
Comparative Analysis of Transcriptional Regulation Patterns: Understanding the Gene Expression Profile in Nucleocytoviricota



Abstract
:1. Introduction
2. Temporal Regulation of Gene Expression
3. Transcription in Nucleocytoviricota
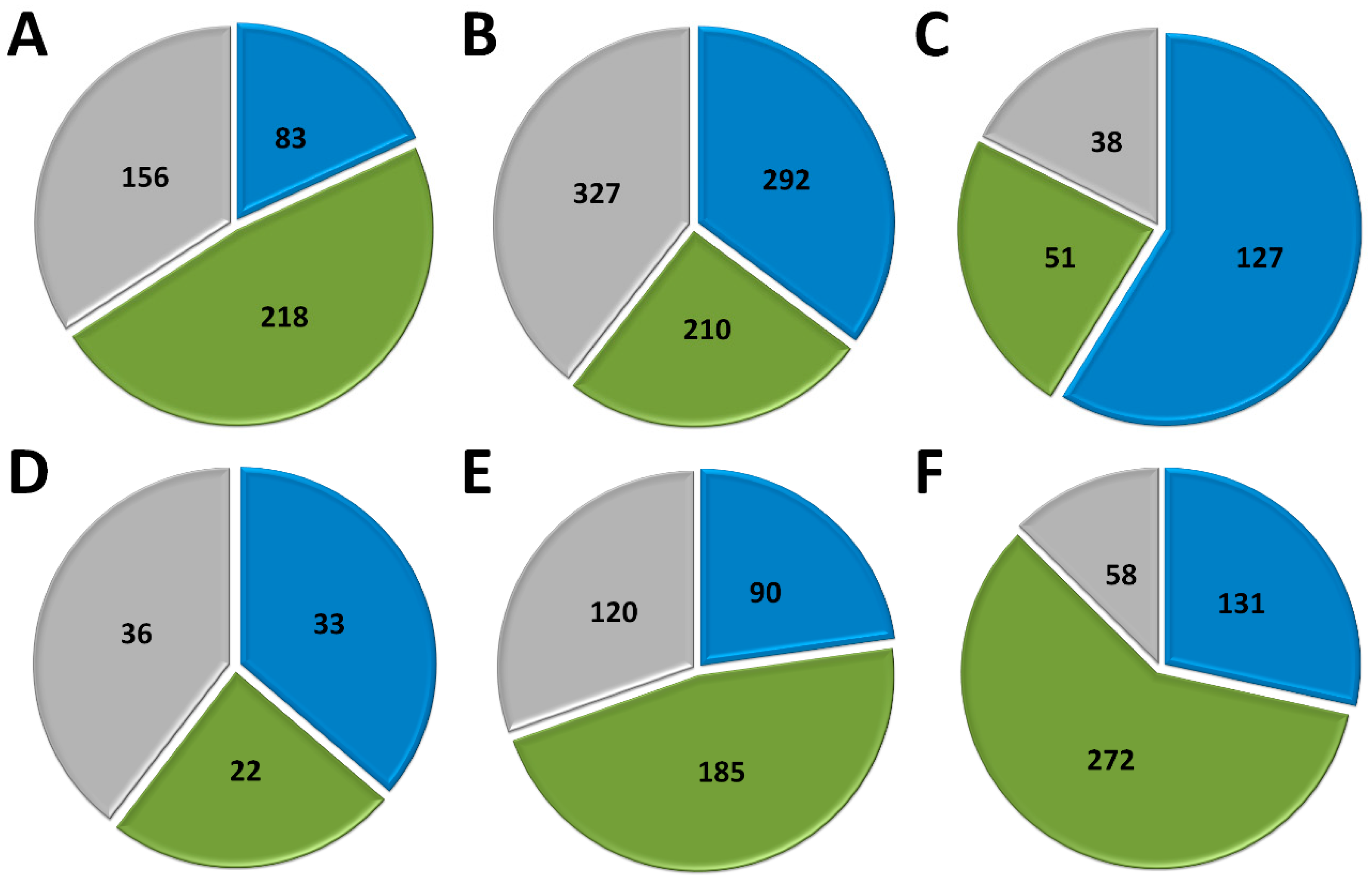
3.1. Poxvirus Gene Transcription
3.2. Asfarvirus Gene Transcription
3.3. Iridovirus Gene Transcription
3.4. Ascovirus Gene Transcription
3.5. Phycodnavirus Gene Transcription
3.6. Transcription of Mimiviruses and Other Giant Amoeba Viruses
4. Functional Comparative Analysis of Temporally Expressed Genes from Pathogens of Nucleocytoviricota
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Yutin, N.; Wolf, Y.I.; Raoult, D.; Koonin, E.V. Eukaryotic large nucleo-cytoplasmic DNA viruses: Clusters of orthologous genes and reconstruction of viral genome evolution. Virol. J. 2009, 6, 223. [Google Scholar] [CrossRef] [Green Version]
- Iyer, L.M.; Aravind, L.; Koonin, E.V. Common Origin of Four Diverse Families of Large Eukaryotic DNA Viruses Common Origin of Four Diverse Families of Large Eukaryotic DNA Viruses. J. Virol. 2001, 75, 11720–11734. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Yutin, N. Evolution of the Large Nucleocytoplasmic DNA Viruses of Eukaryotes and Convergent Origins of Viral Gigantism. Adv. Virus Res. 2019, 103, 167–202. [Google Scholar]
- Guglielmini, J.; Woo, A.; Krupovic, M.; Forterre, P.; Gaia, M. Diversification of giant and large eukaryotic dsDNA viruses predated the origin of modern eukaryotes. Proc. Natl. Acad. Sci. USA 2019, 116, 19585–19592. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef]
- Philippe, N.; Legendre, M.; Doutre, G.; Couté, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reteno, D.G.; Benamar, S.; Khalil, J.B.; Andreani, J.; Armstrong, N.; Klose, T.; Rossmann, M.; Colson, P.; Raoult, D.; La Scola, B. Faustovirus, an asfarvirus-related new lineage of giant viruses infecting amoebae. J. Virol. 2015, 89, 6585–6594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajrai, L.H.; Benamar, S.; Azhar, E.I.; Robert, C.; Levasseur, A.; Raoult, D.; La Scola, B. Kaumoebavirus, a new virus that clusters with Faustoviruses and Asfarviridae. Viruses 2016, 8, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreani, J.; Aherfi, S.; Khalil, J.Y.B.; Di Pinto, F.; Bitam, I.; Raoult, D.; Colson, P.; La Scola, B. Cedratvirus, a double-cork structured giant virus, is a distant relative of pithoviruses. Viruses 2016, 8, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, M.; Lartigue, A.; Bertaux, L.; Jeudy, S.; Bartoli, J.; Lescot, M.; Alempic, J.-M.; Ramus, C.; Bruley, C.; Labadie, K.; et al. In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proc. Natl. Acad. Sci. USA 2015, 112, 5327–5335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 201320670. [Google Scholar] [CrossRef] [Green Version]
- Andreani, J.; Khalil, J.Y.B.; Sevvana, M.; Benamar, S.; Di Pinto, F.; Bitam, I.; Colson, P.; Klose, T.; Rossmann, M.G.; Raoult, D.; et al. Pacmanvirus, a New Giant Icosahedral Virus at the Crossroads between Asfarviridae and Faustoviruses. J. Virol. 2017, 91, e00212-17. [Google Scholar] [CrossRef] [Green Version]
- Andreani, J.; Khalil, J.Y.B.; Baptiste, E.; Hasni, I.; Michelle, C.; Raoult, D.; Levasseur, A.; La Scola, B. Orpheovirus IHUMI-LCC2: A new virus among the giant viruses. Front. Microbiol. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, G.; Blanc-Mathieu, R.; Song, C.; Kayama, Y.; Mochizuki, T.; Murata, K.; Ogata, H.; Takemura, M. Medusavirus, a Novel Large DNA Virus Discovered from Hot Spring Water. J. Virol. 2019, 93, e02130-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abergel, C.; Legendre, M.; Claverie, J.M. The rapidly expanding universe of giant viruses: Mimivirus, Pandoravirus, Pithovirus and Mollivirus. FEMS Microbiol. Rev. 2015, 39, 779–796. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.; Andrade, A.; Rodrigues, R.; Arantes, T.; Boratto, P.; Silva, L.; Dornas, F.; Trindade, G.; Drumond, B.; La Scola, B.; et al. Promoter Motifs in NCLDVs: An Evolutionary Perspective. Viruses 2017, 9, 16. [Google Scholar] [CrossRef]
- Hager, G.L.; McNally, J.G.; Misteli, T. Transcription dynamics. Mol. Cell 2009, 35, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Broyles, S.S.; Knutson, B.A. Poxvirus transcription. Future Virol. 2010, 5, 639–650. [Google Scholar] [CrossRef]
- Kogenaru, S.; Qing, Y.; Guo, Y.; Wang, N. RNA-seq and microarray complement each other in transcriptome profiling. BMC Genom. 2012, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Nagalakshmi, U.; Waern, K.; Snyder, M. RNA-Seq: A method for comprehensive transcriptome analysis. Curr. Protoc. Mol. Biol. 2010, 89, 1–13. [Google Scholar] [CrossRef]
- Broyles, S.S. Vaccinia virus transcription. J. Gen. Virol. 2003, 84, 2293–2303. [Google Scholar] [CrossRef]
- Vorou, R.M.; Papavassiliou, V.G.; Pierroutsakos, I.N. Cowpox virus infection: An emerging health threat. Curr. Opin. Infect. Dis. 2008, 21, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Abergel, C. Mimivirus: The emerging paradox of quasi-autonomous viruses. Trends Genet. 2010, 26, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Reynolds, S.E.; Martens, C.A.; Bruno, D.P.; Porcella, S.F.; Moss, B. Expression profiling of the intermediate and late stages of poxvirus replication. J. Virol. 2011, 85, 9899–9908. [Google Scholar] [CrossRef] [Green Version]
- Moss, B. Poxviridae. In Fields Virology; Knipe, D., Howley, P., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2014; p. 2129. [Google Scholar]
- Cackett, G.; Matelska, D.; Sýkora, M.; Portugal, R.; Malecki, M.; Bähler, J.; Dixon, L.; Werner, F. The African Swine Fever Virus Transcriptome. J. Virol. 2020, 94, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, J.M.; Salas, M.L. African swine fever virus transcription. Virus Res. 2013, 173, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Papp, T.; Marschang, R.E. Detection and characterization of invertebrate iridoviruses found in reptiles and prey insects in Europe over the past two decades. Viruses 2019, 11, 600. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.; Barbosa-Solomieu, V.; Chinchar, V.G. A decade of advances in iridovirus research. Adv. Virus Res. 2005, 65, 173–248. [Google Scholar]
- Asgari, S.; Davis, J.; Wood, D.; Peter Wilson, A.M. Sequence and organization of the Heliothis virescens ascovirus genome. J. Gen. Virol. 2007, 88, 1120–1132. [Google Scholar] [CrossRef]
- Salem, T.Z.; Turney, C.M.; Wang, L.; Xue, J.; Wan, X.F.; Cheng, X.W. Transcriptional analysis of a major capsid protein gene from Spodoptera exigua ascovirus 5a. Arch. Virol. 2008, 153, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Zaghloul, H.A.H.; Hice, R.; Arensburger, P.; Federici, B.A. Transcriptome Analysis of the Spodoptera frugiperda Ascovirus In Vivo Provides Insights into How Its Apoptosis Inhibitors and Caspase Promote Increased Synthesis of Viral Vesicles and Virion Progeny. J. Virol. 2017, 91, e00874-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai-Balser, G.M.; Duncan, G.A.; Eudy, J.D.; Wang, D.; Li, X.; Agarkova, I.V.; Dunigan, D.D.; Van Etten, J.L. Microarray Analysis of Paramecium bursaria Chlorella Virus 1 Transcription. J. Virol. 2010, 84, 532–542. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Tanaka, M.; Fujie, M.; Usami, S.; Yamada, T. Immediate early genes expressed in chlorovirus infections. Virology 2004, 318, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Blanc, G.; Mozar, M.; Agarkova, I.V.; Gurnon, J.R.; Yanai-Balser, G.; Rowe, J.M.; Xia, Y.; Riethoven, J.J.; Dunigan, D.D.; Van Etten, J.L. Deep RNA sequencing reveals hidden features and dynamics of early gene transcription in Paramecium bursaria chlorella virus 1. PLoS ONE 2014, 9, e90989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Etten, J.L.; Graves, M.V.; Müller, D.G.; Boland, W.; Delaroque, N. Phycodnaviridae—Large DNA algal viruses. Arch. Virol. 2002, 147, 1479–1516. [Google Scholar] [CrossRef]
- La Scola, B.; Audic, S.; Robert, C.; Jungang, L.; de Lamballerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.-M.; Raoult, D. A giant virus in amoebae. Science 2003, 299, 2033. [Google Scholar] [CrossRef] [PubMed]
- Zauberman, N.; Mutsafi, Y.; Halevy, D.B.; Shimoni, E.; Klein, E.; Xiao, C.; Sun, S.; Minsky, A. Distinct DNA exit and packaging portals in the virus Acanthamoeba polyphaga mimivirus. PLoS Biol. 2008, 6, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Legendre, M.; Audic, S.; Poirot, O.; Hingamp, P.; Seltzer, V.; Byrne, D.; Lartigue, A.; Lescot, M.; Bernadac, A.; Poulain, J.; et al. mRNA deep sequencing reveals 75 new genes and a complex transcriptional landscape in Mimivirus. Genome Res. 2010, 20, 664–674. [Google Scholar] [CrossRef] [Green Version]
- Boyer, M.; Yutin, N.; Pagnier, I.; Barrassi, L.; Fournous, G.; Espinosa, L.; Robert, C.; Azza, S.; Sun, S.; Rossmann, M.G.; et al. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc. Natl. Acad. Sci. USA 2009, 106, 21848–21853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, R.A.L.; Louazani, A.C.; Picorelli, A.; Oliveira, G.P.; Lobo, F.P.; Colson, P.; La Scola, B.; Abrahão, J.S. Analysis of a Marseillevirus Transcriptome Reveals Temporal Gene Expression Profile and Host Transcriptional Shift. Front. Microbiol. 2020, 11, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Cassetti, M.A.; Moss, B. Interaction of the 82-kDa subunit of the vaccinia virus early transcription factor heterodimer with the promoter core sequence directs downstream DNA binding of the 70-kDa subunit. Proc. Natl. Acad. Sci. USA 1996, 93, 7540–7545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, M.R.; Niles, E.G. The viral RNA polymerase H4L subunit is required for Vaccinia virus early gene transcription termination. J. Biol. Chem. 2001, 276, 20758–20765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, B.Y.; Gershon, P.D.; Moss, B. RNA polymerase-associated protein Rap94 confers promoter specificity for initiating transcription of vaccinia virus early stage genes. J. Biol. Chem. 1994, 269, 7552–7557. [Google Scholar] [CrossRef]
- Rosales, R.; Sutter, G.; Moss, B. A cellular factor is required for transcription of vaccinia viral intermediate-stage genes. Proc. Natl. Acad. Sci. USA 1994, 91, 3794–3798. [Google Scholar] [CrossRef] [Green Version]
- Katsafanas, G.; Moss, B. Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J. Biol. Chem. 2004, 279, 52210–52217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, C.F.; Oswald, B.W.; Dellis, S. Vaccinia Virus Late Transcription Is Activated in Vitro by Cellular Heterogeneous Nuclear Ribonucleoproteins. J. Biol. Chem. 2001, 276, 40680–40686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunasinghe, S.K.; Hubbs, A.E.; Wright, C.F. A vaccinia virus late transcription factor with biochemical and molecular identity to a human cellular protein. J. Biol. Chem. 1998, 273, 27524–27530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourquain, D.; Dabrowski, P.W.; Nitsche, A. Comparison of host cell gene expression in cowpox, monkeypox or vaccinia virus-infected cells reveals virus-specific regulation of immune response genes. Virol. J. 2013, 10, 61. [Google Scholar] [CrossRef] [Green Version]
- Chinchar, V.G.; Yu, K.H.; Jancovich, J.K. The molecular biology of frog virus 3 and other iridoviruses infecting cold-blooded vertebrates. Viruses 2011, 3, 1959–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goorha, R. Frog virus 3 requires RNA polymerase II for its replication. J. Virol. 1981, 37, 496–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majji, S.; Thodima, V.; Sample, R.; Whitley, D.; Deng, Y.; Mao, J.; Chinchar, V.G. Transcriptome analysis of Frog Virus 3, the type species of the genus Ranavirus, family Iridoviridae. Virology 2009, 391, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Lua, D.T.; Yasuike, M.; Hirono, I.; Aoki, T. Transcription program of red sea bream iridovirus as revealed by DNA microarrays. J. Virol. 2005, 79, 15151–15164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Costa, S.M.; Yao, H.J.; Bilimoria, S.L. Transcriptional mapping in Chilo iridescent virus infections. Arch. Virol. 2004, 149, 723–742. [Google Scholar] [CrossRef]
- Cheng, X.-W.; Wang, L.; Carner, G.R.; Arif, B.M. Characterization of three ascovirus isolates from cotton insects. J. Invertebr. Pathol. 2005, 89, 193–202. [Google Scholar] [CrossRef]
- Van Etten, J.L.; Agarkova, I.V.; Dunigan, D.D. Chloroviruses. Viruses 2019, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.J.; Schroeder, D.C.; Holden, M.T.G.; Wilson, W.H. Evolutionary history of the Coccolithoviridae. Mol. Biol. Evol. 2006, 23, 86–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, W.H.; Van Etten, J.L.; Allen, M.J. The Phycodnaviridae: The story of how tiny giants rule the world. Curr. Top. Microbiol. Immunol. 2009, 328, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.; Ku, C.; Sheyn, U.; Sheyn, U.; Sebé-Pedrós, A.; Sebé-Pedrós, A.; Ben-Dor, S.; Schatz, D.; Tanay, A.; Tanay, A.; et al. A single-cell view on alga-virus interactions reveals sequential transcriptional programs and infection states. Sci. Adv. 2020, 6, eaba4137. [Google Scholar] [CrossRef]
- Aylward, F.O.; Moniruzzaman, M.; Ha, A.D.; Koonin, E.V. A Phylogenomic Framework for Charting the Diversity and Evolution of Giant Viruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Xiao, C.; Kuznetso, Y.G.; Sun, S.; Hafenstein, S.L.; Kostyuchenko, V.A.; Chipman, P.R.; Suzan-Monti, M.; Raoult, D.; McPherson, A.; Rossmann, M.G. Structural studies of the giant Mimivirus. PLoS Biol. 2009, 7, 958–966. [Google Scholar] [CrossRef]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; Scola, B.L.; Suzan, M.; Claverie, J.-M. The 1.2-Megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef]
- Legendre, M.; Santini, S.; Rico, A.; Abergel, C.; Claverie, J.-M. Breaking the 1000-gene barrier for Mimivirus using ultra-deep genome and transcriptome sequencing. Virol. J. 2011, 8, 99. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Endo, H.; Takemura, M.; Ogata, H. RNA-seq of the medusavirus suggests remodeling of the host nuclear environment at an early infection stage. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gruffat, H.; Marchione, R.; Manet, E. Herpesvirus Late Gene Expression: A Viral-Specific Pre-initiation Complex Is Key. Front. Microbiol. 2016, 7, 869. [Google Scholar] [CrossRef] [PubMed]
- Friesen, P.D.; Miller, L.K. Temporal regulation of baculovirus RNA: Overlapping early and late transcripts. J. Virol. 1985, 54, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Ma, Y.; Wang, Y.; Yang, H.; Shen, W.; Chen, X. Transcription regulation mechanisms of bacteriophages. Bioengineered 2014, 5, 300–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yutin, N.; Wolf, Y.I.; Koonin, E.V. Origin of giant viruses from smaller DNA viruses not from a fourth domain of cellular life. Virology 2014, 466–467, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasir, A.; Caetano-Anollés, G. A phylogenomic data-driven exploration of viral origins and evolution. Sci. Adv. 2015, 1, e1500527. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Krupovic, M.; Yutin, N. Evolution of double-stranded DNA viruses of eukaryotes: From bacteriophages to transposons to giant viruses. Ann. N. Y. Acad. Sci. 2015, 1341, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 2015, 479–480, 2–25. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, R.A.L.; de Souza, F.G.; de Azevedo, B.L.; da Silva, L.C.; Abrahão, J.S. The morphogenesis of different giant viruses as additional evidence for a common origin of Nucleocytoviricota. Curr. Opin. Virol. 2021, 49, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Fornelos, N.; Browning, D.F.; Pavlin, A.; Podlesek, Z.; Hodnik, V.; Salas, M.; Butala, M. Lytic gene expression in the temperate bacteriophage GIL01 is activated by a phage-encoded LexA homologue. Nucleic Acids Res. 2018, 46, 9432–9443. [Google Scholar] [CrossRef] [PubMed]
- Filée, J. Genomic comparison of closely related Giant Viruses supports an accordion-like model of evolution. Front. Microbiol. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, C.; Shen, W.; Huang, G.; Le, S.; Lu, S.; Li, M.; Zhao, Y.; Wang, J.; Rao, X.; et al. Global Transcriptomic Analysis of Interactions between Pseudomonas aeruginosa and Bacteriophage PaP3. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Family | Virus | Particle Size (nm) | Genome Size (Kbp) | Proteins Encoded | Temporal Classification | Time Range | Host Range of the Family | Refs. |
|---|---|---|---|---|---|---|---|---|
| Poxiviridae | VACV | 200–300 | 195 | 218 | E, I, L | E (0.5–1 h); I (1–2 h); L (4 h) | Mammals, birds, reptiles, fish, insects | [22,23,26] |
| Asfarviridae | ASFV | 200 | 170 | 152 | IE, E, I, L | NA | Pigs and wild boars | [27,28] |
| Iridoviridae | FV-3 | 300 | 105 | 91 | E, I, L | E (2 h); I (4 h); L (9 h) | Reptiles, fish, insects, crustaceans | [29,30] |
| Ascoviridae | HVaV-3g | 300–400 | 199 | 194 | E, L, VL | NA | Arthropods | [31,32,33] |
| Phycodnaviridae | PBCV | 100–220 | 350 | 376 | E, L | E (5–10 min); L (60–90 min) | Eukaryotic algae | [34,35,36,37] |
| Mimiviridae | APMV | 750 | 1180 | 979 | E, I, L | E (0–3 h) I (3–6 h) L (>6 h) | Acanthamoeba sp. | [38,39,40] |
| Marseilleviridae | MRSV | 200–250 | 368 | 457 | E, I, L | E (0–1 h) I (1–2 h) L (>4 h) | Acanthamoeba sp. | [41,42] |
| Pandoraviridae * | PANDV | 1000 | 2470 | 1430 | NA | NA | Acanthamoeba sp. | [6] |
| Pithoviridae * | PITHV | 1500 | 610 | 467 | NA | NA | Acanthamoeba sp. | [11] |
| Molliviridae * | MOLLV | 600 | 651 | 523 | NA | NA | Acanthamoeba sp. | [10] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, F.G.; Abrahão, J.S.; Rodrigues, R.A.L. Comparative Analysis of Transcriptional Regulation Patterns: Understanding the Gene Expression Profile in Nucleocytoviricota. Pathogens 2021, 10, 935. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10080935
de Souza FG, Abrahão JS, Rodrigues RAL. Comparative Analysis of Transcriptional Regulation Patterns: Understanding the Gene Expression Profile in Nucleocytoviricota. Pathogens. 2021; 10(8):935. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10080935
Chicago/Turabian Stylede Souza, Fernanda Gil, Jônatas Santos Abrahão, and Rodrigo Araújo Lima Rodrigues. 2021. "Comparative Analysis of Transcriptional Regulation Patterns: Understanding the Gene Expression Profile in Nucleocytoviricota" Pathogens 10, no. 8: 935. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10080935