R.; et al. Screening for Carbapenemases in Ertapenem-Resistant Enterobacteriaceae Collected at a Tunisian Hospital Between 2014 and 2018. Eur. J. Microbiol. Immunol. 2019, 9, 9–13. [Google Scholar] [CrossRef]
- Marathe, N.P.; Berglund, F.; Razavi, M.; Pal, C.; Droge, J.; Samant, S.; Kristiansson, E.; Larsson, D.G.J. Sewage effluent from an Indian hospital harbors novel carbapenemases and integron-borne antibiotic resistance genes. Microbiome 2019, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Hammoudi Halat, D.; Ayoub Moubareck, C. The current burden of carbapenemases: Review of significant properties and dissemination among gram-negative bacteria. Antibiotics 2020, 9, 186. [Google Scholar] [CrossRef]
- Lahiri, S.D.; Johnstone, M.R.; Ross, P.L.; McLaughlin, R.E.; Olivier, N.B.; Alm, R.A. Avibactam and class C β-lactamases: Mechanism of inhibition, conservation of the binding pocket, and implications for resistance. Antimicrob. Agents Chemother. 2014, 58, 5704–5713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.P.; Saraiva, L.; Pinto, M.; Sousa, M.E. Boronic acids and their derivatives in medicinal chemistry: Synthesis and biological applications. Molecules 2020, 25, 4323. [Google Scholar] [CrossRef]
- Rodríguez, D.; Maneiro, M.; Vázquez-Ucha, J.C.; Beceiro, A.; González-Bello, C. 6-arylmethylidene penicillin-based sulfone inhibitors for repurposing antibiotic efficiency in priority pathogens. J. Med. Chem. 2020, 63, 3737–3755. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bethel, C.R.; Doppalapudi, V.R.; Sheri, A.; Pagadala, S.R.; Hujer, A.M.; Skalweit, M.J.; Anderson, V.E.; Chen, S.G.; Buynak, J.D.; et al. Penicillin sulfone inhibitors of class D β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 1414–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, L.C.; Cheng, Z.; Fast, W.; Bonomo, R.A.; Crowder, M.W. The continuing challenge of metallo-β-lactamase inhibition: Mechanism matters. Trends Pharmacol. Sci. 2018, 39, 635–647. [Google Scholar] [CrossRef]
- Poole, K. Pseudomonas aeruginosa: Resistance to the max. Front. Microbiol. 2011, 2, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Gallego, M.; Torrens, G.; Castillo-Vera, J.; Moya, B.; Zamorano, L.; Cabot, G.; Hultenby, K.; Albertí, S.; Mellroth, P.; Henriques-Normark, B. Impact of AmpC derepression on fitness and virulence: The mechanism or the pathway? mBio 2016, 7, e01783-16. [Google Scholar] [CrossRef] [Green Version]
- Rafiee, R.; Eftekhar, F.; Tabatabaei, S.A.; Minaee Tehrani, D. Prevalence of extended-spectrum and metallo β-lactamase production in AmpC β-lactamase producing Pseudomonas aeruginosa isolates from burns. Jundishapur J. Microbiol. 2014, 7, e16436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, J.F.; Mobashery, S. Constructing and deconstructing the bacterial cell wall. Protein Sci. 2020, 29, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Vadlamani, G.; Thomas, M.D.; Patel, T.R.; Donald, L.J.; Reeve, T.M.; Stetefeld, J.; Standing, K.G.; Vocadlo, D.J.; Mark, B.L. The β-lactamase gene regulator AmpR is a tetramer that recognizes and binds the d-Ala-d-Ala motif of its repressor UDP-N-acetylmuramic ccid (MurNAc)-pentapeptide. J. Biol. Chem. 2015, 290, 2630–2643. [Google Scholar] [CrossRef] [Green Version]
- Dik, D.A.; Domínguez-Gil, T.; Lee, M.; Hesek, D.; Byun, B.; Fishovitz, J.; Boggess, B.; Hellman, L.M.; Fisher, J.F.; Hermoso, J.A.; et al. Muropeptide Binding and the X-ray Structure of the Effector Domain of the Transcriptional Regulator AmpR of Pseudomonas aeruginosa. J. Am. Chem. Soc. 2017, 139, 1448–1451. [Google Scholar] [CrossRef] [Green Version]
- Juan, C.; Macia, M.D.; Gutierrez, O.; Vidal, C.; Perez, J.L.; Oliver, A. Molecular mechanisms of β-lactam resistance mediated by AmpC hyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob. Agents Chemother. 2005, 49, 4733–4738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtke, A.J.; Hanson, N.D. Role of ampD homologs in overproduction of AmpC in clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2008, 52, 3922–3927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, K.F.; Jayawardena, S.R.; Indulkar, S.D.; Del Puerto, A.; Koh, C.L.; Hoiby, N.; Mathee, K. Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB β-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob. Agents Chemother. 2005, 49, 4567–4575. [Google Scholar] [CrossRef] [Green Version]
- Caille, O.; Zincke, D.; Merighi, M.; Balasubramanian, D.; Kumari, H.; Kong, K.-F.; Silva-Herzog, E.; Narasimhan, G.; Schneper, L.; Lory, S.; et al. Structural and functional characterization of Pseudomonas aeruginosa global regulator AmpR. J. Bacteriol. 2014, 196, 3890–3902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domitrovic, T.N.; Hujer, A.M.; Perez, F.; Marshall, S.H.; Hujer, K.M.; Woc-Colburn, L.E.; Parta, M.; Bonomo, R.A. Multidrug resistant Pseudomonas aeruginosa causing prosthetic valve endocarditis: A genetic-based chronicle of evolving antibiotic resistance. Open Forum Infect Dis. 2016, 3, ofw188. [Google Scholar] [CrossRef] [Green Version]
- Bagge, N.; Ciofu, O.; Hentzer, M.; Campbell, J.I.; Givskov, M.; Hoiby, N. Constitutive high expression of chromosomal β-lactamase in Pseudomonas aeruginosa caused by a new insertion sequence (IS1669) located in ampD. Antimicrob. Agents Chemother. 2002, 46, 3406–3411. [Google Scholar] [CrossRef] [Green Version]
- Moya, B.; Dötsch, A.; Juan, C.; Blázquez, J.; Zamorano, L.; Haussler, S.; Oliver, A. β-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 2009, 5, e1000353. [Google Scholar] [CrossRef] [Green Version]
- Zamorano, L.; Moya, B.; Juan, C.; Mulet, X.; Blazquez, J.; Oliver, A. The Pseudomonas aeruginosa CreBC two-component system plays a major role in the response to β-lactams, fitness, biofilm growth, and global regulation. Antimicrob. Agents Chemother. 2014, 58, 5084–5095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simner, P.J.; Beisken, S.; Bergman, Y.; Posch, A.E.; Cosgrove, S.E.; Tamma, P.D. Cefiderocol activity against clinical Pseudomonas aeruginosa isolates exhibiting ceftolozane-tazobactam resistance. Open Forum Infect Disease. 2021, 8, ofab311. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.H.; Schilling, A.N.; LaRocco, M.T.; Gentry, L.O.; Lolans, K.; Quinn, J.P.; Garey, K.W. Prevalence of AmpC over-expression in bloodstream isolates of Pseudomonas aeruginosa. Clin. Microbiol. Infect. 2007, 13, 413–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slater, C.L.; Winogrodzki, J.; Fraile-Ribot, P.A.; Oliver, A.; Khajehpour, M.; Mark, B.L. Adding insult to injury: Mechanistic basis for how AmpC mutations allow Pseudomonas aeruginosa to accelerate cephalosporin hydrolysis and evade avibactam. Antimicrob. Agents Chemother. 2020, 64, e00894-20. [Google Scholar] [CrossRef]
- Berrazeg, M.; Jeannot, K.; Ntsogo Enguene, V.Y.; Broutin, I.; Loeffert, S.; Fournier, D.; Plesiat, P. Mutations in β-lactamase AmpC increase resistance of Pseudomonas aeruginosa isolates to antipseudomonal cephalosporins. Antimicrob. Agents Chemother. 2015, 59, 6248–6255. [Google Scholar] [CrossRef] [Green Version]
- Compain, F.; Debray, A.; Adjadj, P.; Dorchene, D.; Arthur, M. Ceftazidime-avibactam resistance mediated by the N346Y substitution in various AmpC β-lactamases. Antimicrob. Agents Chemother. 2020, 64, e02311-19. [Google Scholar] [CrossRef]
- Fernandez-Esgueva, M.; Lopez-Calleja, A.I.; Mulet, X.; Fraile-Ribot, P.A.; Cabot, G.; Huarte, R.; Rezusta, A.; Oliver, A. Characterization of AmpC β-lactamase mutations of extensively drug-resistant Pseudomonas aeruginosa isolates that develop resistance to ceftolozane/tazobactam during therapy. Enferm. Infecc. Microbiol. Clínica 2020, 38, 474–478. [Google Scholar] [CrossRef]
- Fajardo, A.; Hernando-Amado, S.; Oliver, A.; Ball, G.; Filloux, A.; Martinez, J.L. Characterization of a novel Zn(2)(+)-dependent intrinsic imipenemase from Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2014, 69, 2972–2978. [Google Scholar] [CrossRef] [PubMed]
- Girlich, D.; Naas, T.; Nordmann, P. Biochemical characterization of the naturally occurring oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2004, 48, 2043–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zincke, D.; Balasubramanian, D.; Silver, L.L.; Mathee, K. Characterization of a Carbapenem-Hydrolyzing Enzyme, PoxB, in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 2016, 60, 936–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahiri, S.D.; Mangani, S.; Durand-Reville, T.; Benvenuti, M.; Luca, F.D.; Sanyal, G.; Docquier, J.-D. Structural Insight into Potent Broad-Spectrum Inhibition with Reversible Recyclization Mechanism: Avibactam in Complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-Lactamases. Antimicrob. Agents Chemother. 2013, 57, 2496–2505. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Iyobe, S.; Inoue, M.; Mitsuhashi, S. Transferable imipenem resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1991, 35, 147–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dortet, L.; Flonta, M.; Boudehen, Y.M.; Creton, E.; Bernabeu, S.; Vogel, A.; Naas, T. Dissemination of carbapenemase-producing Enterobacteriaceae and Pseudomonas aeruginosa in Romania. Antimicrob. Agents Chemother. 2015, 59, 7100–7103. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.A.; Amyes, S.G. OXA β-lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehbashi, S.; Tahmasebi, H.; Alikhani, M.Y.; Keramat, F.; Arabestani, M.R. Distribution of Class B and Class A β-Lactamases in Clinical Strains of Pseudomonas aeruginosa: Comparison of Phenotypic Methods and High-Resolution Melting Analysis (HRMA) Assay. Infect. Drug Resist. 2020, 13, 2037–2052. [Google Scholar] [CrossRef]
- De Champs, C.; Chanal, C.; Sirot, D.; Baraduc, R.; Romaszko, J.P.; Bonnet, R.; Plaidy, A.; Boyer, M.; Carroy, E.; Gbadamassi, M.C.; et al. Frequency and diversity of Class A extended-spectrum β-lactamases in hospitals of the Auvergne, France: A 2 year prospective study. J. Antimicrob. Chemother. 2004, 54, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Schauer, J.; Gatermann, S.G.; Hoffmann, D.; Hupfeld, L.; Pfennigwerth, N. GPC-1, a novel class A carbapenemase detected in a clinical Pseudomonas aeruginosa isolate. J. Antimicrob. Chemother. 2020, 75, 911–916. [Google Scholar] [CrossRef]
- Zhao, W.-H.; Hu, Z.-Q. β-Lactamases identified in clinical isolates of Pseudomonas aeruginosa. Crit. Rev. Microbiol. 2010, 36, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Bradford, P.A. Epidemiology of β-Lactamase-Producing Pathogens. Clin. Microbiol. Rev. 2020, 33, e00047-19. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X. Prevalence of metallo-β-lactamase genes among Pseudomonas aeruginosa isolated from various clinical samples in China. J. Lab. Med. 2020, 44, 197–203. [Google Scholar] [CrossRef]
- Philippon, A.; Arlet, G.; Jacoby, G.A. Plasmid-determined AmpC-type β-lactamases. Antimicrob. Agents Chemother. 2002, 46, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Doi, Y.; Paterson, D.L. Detection of plasmid-mediated class C β-lactamases. Int. J. Infect. Dis. 2007, 11, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraile-Ribot, P.A.; Del Rosario-Quintana, C.; López-Causapé, C.; Gomis-Font, M.A.; Ojeda-Vargas, M.; Oliver, A. Emergence of resistance to novel β-lactam-β-lactamase inhibitor combinations due to horizontally acquired AmpC (FOX-4) in Pseudomonas aeruginosa sequence type 308. Antimicrob. Agents Chemother. 2019, 64, e02112-19. [Google Scholar] [CrossRef]
- Upadhyay, S.; Mishra, S.; Sen, M.R.; Banerjee, T.; Bhattacharjee, A. Co-existence of Pseudomonas-derived cephalosporinase among plasmid encoded CMY-2 harbouring isolates of Pseudomonas aeruginosa in north India. Indian J. Med Microbiol. 2013, 31, 257–260. [Google Scholar] [CrossRef]
- Antunes, N.T.; Fisher, J.F. Acquired Class D β-lactamases. Antibiotics 2014, 3, 398–434. [Google Scholar] [CrossRef]
- Nasser, M.; Gayen, S.; Kharat, A.S. Prevalence of β-lactamase and antibiotic-resistant Pseudomonas aeruginosa in the Arab region. J. Glob. Antimicrob. Resist. 2020, 22, 152–160. [Google Scholar] [CrossRef]
- Amirkamali, S.; Naserpour-Farivar, T.; Azarhoosh, K.; Peymani, A. Distribution of the bla OXA, bla VEB-1, and bla GES-1 genes and resistance patterns of ESBL-producing Pseudomonas aeruginosa isolated from hospitals in Tehran and Qazvin, Iran. Rev. Soc. Bras. Med. Trop. 2017, 50, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Esenkaya Taşbent, F.; Özdemir, M. The presence of OXA type carbapenemases in Pseudomonas strains: First report from Turkey. Mikrobiyoloji Bul. 2015, 49, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, E.-J.; Jeong, S.H. Mobile carbapenemase genes in Pseudomonas aeruginosa. Front. Microbiol. 2021, 12, 30. [Google Scholar] [CrossRef]
- Perez-Vazquez, M.; Sola-Campoy, P.J.; Zurita, Á.M.; Avila, A.; Gomez-Bertomeu, F.; Solis, S.; Lopez-Urrutia, L.; GÓnzalez-BarberÁ, E.M.; Cercenado, E.; Bautista, V. Carbapenemase-producing Pseudomonas aeruginosa in Spain: Interregional dissemination of the high-risk clones ST175 and ST244 carrying blaVIM-2, blaVIM-1, blaIMP-8, blaVIM-20 and blaKPC-2. Int. J. Antimicrob. Agents 2020, 56, 106026. [Google Scholar] [CrossRef]
- Bonnin, R.A.; Bogaerts, P.; Girlich, D.; Huang, T.-D.; Dortet, L.; Glupczynski, Y.; Naas, T. Molecular characterization of OXA-198 carbapenemase-producing Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 2018, 62, e02496-17. [Google Scholar] [CrossRef] [Green Version]
- Botelho, J.; Grosso, F.; Peixe, L. Unravelling the genome of a Pseudomonas aeruginosa isolate belonging to the high-risk clone ST235 reveals an integrative conjugative element housing a blaGES-6 carbapenemase. J. Antimicrob. Chemother. 2018, 73, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Botelho, J.; Schulenburg, H. The role of integrative and conjugative elements in antibiotic resistance evolution. Trends Microbiol. 2021, 29, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Gniadkowski, M.; Giske, C.G.; Poirel, L.; Woodford, N.; Miriagou, V.; European Network on, C. Identification and screening of carbapenemase-producing Enterobacteriaceae. Clin. Microbiol. Infect. 2012, 18, 432–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Yang, H.; Li, J.; Chen, F.; Hu, L.; Jing, Y.; Luo, X.; Yin, Z.; Zou, M.; Zhou, D. Novel chromosome-borne accessory genetic elements carrying multiple antibiotic resistance genes in Pseudomonas aeruginosa. Front. Cell. Infect. Microbiol. 2021, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Botelho, J.; Roberts, A.P.; León-Sampedro, R.; Grosso, F.; Peixe, L. Carbapenemases on the move: It’s good to be on ICEs. Mob. DNA 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Domingues, S.; da Silva, G.J.; Nielsen, K.M. Integrons: Vehicles and pathways for horizontal dissemination in bacteria. Mob. Genet. Elem. 2012, 2, 211–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Ma, J.; Jia, W.; Li, W. Antimicrobial resistance and molecular characterization of gene cassettes from class 1 integrons in Pseudomonas aeruginosa strains. Microb. Drug Resist. 2020, 26, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, L.; Haghshenas, M.R.; Mirzaei, B.; Norouzi Bazgir, Z.; Goli, H.R. Distribution and molecular characterization of resistance gene cassettes containing class 1 integrons in multi-drug resistant (MDR) clinical isolates of Pseudomonas aeruginosa. Infect. Drug Resist. 2020, 13, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Jamal, A.J.; Mataseje, L.F.; Brown, K.A.; Katz, K.; Johnstone, J.; Muller, M.P.; Allen, V.G.; Borgia, S.; Boyd, D.A.; Ciccotelli, W.; et al. Carbapenemase-producing Enterobacterales in hospital drains in Southern Ontario, Canada. J. Hosp. Infect. 2020, 106, 820–827. [Google Scholar] [CrossRef]
- Park, S.C.; Parikh, H.; Vegesana, K.; Stoesser, N.; Barry, K.E.; Kotay, S.M.; Dudley, S.; Peto, T.E.A.; Crook, D.W.; Walker, A.S.; et al. Risk factors associated with carbapenemase-producing enterobacterales (CPE) positivity in the hospital wastewater environment. Appl. Environ. Microbiol. 2020, 86, e01715-20. [Google Scholar] [CrossRef] [PubMed]
- Snitkin, E.S. Contamination of Hospital Plumbing: A Source or a Sink for Antibiotic-Resistant Organisms? JAMA Netw. Open 2019, 2, e187660. [Google Scholar] [CrossRef]
- Cahill, N.; O’Connor, L.; Mahon, B.; Varley, A.; McGrath, E.; Ryan, P.; Cormican, M.; Brehony, C.; Jolley, K.A.; Maiden, M.C.; et al. Hospital effluent: A reservoir for carbapenemase-producing Enterobacterales? Sci. Total Environ. 2019, 672, 618–624. [Google Scholar] [CrossRef]
- Cherak, Z.; Loucif, L.; Moussi, A.; Rolain, J.-M. Carbapenemase-producing Gram-negative bacteria in aquatic environments: A review. J. Glob. Antimicrob. Resist. 2021, 25, 287–309. [Google Scholar] [CrossRef] [PubMed]
- Bleichenbacher, S.; Stevens, M.J.A.; Zurfluh, K.; Perreten, V.; Endimiani, A.; Stephan, R.; Nüesch-Inderbinen, M. Environmental dissemination of carbapenemase-producing Enterobacteriaceae in rivers in Switzerland. Environ. Pollut. 2020, 265, 115081. [Google Scholar] [CrossRef]
- Colosi, I.A.; Baciu, A.M.; Opris, R.V.; Peca, L.; Gudat, T.; Simon, L.M.; Colosi, H.A.; Costache, C. Prevalence of ESBL, AmpC and carbapenemase-producing enterobacterales isolated from raw vegetables retailed in Romania. Foods 2020, 9, 1726. [Google Scholar] [CrossRef]
- Borlee, B.R.; Goldman, A.D.; Murakami, K.; Samudrala, R.; Wozniak, D.J.; Parsek, M.R. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol. Microbiol. 2010, 75, 827–842. [Google Scholar] [CrossRef] [Green Version]
- Gloag, E.S.; Marshall, C.W.; Snyder, D.; Lewin, G.R.; Harris, J.S.; Santos-Lopez, A.; Chaney, S.B.; Whiteley, M.; Cooper, V.S.; Wozniak, D.J. Pseudomonas aeruginosa Interstrain Dynamics and Selection of Hyperbiofilm Mutants during a Chronic Infection. mBio 2019, 10, e01698-19. [Google Scholar] [CrossRef] [Green Version]
- Kovach, K.; Davis-Fields, M.; Irie, Y.; Jain, K.; Doorwar, S.; Vuong, K.; Dhamani, N.; Mohanty, K.; Touhami, A.; Gordon, V.D. Evolutionary adaptations of biofilms infecting cystic fibrosis lungs promote mechanical toughness by adjusting polysaccharide production. npj Biofilms Microbiomes 2017, 3, 1–9. [Google Scholar] [CrossRef]
- Starkey, M.; Hickman, J.H.; Ma, L.; Zhang, N.; De Long, S.; Hinz, A.; Palacios, S.; Manoil, C.; Kirisits, M.J.; Starner, T.D.; et al. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J. Bacteriol. 2009, 191, 3492–3503. [Google Scholar] [CrossRef] [Green Version]
- Hickman, J.W.; Tifrea, D.F.; Harwood, C.S. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. USA 2005, 102, 14422–14427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, X.; Pan, Y.; Cai, Z.; Liu, Y.; Zhang, Y.; Liu, M.; Liu, Y.; Wang, K.; Zhang, L.; Yang, L. rpoS-mutation variants are selected in Pseudomonas aeruginosa biofilms under imipenem pressure. Cell Biosci. 2021, 11, 138. [Google Scholar] [CrossRef]
- Cochran, W.L.; Suh, S.J.; McFeters, G.A.; Stewart, P.S. Role of RpoS and AlgT in Pseudomonas aeruginosa biofilm resistance to hydrogen peroxide and monochloramine. J. Appl. Microbiol. 2000, 88, 546–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, L.; Álvarez-Ortega, C.; Wiegand, I.; Olivares, J.; Kocíncová, D.; Lam, J.S.; Martínez, J.L.; Hancock, R.E.W. Characterization of the polymyxin B resistome of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Groote, V.N.; Verstraeten, N.; Fauvart, M.; Kint, C.I.; Verbeeck, A.M.; Beullens, S.; Cornelis, P.; Michiels, J. Novel persistence genes in Pseudomonas aeruginosa identified by high-throughput screening. FEMS Microbiol. Lett. 2009, 297, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Wright, R.C.T.; Friman, V.-P.; Smith, M.C.M.; Brockhurst, M.A. Resistance evolution against phage combinations depends on the timing and order of exposure. mBio 2019, 10, e01652-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rosa, R.; Rossi, E.; Feist, A.M.; Johansen, H.K.; Molin, S. Compensatory evolution of Pseudomonas aeruginosa’s slow growth phenotype suggests mechanisms of adaptation in cystic fibrosis. Nat. Commun. 2021, 12, 3186. [Google Scholar] [CrossRef]
- Lopatkin Allison, J.; Bening Sarah, C.; Manson Abigail, L.; Stokes Jonathan, M.; Kohanski Michael, A.; Badran Ahmed, H.; Earl Ashlee, M.; Cheney Nicole, J.; Yang Jason, H.; Collins James, J. Clinically relevant mutations in core metabolic genes confer antibiotic resistance. Science 2021, 371, eaba0862. [Google Scholar] [CrossRef]
- Malone, J.G. Role of small colony variants in persistence of Pseudomonas aeruginosa infections in cystic fibrosis lungs. Infect. Drug Resist. 2015, 8, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Al Ahmar, R.; Kirby, B.D.; Yu, H.D. Pyrimidine biosynthesis regulates the small-colony variant and mucoidy in Pseudomonas aeruginosa through sigma factor competition. J. Bacteriol. 2019, 201, e00575-18. [Google Scholar] [CrossRef] [Green Version]
- Jeukens, J.; Freschi, L.; Kukavica-Ibrulj, I.; Emond-Rheault, J.-G.; Tucker, N.P.; Levesque, R.C. Genomics of antibiotic-resistance prediction in Pseudomonas aeruginosa. Ann. N. Y. Acad. Sci. 2019, 1435, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaledi, A.; Weimann, A.; Schniederjans, M.; Asgari, E.; Kuo, T.-H.; Oliver, A.; Cabot, G.; Kola, A.; Gastmeier, P.; Hogardt, M.; et al. Predicting antimicrobial resistance in Pseudomonas aeruginosa with machine learning-enabled molecular diagnostics. EMBO Mol. Med. 2020, 12, e10264. [Google Scholar] [CrossRef] [PubMed]
- Geha, D.J.; Uhl, J.R.; Gustaferro, C.A.; Persing, D.H. Multiplex PCR for identification of methicillin-resistant staphylococci in the clinical laboratory. J. Clin. Microbiol. 1994, 32, 1768–1772. [Google Scholar] [CrossRef] [Green Version]
- Pournajaf, A.; Ardebili, A.; Goudarzi, L.; Khodabandeh, M.; Narimani, T.; Abbaszadeh, H. PCR-based identification of methicillin-resistant Staphylococcus aureus strains and their antibiotic resistance profiles. Asian Pac. J. Trop. Biomed. 2014, 4, S293–S297. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.N.A.; Anton-Le Berre, V.; Bañuls, A.-L.; Nguyen, T.V.A. Molecular Diagnosis of Drug-Resistant Tuberculosis; A Literature Review. Front. Microbiol. 2019, 10, 794. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.A.; Rivard, K.R.; Dumkow, L.E. Antimicrobial stewardship interventions to combat antibiotic resistance: An update on targeted strategies. Curr. Infect. Dis. Rep. 2019, 21, 33. [Google Scholar] [CrossRef]
- Wang, M.; Wei, H.; Zhao, Y.; Shang, L.; Di, L.; Lyu, C.; Liu, J. Analysis of multidrug-resistant bacteria in 3223 patients with hospital-acquired infections (HAI) from a tertiary general hospital in China. Bosn. J. Basic Med. Sci. 2019, 19, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Stultz, J.S.; Arnold, S.R.; Shelton, C.M.; Bagga, B.; Lee, K.R. Antimicrobial stewardship impact on Pseudomonas aeruginosa susceptibility to meropenem at a tertiary pediatric institution. Am. J. Infect. Control 2019, 47, 1513–1515. [Google Scholar] [CrossRef] [PubMed]
- Raymond, B. Five rules for resistance management in the antibiotic apocalypse, a road map for integrated microbial management. Evol. Appl. 2019, 12, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shen, W.; Zhong, Q.; Chen, Q.; He, X.; Baker, J.L.; Xiong, K.; Jin, X.; Wang, J.; Hu, F.; et al. Development of a Bacteriophage Cocktail to Constrain the Emergence of Phage-Resistant Pseudomonas aeruginosa. Front. Microbiol. 2020, 11, 327. [Google Scholar] [CrossRef] [Green Version]
- El Haddad, L.; Harb, C.P.; Gebara, M.A.; Stibich, M.A.; Chemaly, R.F. A systematic and critical review of bacteriophage therapy against multidrug-resistant ESKAPE organisms in humans. Clin. Infect. Dis. 2019, 69, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, Y.; Wang, Z.; Wei, J.; Hu, T.; Si, J.; Tao, G.; Zhang, L.; Xie, L.; Abdalla, A.E. A combination therapy of phages and antibiotics: Two is better than one. Int. J. Biol. Sci. 2021, 17, 3573. [Google Scholar] [CrossRef]
- Oechslin, F.; Piccardi, P.; Mancini, S.; Gabard, J.; Moreillon, P.; Entenza, J.M.; Resch, G.; Que, Y.-A. Synergistic interaction between phage therapy and antibiotics clears Pseudomonas aeruginosa infection in endocarditis and reduces virulence. J. Infect. Dis. 2017, 215, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Gu Liu, C.; Green, S.I.; Min, L.; Clark, J.R.; Salazar, K.C.; Terwilliger, A.L.; Kaplan, H.B.; Trautner, B.W.; Ramig, R.F.; Maresso, A.W. Phage-antibiotic synergy is driven by a unique combination of antibacterial mechanism of action and stoichiometry. mBio 2020, 11, e01462-20. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chang, R.Y.K.; Lin, Y.; Morales, S.; Kutter, E.; Chan, H.-K. Phage cocktail powder for Pseudomonas aeruginosa respiratory infections. Int. J. Pharm. 2021, 596, 120200. [Google Scholar] [CrossRef] [PubMed]
- Pletzer, D.; Coleman, S.R.; Hancock, R.E. Anti-biofilm peptides as a new weapon in antimicrobial warfare. Curr. Opin. Microbiol. 2016, 33, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.S.; Paterson, D.L. Antibiotics in the clinical pipeline in October 2019. J. Antibiot. 2020, 73, 329–364. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.-Y.; Lv, Y.; Zhu, Y.; Wei, M.-J.; Liu, M.-Y.; Ji, X.-W.; Kang, Z.-S.; Xia, Y.-H.; Tian, J.-H.; Ma, Y. A first-in-human safety, tolerability, and pharmacokinetics study of benapenem in healthy Chinese volunteers. Antimicrob. Agents Chemother. 2019, 63, e02188-18. [Google Scholar] [CrossRef] [Green Version]
- Reck, F.; Bermingham, A.; Blais, J.; Capka, V.; Cariaga, T.; Casarez, A.; Colvin, R.; Dean, C.R.; Fekete, A.; Gong, W. Optimization of novel monobactams with activity against carbapenem-resistant Enterobacteriaceae–identification of LYS228. Bioorganic Med. Chem. Lett. 2018, 28, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Tümmler, B. Emerging therapies against infections with Pseudomonas aeruginosa. F1000Research 2019, 8, 1371. [Google Scholar] [CrossRef]
- Vázquez-Ucha, J.C.; Arca-Suárez, J.; Bou, G.; Beceiro, A. New Carbapenemase Inhibitors: Clearing the Way for the β-lactams. Int. J. Mol. Sci. 2020, 21, 9308. [Google Scholar] [CrossRef] [PubMed]
- Everett, M.; Sprynski, N.; Coelho, A.; Castandet, J.; Bayet, M.; Bougnon, J.; Lozano, C.; Davies, D.T.; Leiris, S.; Zalacain, M.; et al. Discovery of a novel metallo-β-lactamase inhibitor that potentiates meropenem activity against carbapenem-resistant enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62, e00074-18. [Google Scholar] [CrossRef] [Green Version]
- Davies, D.T.; Leiris, S.; Sprynski, N.; Castandet, J.; Lozano, C.; Bousquet, J.; Zalacain, M.; Vasa, S.; Dasari, P.K.; Pattipati, R.; et al. ANT2681: SAR studies leading to the identification of a metallo-β-lactamase inhibitor with potential for clinical use in combination with meropenem for the treatment of infections caused by NDM-producing enterobacteriaceae. ACS Infect. Dis. 2020, 6, 2419–2430. [Google Scholar] [CrossRef]
- Bush, K.; Page, M.G.P. What we may expect from novel antibacterial agents in the pipeline with respect to resistance and pharmacodynamic principles. J. Pharmacokinet. Pharmacodyn. 2017, 44, 113–132. [Google Scholar] [CrossRef]
- Durand-Reville, T.F.; Miller, A.A.; O’Donnell, J.P.; Wu, X.; Sylvester, M.A.; Guler, S.; Iyer, R.; Shapiro, A.B.; Carter, N.M.; Velez-Vega, C.; et al. Rational design of a new antibiotic class for drug-resistant infections. Nature 2021, 597, 698–702. [Google Scholar] [CrossRef]
- Möllmann, U.; Heinisch, L.; Bauernfeind, A.; Köhler, T.; Ankel-Fuchs, D. Siderophores as drug delivery agents: Application of the “Trojan Horse” strategy. BioMetals 2009, 22, 615–624. [Google Scholar] [CrossRef]
- Miller, M.J.; Zhu, H.; Xu, Y.; Wu, C.; Walz, A.J.; Vergne, A.; Roosenberg, J.M.; Moraski, G.; Minnick, A.A.; McKee-Dolence, J.; et al. Utilization of microbial iron assimilation processes for the development of new antibiotics and inspiration for the design of new anticancer agents. BioMetals 2009, 22, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhanel, G.G.; Golden, A.R.; Zelenitsky, S.; Wiebe, K.; Lawrence, C.K.; Adam, H.J.; Idowu, T.; Domalaon, R.; Schweizer, F.; Zhanel, M.A. Cefiderocol: A siderophore cephalosporin with activity against carbapenem-resistant and multidrug-resistant Gram-negative bacilli. Drugs 2019, 79, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Owusu, Y.B.; Sun, D. US FDA-Approved Antibiotics During the 21st Century. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Kresken, M.; Korte-Berwanger, M.; Gatermann, S.G.; Pfeifer, Y.; Pfennigwerth, N.; Seifert, H.; Werner, G. In vitro activity of cefiderocol against aerobic Gram-negative bacterial pathogens from Germany. Int. J. Antimicrob. Agents 2020, 56, 106128. [Google Scholar] [CrossRef] [PubMed]
- Luscher, A.; Moynié, L.; Auguste, P.S.; Bumann, D.; Mazza, L.; Pletzer, D.; Naismith, J.H.; Köhler, T. TonB-dependent receptor repertoire of Pseudomonas aeruginosa for uptake of siderophore-drug conjugates. Antimicrob. Agents Chemother. 2018, 62, e00097-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streling, A.P.; Al Obaidi, M.M.; Lainhart, W.D.; Zangeneh, T.; Khan, A.; Dinh, A.Q.; Hanson, B.; Arias, C.A.; Miller, W.R. Evolution of Cefiderocol Non-Susceptibility in Pseudomonas aeruginosa in a Patient Without Previous Exposure to the Antibiotic. Clin. Infect. Dis. 2021, 73, e4472–e4474. [Google Scholar] [CrossRef]
- Gonzalez Gomez, A.; Hosseinidoust, Z. Liposomes for antibiotic encapsulation and delivery. ACS Infect. Dis. 2020, 6, 896–908. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, Q.; Feng, W.; Pu, W.; Ding, J.; Zhang, H.; Li, X.; Yang, B.; Dai, Q.; Cheng, L. Targeted delivery of antibiotics to the infected pulmonary tissues using ROS-responsive nanoparticles. J. Nanobiotechnology 2019, 17, 103. [Google Scholar] [CrossRef] [Green Version]
- Lamers, R.P.; Cavallari, J.F.; Burrows, L.L. The efflux inhibitor phenylalanine-arginine β-naphthylamide (PAβN) permeabilizes the outer membrane of gram-negative bacteria. PLoS ONE 2013, 8, e60666. [Google Scholar] [CrossRef] [Green Version]
- Rampioni, G.; Pillai, C.R.; Longo, F.; Bondì, R.; Baldelli, V.; Messina, M.; Imperi, F.; Visca, P.; Leoni, L. Effect of efflux pump inhibition on Pseudomonas aeruginosa transcriptome and virulence. Sci. Rep. 2017, 7, 11392. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Roles of penicillin-binding proteins (PBPs) in peptidoglycan synthesis. (A) NAG-NAM chains are not cross-linked before processing by PBPs. (B) LMM PBPs 4, 5 and 7 (DD-carboxypeptidases) cleave terminal D-alanyl residues from some pentapeptides, regulating levels of cross-linking. (C) HMM PBP 1a, 1b, 2, 3, 3a (DD-transpeptidases) cross-link pentapeptide-containing side chains to penta-, tetra-, or tri-peptides of adjacent NAG-NAM chains while simultaneously removing terminal D-alanyl residues. (D) Mature peptidoglycan contains a mixture of cross-linked and unlinked peptides. NAG, N-acetyl glucosamine; NAM, N-acetyl muramic acid.
Figure 1.
Roles of penicillin-binding proteins (PBPs) in peptidoglycan synthesis. (A) NAG-NAM chains are not cross-linked before processing by PBPs. (B) LMM PBPs 4, 5 and 7 (DD-carboxypeptidases) cleave terminal D-alanyl residues from some pentapeptides, regulating levels of cross-linking. (C) HMM PBP 1a, 1b, 2, 3, 3a (DD-transpeptidases) cross-link pentapeptide-containing side chains to penta-, tetra-, or tri-peptides of adjacent NAG-NAM chains while simultaneously removing terminal D-alanyl residues. (D) Mature peptidoglycan contains a mixture of cross-linked and unlinked peptides. NAG, N-acetyl glucosamine; NAM, N-acetyl muramic acid.

Figure 2.
Core structures of β-lactam subclasses used in P. aeruginosa treatment, and the terminal D-alanine-D-alanyl residues of peptidoglycan pentapeptide. (A) Penicillins. (B) Cephalosporins. (C) Carbapenems. (D) Monobactams. (E) D-alanine-D-alanyl residues. The β-lactam ring is indicated in red and mimics the terminal D-alanine-D-alanyl of the peptidoglycan pentapeptide precursor. Figure adapted from [101,107].
Figure 2.
Core structures of β-lactam subclasses used in P. aeruginosa treatment, and the terminal D-alanine-D-alanyl residues of peptidoglycan pentapeptide. (A) Penicillins. (B) Cephalosporins. (C) Carbapenems. (D) Monobactams. (E) D-alanine-D-alanyl residues. The β-lactam ring is indicated in red and mimics the terminal D-alanine-D-alanyl of the peptidoglycan pentapeptide precursor. Figure adapted from [101,107].

Figure 3.
Mechanisms of β-lactam resistance in P. aeruginosa.

Figure 4.
Structure of PBP3 in complex with meropenem. The transpeptidase domain is shown in red. The domain shown in blue is thought to play a role in protein-protein interactions. The membrane-spanning helix and small cytoplasmic part of PBP3 are not included in the structure. Meropenem shown in yellow is bound to the catalytic serine S294 (in black). Amino acid residues that are commonly substituted in clinical isolates are coloured green with side chains displayed. The image is based on protein structure PDB 3PBR_1 [102].
Figure 4.
Structure of PBP3 in complex with meropenem. The transpeptidase domain is shown in red. The domain shown in blue is thought to play a role in protein-protein interactions. The membrane-spanning helix and small cytoplasmic part of PBP3 are not included in the structure. Meropenem shown in yellow is bound to the catalytic serine S294 (in black). Amino acid residues that are commonly substituted in clinical isolates are coloured green with side chains displayed. The image is based on protein structure PDB 3PBR_1 [102].

Figure 5.
Regulation of ampC expression. (A) Under normal cellular conditions expression of ampC is repressed. LMM PBPs such as PBP4 hydrolyse uncross-linked peptidoglycan pentapeptides to tetrapeptides. During recycling of peptidoglycan, peptidoglycan fragments (the majority being NAG-NAM tetrapeptide cleaved from the NAG-NAM chains by lytic transglycosylases) are imported into the cytoplasm. NAG is removed by NagZ, after which NAM is cleaved from the peptide side chain by AmpD. NAG, NAM and the peptide side chains are used in synthesis of new peptidoglycan. Excess peptidoglycan precursor UDP-NAM pentapeptide formed through recycling as well as de novo synthesis binds to the AmpR regulator protein, which acts as a repressor inhibiting ampC expression. (B) β-lactams cause upregulation of ampC. Increased peptidoglycan recycling occurs because of the presence of β-lactams, which also inhibit conversion of tetrapeptides to pentapeptides by LMMs PBPs. The resulting peptidoglycan fragments (primarily NAG-NAM pentapeptide but also NAG-NAM tripeptide [not shown]) are imported into the cytoplasm. In the recycling pathway, AmpD becomes saturated because of increased amounts of peptidoglycan fragments, increasing the intracellular concentrations of the AmpR activator molecules NAG-NAM pentapeptide, NAM-pentapeptide and NAG-NAM tripeptide. Increased export of UDP-NAM pentapeptide for peptidoglycan synthesis also occurs. The activator molecules outcompete UDP-NAM pentapeptide for binding to AmpR and the AmpR-activator complexes trigger increased expression of ampC. UDP, uridine diphosphate; NAG, N-acetyl glucosamine; NAM, N-acetyl muramic acid; pentapeptide, L-alanine-γ-D-Glutamate-meso-DAP-D-Ala-D-Ala.
Figure 5.
Regulation of ampC expression. (A) Under normal cellular conditions expression of ampC is repressed. LMM PBPs such as PBP4 hydrolyse uncross-linked peptidoglycan pentapeptides to tetrapeptides. During recycling of peptidoglycan, peptidoglycan fragments (the majority being NAG-NAM tetrapeptide cleaved from the NAG-NAM chains by lytic transglycosylases) are imported into the cytoplasm. NAG is removed by NagZ, after which NAM is cleaved from the peptide side chain by AmpD. NAG, NAM and the peptide side chains are used in synthesis of new peptidoglycan. Excess peptidoglycan precursor UDP-NAM pentapeptide formed through recycling as well as de novo synthesis binds to the AmpR regulator protein, which acts as a repressor inhibiting ampC expression. (B) β-lactams cause upregulation of ampC. Increased peptidoglycan recycling occurs because of the presence of β-lactams, which also inhibit conversion of tetrapeptides to pentapeptides by LMMs PBPs. The resulting peptidoglycan fragments (primarily NAG-NAM pentapeptide but also NAG-NAM tripeptide [not shown]) are imported into the cytoplasm. In the recycling pathway, AmpD becomes saturated because of increased amounts of peptidoglycan fragments, increasing the intracellular concentrations of the AmpR activator molecules NAG-NAM pentapeptide, NAM-pentapeptide and NAG-NAM tripeptide. Increased export of UDP-NAM pentapeptide for peptidoglycan synthesis also occurs. The activator molecules outcompete UDP-NAM pentapeptide for binding to AmpR and the AmpR-activator complexes trigger increased expression of ampC. UDP, uridine diphosphate; NAG, N-acetyl glucosamine; NAM, N-acetyl muramic acid; pentapeptide, L-alanine-γ-D-Glutamate-meso-DAP-D-Ala-D-Ala.

Figure 6.
Locations of amino acid variants in AmpC that contribute to β-lactam resistance.


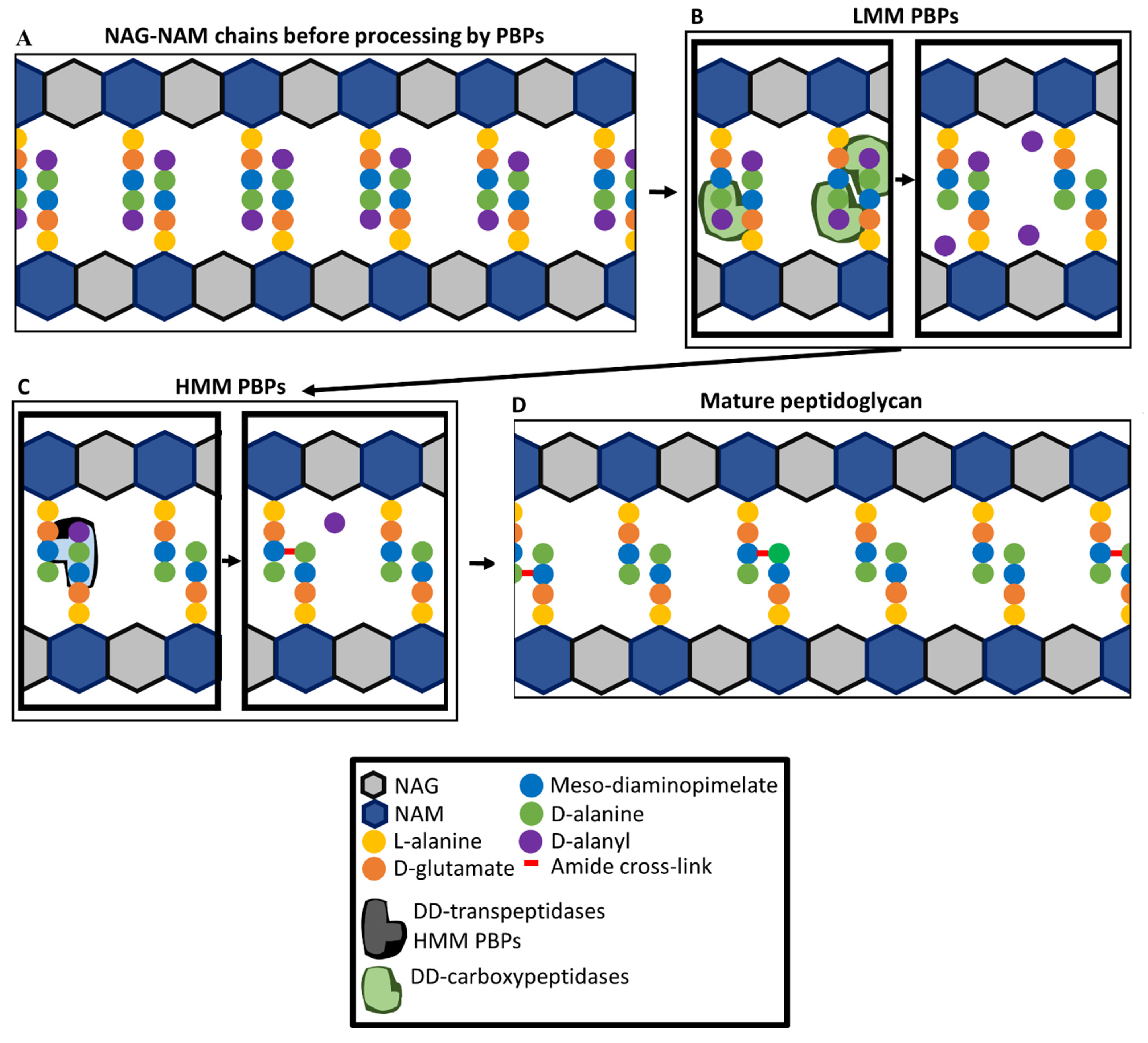{kind=link}
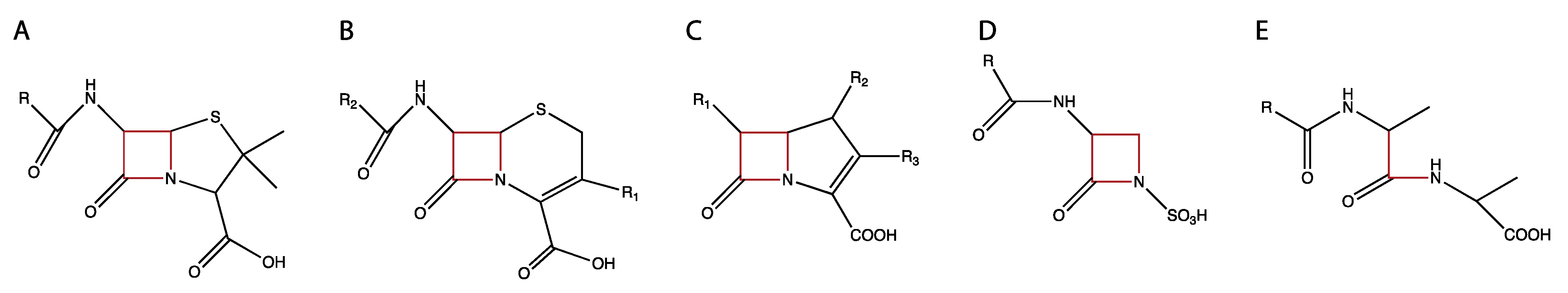{kind=link}
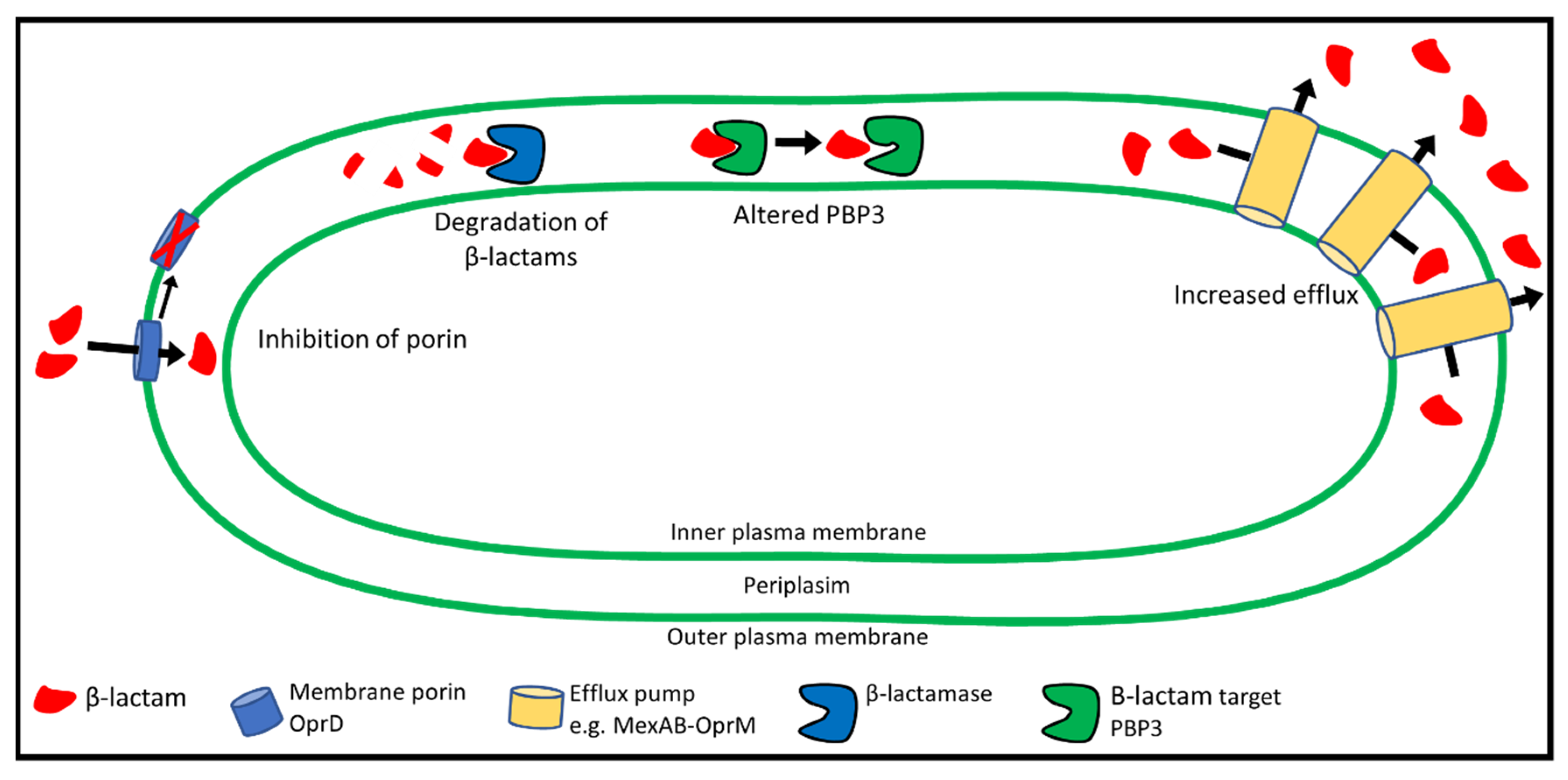{kind=link}
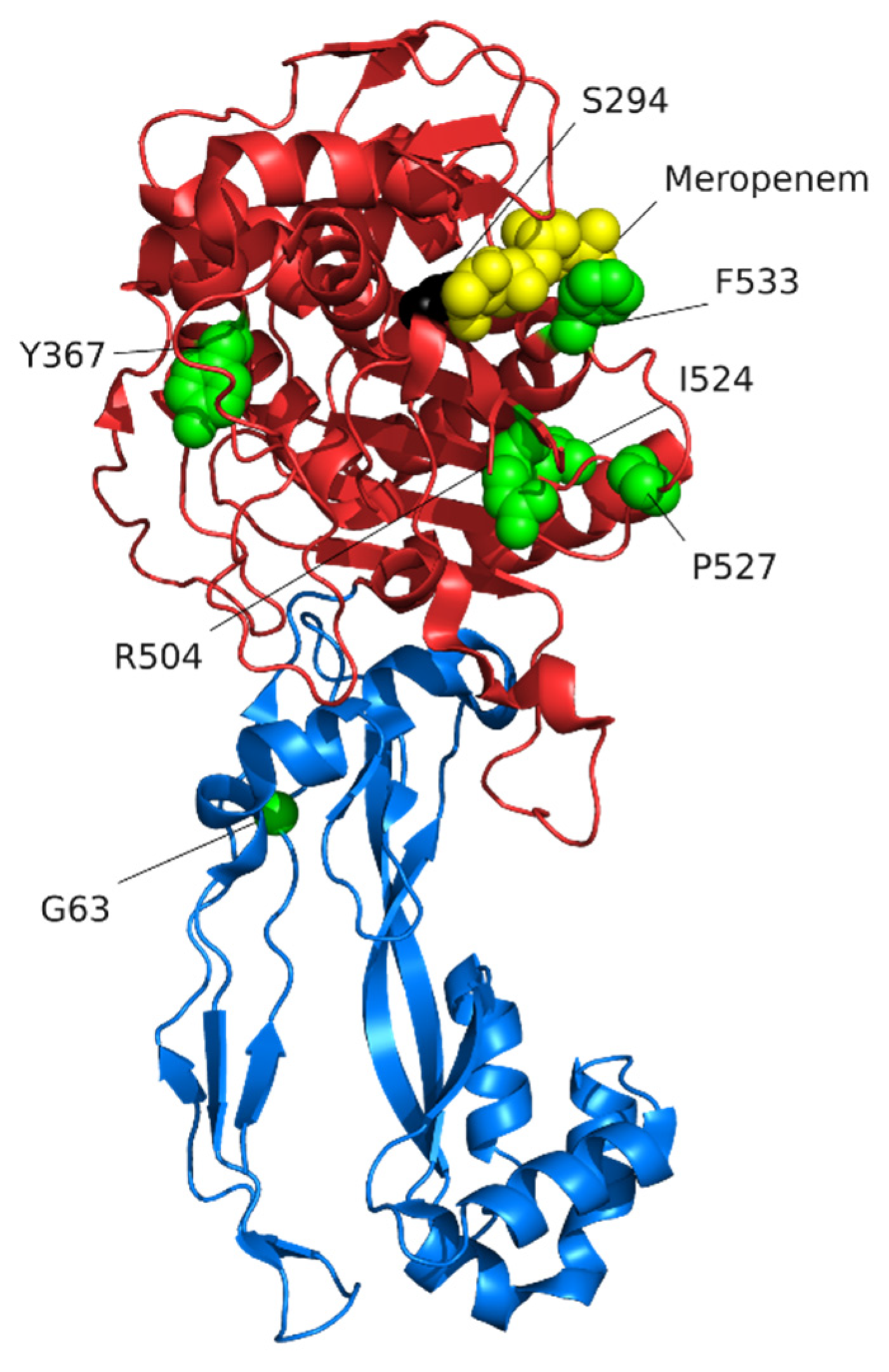{kind=link}
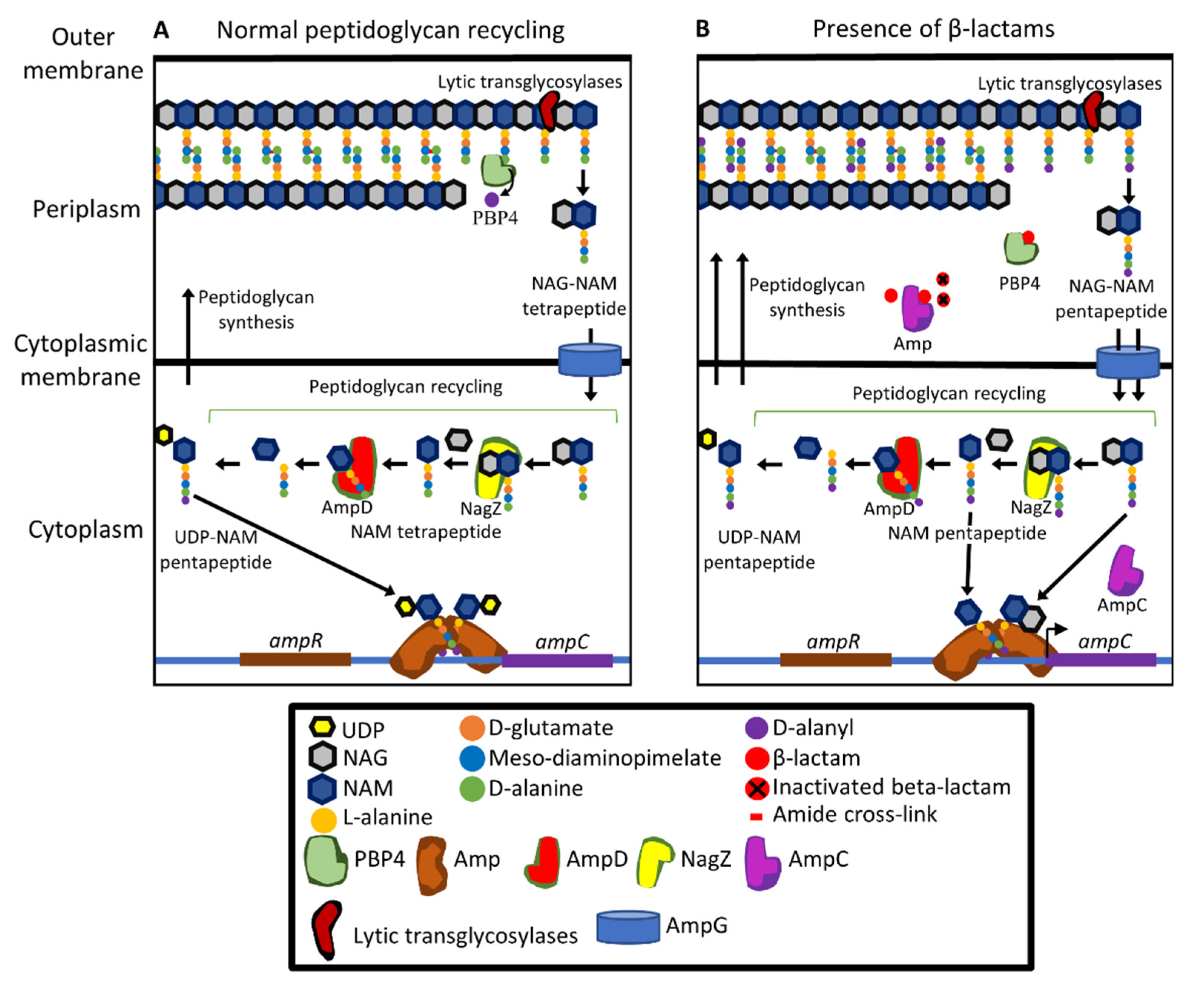{kind=link}
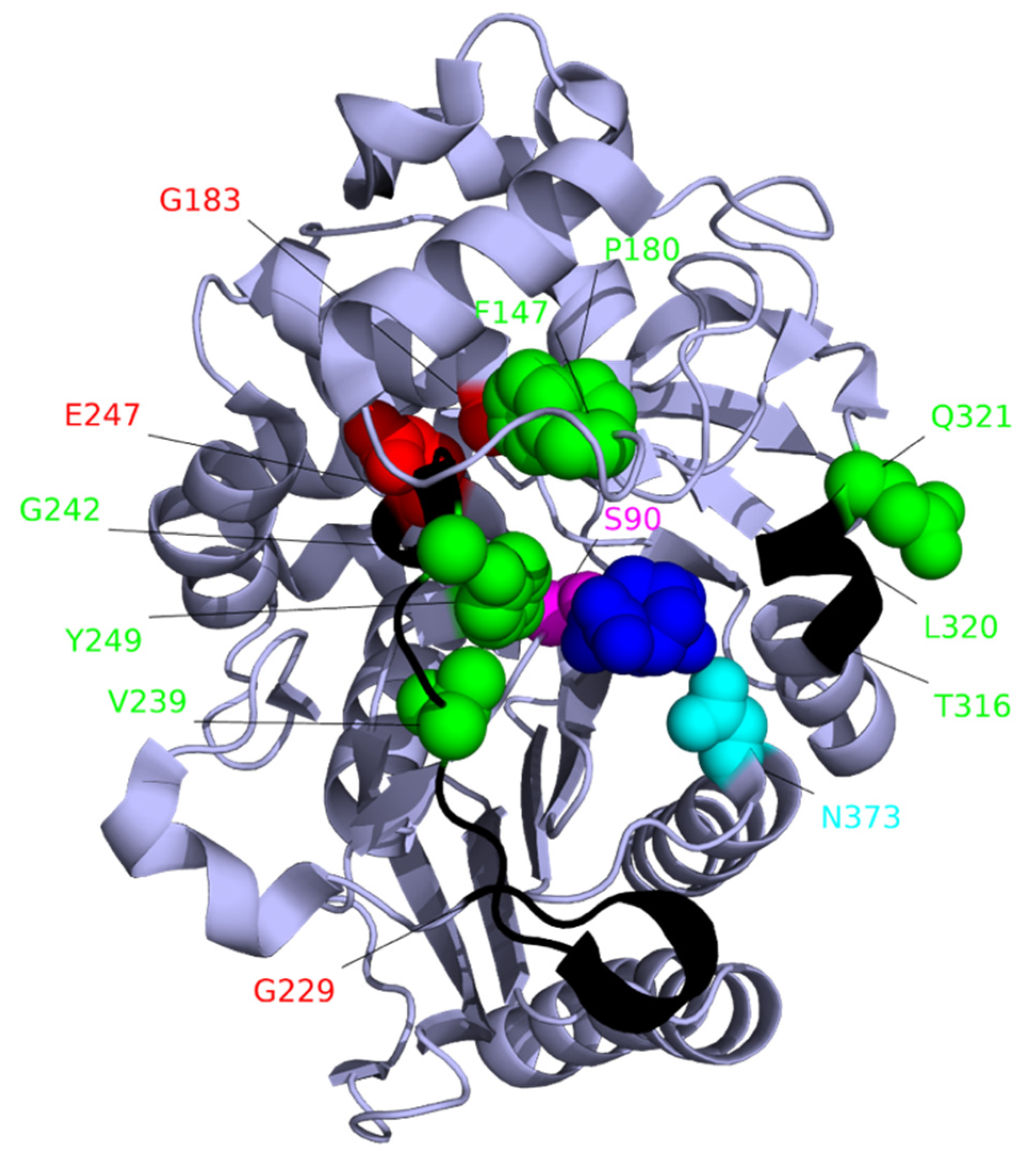{kind=link}
Table 1.
Key antibiotics used in the treatment of P. aeruginosa [2,42,43,44,45,46,47,48,49,50,51,52,53].
| Antibiotic Class | Antibiotic Subclass | Antibiotic | Antibiotic Use |
|---|---|---|---|
| β-lactams a | Penicillins | Ticarcillin Piperacillin | Parenteral or intravenous for treatment of infections |
| Monobactams | Aztreonam | Inhalation for long-term treatment of chronic lung infections and intramuscular injection for the treatment of acute infections | |
| Carbapenems | Imipenem b meropenem | Intravenous for treatment of acute or chronic infections | |
| Cephalosporins | Ceftazidime Cefepime Ceftolozone | Inhalation or intravenous for treatment of acute or chronic infections | |
| Aminoglycosides | 4,6-di-substituted deoxystreptamine ring | Tobramycin Gentamicin Amikacin | Inhalation or intravenous for treatment of acute or chronic infections |
| Quinolones | Fluoroquinolones | Ciprofloxacin Levofloxacin | Oral or intravenous intake for treatment of acute infections |
| Lipopeptides | Polymyxins | Colistin | Inhalation for treatment of chronic lung infections |
a β-lactams are often paired with a β-lactamase inhibitor. b Imipenem is generally administered with cilastatin (inhibitor of DHP-1 an enzyme that metabolises imipenem).
Table 2.
Binding affinities of β-lactams for PBPs of P. aeruginosa.
| Antibiotic a | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DOR | IMI | MER | CEF | AZT | CEN | CEP | FAR | |||||
| PBP | Binding Affinity IC50 (μg/mL) b | |||||||||||
| 1a | 0.5 | 0.1 | 0.5 | 0.5 | 0.2 | 0.2 | 0.8 | 2 | 2 | 19 | 35 | 0.23 |
| 1b | 0.6 | 0.2 | 0.5 | 0.5 | 3 | 5 | 6 | 2 | 2 | 2 | 0.7 | 0.15 |
| 2 | 0.06 | 0.1 | 0.1 | 0.05 | >32 | >32 | 25 | 16 | 16 | >250 | ND | 0.19 |
| 3 | 0.07 | 0.09 | 0.1 | 0.08 | 0.1 | 0.1 | 0.1 | 0.03 | 0.03 | 0.3 | 41 | 0.20 |
| 4 | 0.008 | 0.008 | 0.01 | 0.008 | 2 | 2 | ND | 16 | 16 | ND | ND | 0.13 |
| 5 | 8 | 2 | 2 | 16 | >32 | >32 | ND | >16 | >16 | ND | ND | >1 |
| MIC (μg/mL) for P. aeruginosa c | ||||||||||||
| 0.25 | 1 | 1 | 0.5 | 1 | 1 | 0.5 | 4 | 4 | 0.03 | 16 | 512 | |
| Reference | [110] | [110] | [111] | [110] | [110] | [111] | [112] | [110] | [111] | [112] | [112] | [109] |
a DOR, doripenem; IMI, imipenem; MER, meropenem; CEF, ceftazidime; AZT, aztreonam; CEN, Cefsulodin; CEP, Cephalexin; FAR, Faropenem; b IC50: Half maximal inhibitory concentration, determined through competition assays with the fluorescent β-lactam bocillin FL. ND, not determined. c MIC: Minimum inhibitory concentration (μg/mL).
Table 3.
Antibiotic resistance associated with increased expression of efflux pumps.
| Efflux System | Antibiotics Affected by Increased Expression |
|---|---|
| MexAB-OprM | Aztreonam, other β-lactams a, quinolones, tetracyclines, macrolides, novobiocin and chloramphenicol [172,173] |
| MexXY-OprM | Aminoglycosides, tetracyclines, β-lactams b and macrolides [171,172,174,180,181] |
| MexCD-OprJ | β-lactams c and fluoroquinolones [170,174] |
| MexEF-OprN | Imipenem d and fluoroquinolones [172,177] |
a MexAB-OprM exports all β-lactams except imipenem [174,182]. b MexXY-OprM exports all β-lactams except carbenicillin, sulbenicillin, cefsulodin, ceftazidime, moxalactam, flomoxef, aztreonam, and imipenem and has low substrate specificity for other carbapenems [174]. c MexCD-OprJ exports all β-lactams except carbenicillin, sulbenicillin, ceftazidime, moxalactam, aztreonam, and imipenem and has low substrate specificity for other carbapenems [174]. d Increased expression of mexEFoprN reduces oprD expression [183,184].
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Glen, K.A.; Lamont, I.L. β-lactam Resistance in Pseudomonas aeruginosa: Current Status, Future Prospects. Pathogens 2021, 10, 1638. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10121638
AMA Style
Glen KA, Lamont IL. β-lactam Resistance in Pseudomonas aeruginosa: Current Status, Future Prospects. Pathogens. 2021; 10(12):1638. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10121638
Chicago/Turabian StyleGlen, Karl A., and Iain L. Lamont. 2021. "β-lactam Resistance in Pseudomonas aeruginosa: Current Status, Future Prospects" Pathogens 10, no. 12: 1638. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10121638
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.