
SARS-CoV-2 Antibody Effectiveness Is Influenced by Non-Epitope Mutation/Binding-Induced Denaturation of the Epitope 3D Architecture


Abstract
:1. Introduction
2. Materials and Methods
2.1. Acquisition of 3D Structures
2.2. Analysis of 3D Structures
2.3. Analysis of Immune-Pressured AASCs
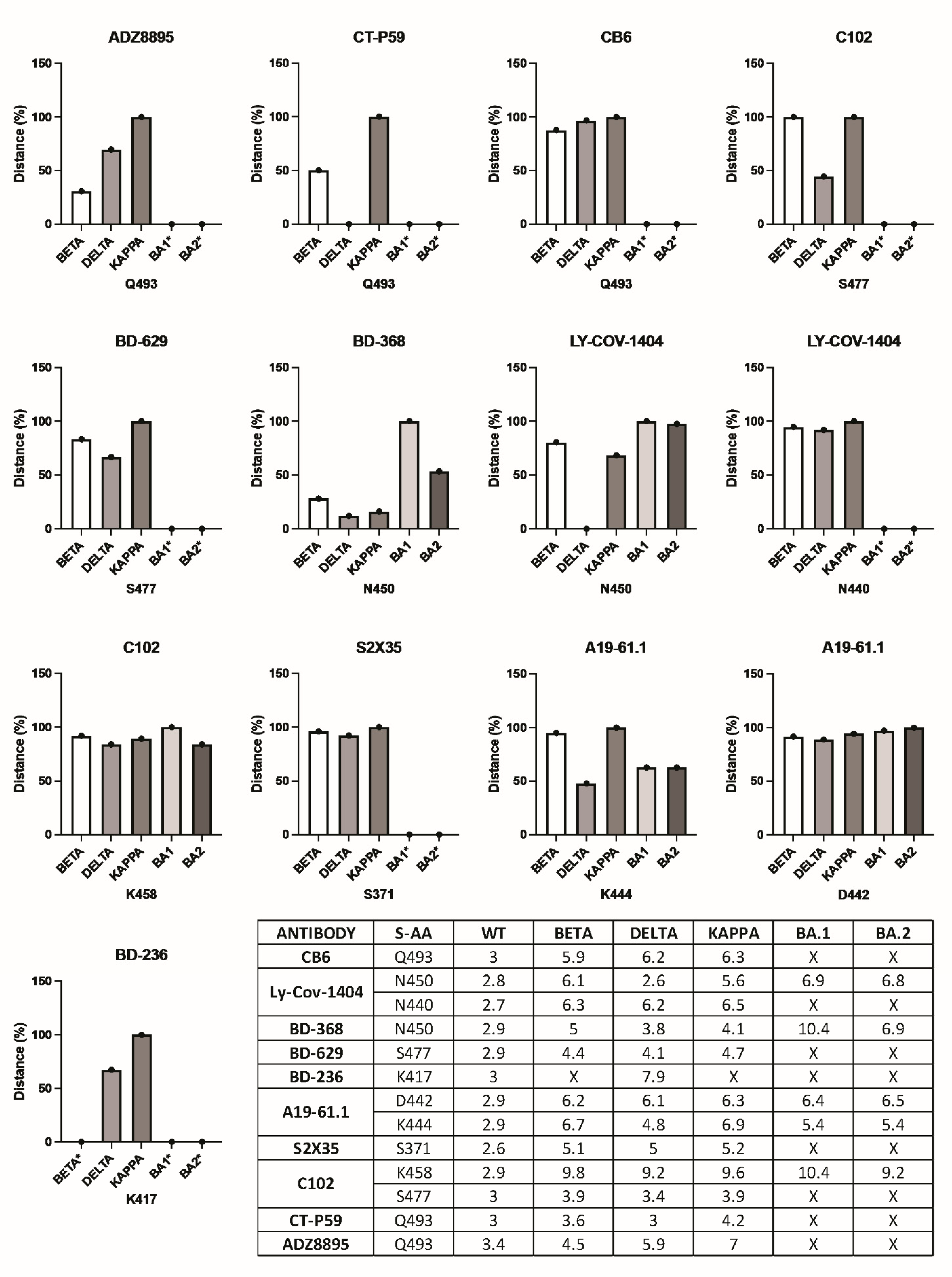
3. Results
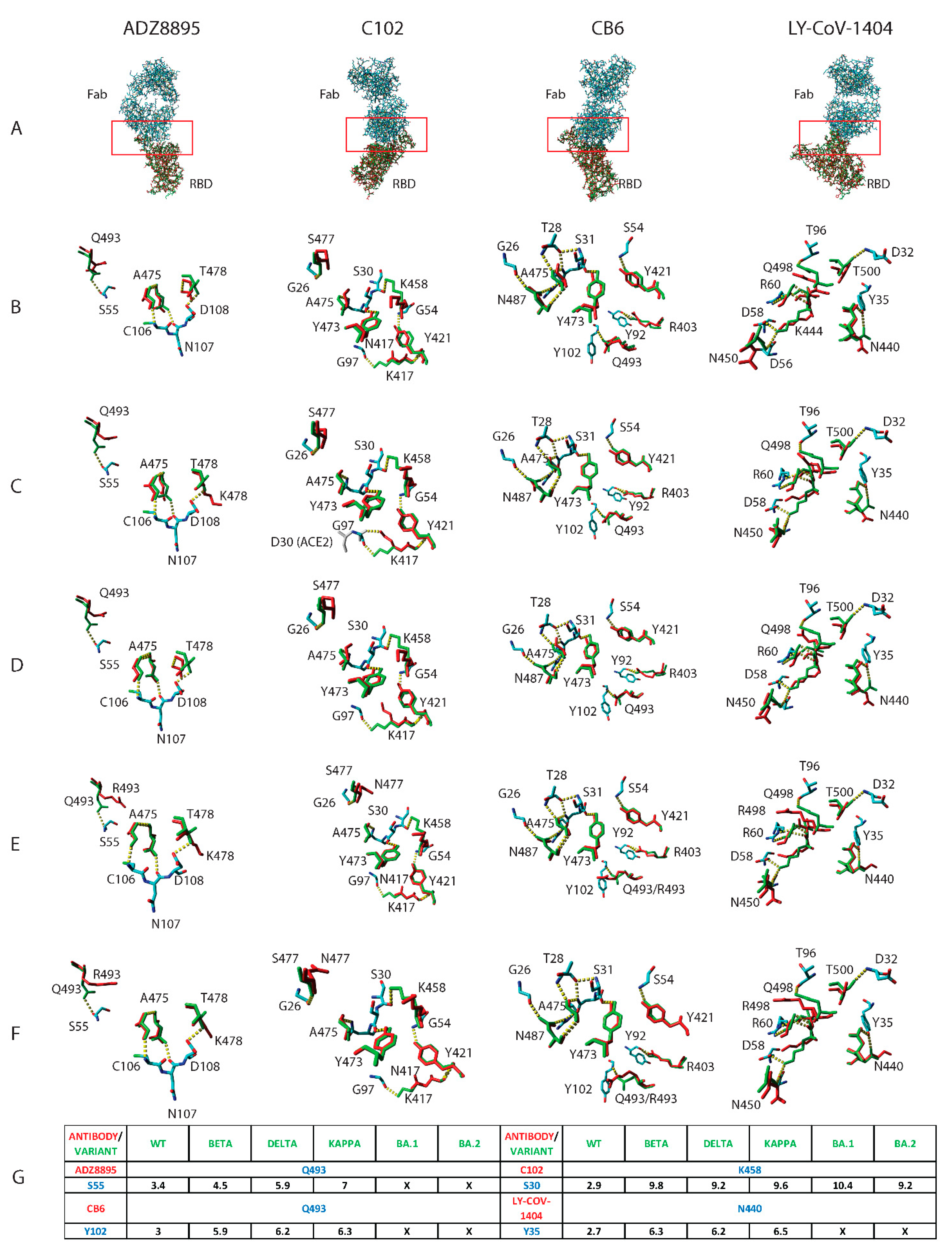
3.1. Mutation-Driven AASC Repositioning Influences the Formation of Hydrogen Bonds between the Paratope and RBD Complementary Donor/Acceptor Atoms
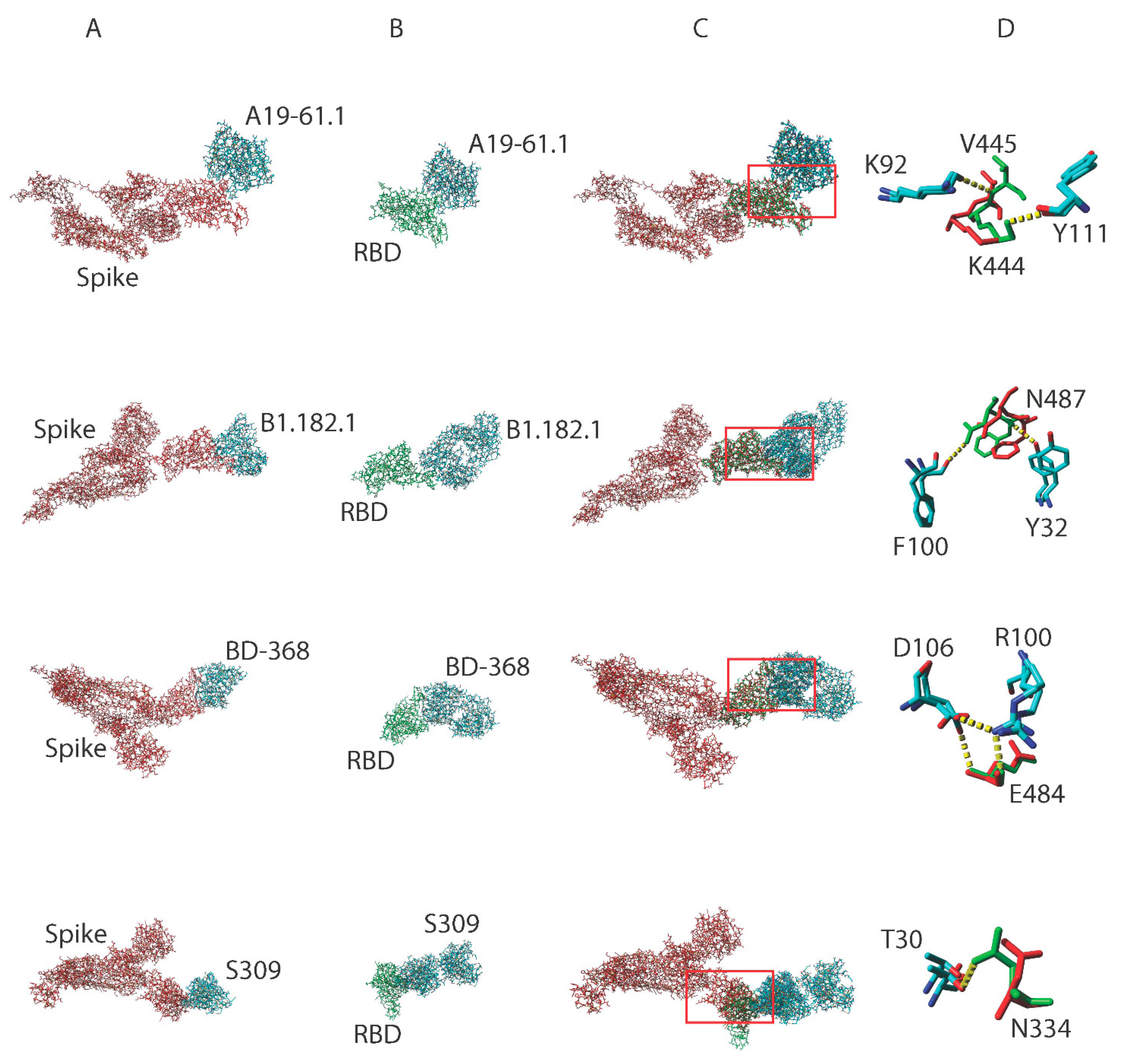
3.2. Truncating the Spike Protein Repositions Epitope AASCs Which Influences the Formation of Hydrogen Bonds
3.3. Interactions by the Same Antibody Vary Depending on Whether They Are Used Individually or as an Antibody Cocktail
3.4. Antibody Pressure Drives the Repositioning of Epitope AASCs and Their Subsequent Substitution
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eckersall, P.D. Proteins, Proteomics, and the Dysproteinemias. 2008. Available online: www.ebi.uniprot.org (accessed on 25 October 2022).
- Narayan, A.; Bhattacharjee, K.; Naganathan, A.N. Thermally versus Chemically Denatured Protein States. Biochemistry 2019, 58, 2519–2523. [Google Scholar] [CrossRef] [Green Version]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Saunders, K.; Edwards, R.J.; et al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation and antigenicity. Science 2021, 373, eabi6226. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subruamoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar]
- Jiang, S.; Hillyer, C.; Du, L. Neutralizing Antibodies against SARS-CoV-2 and Other Human Coronaviruses. Trends Immunol. 2020, 41, 355–359. [Google Scholar]
- Huang, H.Y.; Liao, H.Y.; Chen, X.; Wang, S.W.; Cheng, C.W.; Shahed-Al-Mahmud, M.; Liu, Y.M.; Mohapatra, A.; Chen, T.H.; Lo, J.M.; et al. Vaccination with SARS-CoV-2 spike protein lacking glycan shields elicits enhanced protective responses in animal models. Sci. Transl. Med. 2022, 14, 639. [Google Scholar]
- McRee, D.E. 3—Computational Techniques. In Practical Protein Crystallography, 2nd ed.; McRee, D.E., Ed.; Academic Press: San Diego, CA, USA, 1999; pp. 91–269. [Google Scholar]
- Xu, D.; Tsai, C.J.; Nussinov, R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. 1997, 10, 999–1012. [Google Scholar]
- Harvey, W.T.; Carabelli, A.; Jackson, B.; Gupta, R.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Li, L.; Liao, H.; Meng, Y.; Li, W.; Han, P.; Liu, K.; Wang, Q.; Li, D.; Zhang, Y.; Wang, L.; et al. Structural basis of human ACE2 higher binding affinity to currently circulating Omicron SARS-CoV-2 sub-variants BA.2 and BA.1.1. Cell 2022, 186, 2952–2960. [Google Scholar]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar]
- Konagurthu, A.; Whisstock, J.; Stuckey, P.; Lesk, A. MUSTANG: A multiple structural alignment algorithm. Proteins Struct. Funct. Bioinform. 2006, 64, 559–574. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, L.; Misasi, J.; Pegu, A.; Zhang, Y.; Harris, D.R.; Olia, A.S.; Talama, C.A.; Yang, E.S.; Chen, M.; et al. Structural basis for potent antibody neutralization of SARS-CoV-2 variants including B.1.1.529. Science 2022, 376. [Google Scholar] [CrossRef]
- VanBlargan, L.A.; Errico, J.M.; Halfmann, P.J.; Zost, S.J.; Crowe, J.E.; Purcell, L.A.; Kawaoka, Y.; Corti, D.; Fremont, D.H.; Diamond, M.S. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 2022, 28, 490–495. [Google Scholar] [CrossRef]
- Nabel, K.G.; Clark, S.A.; Shankar, S.; Pan, J.; Clark, L.E.; Yang, P.; Coscia, A.; Mckay, L.G.A.; Varnum, H.H.; Brusic, V.; et al. Structural basis for continued antibody evasion by the SARS-CoV-2 receptor binding domain. Science 2022, 375, 6578. [Google Scholar]
- Bruel, T.; Hadjadj, J.; Maes, P.; Planas, D.; Seve, A.; Staropoli, I.; Guivel-Benhassine, F.; Porrot, F.; Bolland, W.-H.; Neguyen, Y.; et al. Serum neutralization of SARS-CoV-2 Omicron sublineages BA.1 and BA.2 in patients receiving monoclonal antibodies. Nat. Med. 2022, 28, 1297–1302. [Google Scholar]
- Tortorici, M.A.; Beltramello, M.; Lempp, F.A.; Pinto, D.; Dang, H.V.; Rosen, L.E.; McCallum, M.; Bowen, J.; Minola, A.; Jaconi, S.; et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science 2020, 370, 950–957. [Google Scholar] [CrossRef]
- McInerney, T.L.; McLain, L.; Armstrong, S.J.; Dimmock, N.J. A Human IgG1 (b12) Specific for the CD4 Binding Site of HIV-1 Neutralizes by Inhibiting the Virus Fusion Entry Process, but b12 Fab Neutralizes by Inhibiting a Postfusion Event. Virology 1997, 233, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.S.; Ohshima, N.; Stanfield, R.L.; Yu, W.; Iba, Y.; Okuno, Y.; Kurosawa, Y.; Wilson, I.A. Receptor mimicry by antibody F045–092 facilitates universal binding to the H3 subtype of influenza virus. Nat. Commun. 2014, 5, 3614. [Google Scholar] [CrossRef]
- Wu, X.; Sereno, A.J.; Huang, F.; Lewis, S.M.; Lieu, R.L.; Weldon, C.; Torres, C.; Fine, C.; Batt, M.A.; Fitchett, J.R.; et al. Fab-Based Bispecific Antibody Formats with Robust Biophysical Properties and Biological Activity; Taylor & Francis: Oxford, UK, 2015; Volume 7, pp. 470–482. [Google Scholar] [CrossRef] [Green Version]
- Al Qaraghuli, M.M.; Kubiak-Ossowska, K.; Ferro, V.A.; Mulheran, P.A. Antibody-protein binding and conformational changes: Identifying allosteric signalling pathways to engineer a better effector response. Sci. Rep. 2020, 10, 13696. [Google Scholar]
- Hammes, G.G.; Chang, Y.-C.; Oas, T.G. Conformational selection or induced fit: A flux description of reaction mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 13737–13741. [Google Scholar] [CrossRef] [Green Version]
- Rockett, R.; Basile, K.; Maddocks, S.; Fong, W.; Agius, J.E.; Johnson-Mackinnon, J.; Arnott, A.; Chandra, S.; Gall, M.; Draper, J.; et al. Resistance Mutations in SARS-CoV-2 Delta Variant after Sotrovimab Use. N. Engl. J. Med. 2022, 386, 1477–1479. [Google Scholar] [CrossRef]
- Du, S.; Cao, Y.; Zhu, Q.; Yu, P.; Qi, F.; Wang, G.; Du, X.; Bao, L.; Deng, W.; Zhu, H.; et al. Structurally Resolved SARS-CoV-2 Antibody Shows High Efficacy in Severely Infected Hamsters and Provides a Potent Cocktail Pairing Strategy. Cell 2020, 183, 1013–1023.e13. [Google Scholar]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef]
- Lang, S.; Xie, J.; Zhu, X.; Wu, N.; Lerner, R.A.; Wilson, I.A. Antibody 27F3 Broadly Targets Influenza A Group 1 and 2 Hemagglutinins through a Further Variation in VH1-69 Antibody Orientation on the HA Stem. Cell Rep. 2017, 20, 2935–2943. [Google Scholar] [CrossRef] [Green Version]
- Maun, H.R.; Vij, R.; Walters, B.T.; Morando, A.; Jackman, J.K.; Wu, P.; Estevez, A.; Chen, X.; Franke, Y.; Lipari, M.T.; et al. Bivalent antibody pliers inhibit β-tryptase by an allosteric mechanism dependent on the IgG hinge. Nat. Commun. 2020, 11, 6435. [Google Scholar] [CrossRef]
- Yan, R.; Wang, R.; Ju, B.; Yu, J.; Zhang, Y.; Liu, N.; Wang, J.; Zhang, Q.; Chen, P.; Zhou, B.; et al. Structural basis for bivalent binding and inhibition of SARS-CoV-2 infection by human potent neutralizing antibodies. Cell Res. 2021, 31, 517–525. [Google Scholar]
- Suryadevara, N.; Shrihari, S.; Gilchuk, P.; VanBlargan, L.A.; Binshtein, E.; Zost, S.J.; Nargi, R.S.; Sutton, R.E.; Winkler, E.S.; Chen, E.C.; et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell 2021, 184, 2316–2331.e15. [Google Scholar]
- Astronomo, R.D.; Santra, S.; Ballweber-Fleming, L.; Westerberg, K.G.; Mach, L.; Hensley-McBain, T.; Sutherland, L.; Mildenberg, B.; Morton, G.; Yates, N.L.; et al. Neutralization Takes Precedence Over IgG or IgA Isotype-related Functions in Mucosal HIV-1 Antibody-mediated Protection. eBioMedicine 2016, 14, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Sterlin, D.; Mathian, A.; Miyara, M.; Mohr, A.; Anna, F.; Claër, L.; Quentric, P.; Fadlallah, J.; Devilliers, H.; Ghillani, P.; et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci. Transl. Med. 2021, 13, 577. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Coronavirus (COVID-19) Update: FDA Authorizes Moderna, Pfizer-BioNTech Bivalent COVID-19 Vaccines for Use as a Booster Dose. Food and Drug Administration; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2022. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-moderna-pfizer-biontech-bivalent-covid-19-vaccines-use (accessed on 19 September 2022).
- Bar-On, Y.M.; Goldberg, Y.; Mandel, M.; Bodenheimer, O.; Freedman, L.; Kalkstein, N.; Mizrahi, B.; Alroy-Preis, S.; Ash, N.; Milo, R.; et al. Protection of BNT162b2 Vaccine Booster against COVID-19 in Israel. N. Engl. J. Med. 2021, 385, 1393–1400. [Google Scholar] [CrossRef]
- Magen, O.; Waxman, J.G.; Makov-Assif, M.; Vered, R.; Dicker, D.; Hernán, M.A.; Lipsitch, M.; Reis, B.Y.; Balicer, R.D.; Dagan, N. Fourth Dose of BNT162b2 mRNA COVID-19 Vaccine in a Nationwide Setting. N. Engl. J. Med. 2022, 386, 1603–1614. [Google Scholar] [CrossRef]
- Andrews, N.; Stowe, J.; Kirsebom, F.; Toffa, S.; Rickeard, T.; Gallagher, E.; Gower, C.; Kall, M.; Groves, N.; O’Connell, A.-M.; et al. COVID-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 2022, 386, 1532–1546. [Google Scholar] [CrossRef]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.E.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433. [Google Scholar]
- Chang, M.R.; Ke, H.; Coherd, C.D.; Wang, Y.; Mashima, K.; Kastrunes, G.M.; Huang, C.Y.; Marasco, W.A. Analysis of a SARS-CoV-2 convalescent cohort identified a common strategy for escape of vaccine-induced anti-RBD antibodies by Beta and Omicron variants. EBioMedicine 2022, 80, 104025. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4, & BA.5. Nature 2022, 608, 603–608. [Google Scholar]
- Dejnirattisai, W.; Huo, J.; Zhou, D.; Zahradník, J.; Supasa, P.; Liu, C.; Duyvesteyn, H.M.E.; Gin, H.M.; Mentzer, A.J.; Tuekprakhon, A.; et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 2022, 185, 467–484.e15. [Google Scholar]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Protein/Antibody Name (Mutations) | Target Protein | Antibody Formulation | Virus/Variant | Expression System | Method |
|---|---|---|---|---|---|---|
| 7MMO | LY-CoV1404 | RBD | Monoclonal | SARS-CoV-2 wild type | Cricetulus griseus | X-ray diffraction |
| 6WPS | S309 | Spike | Monoclonal | Homo sapiens | Electron microscopy | |
| 7CHH | BD-368-2 | Monoclonal | ||||
| 7TB8 | B1-182.1 and A19-61.1 | Cocktail | ||||
| 7C01 | CB6 | RBD | Monoclonal | X-ray diffraction | ||
| 7K8M | C102 | Monoclonal | ||||
| 7L7D | AZD8895 | Monoclonal | ||||
| 7CH4 | BD-604 | Monoclonal | ||||
| 7CH5 | BD-629 | Monoclonal | ||||
| 7CHB | BD-236 | Monoclonal | ||||
| 7L7E | AZD8895 and AZD1061 | Cocktail | ||||
| 7R6W | S2X35 and S309 | Cocktail | ||||
| 7R6X | S2E12, S309, and S304 | Cocktail | ||||
| 7TBF | B1-182.1 and A19-61.1 | Cocktail | ||||
| 7CHC | BD-629 and BD-368-2 | Cocktail | ||||
| 7CHE | BD-236 and BD-368-2 | Cocktail | ||||
| 7CHF | BD-604 and BD-368-2 | Cocktail | ||||
| 7TN0 | ACE2, S304, and S309 | Cocktail | SARS-CoV-2 Omicron | |||
| 7VX4 | ACE2-RBD (K417N, E484K, N501Y) | Monoclonal | SARS-CoV-2 Beta | Electron microscopy | ||
| 7VX5 | ACE2-RBD (L452R and E484Q) | SARS-CoV-2 Kappa | ||||
| 7WBP | ACE2-RBD (G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H) | SARS-CoV-2 BA.1 | X-ray diffraction | |||
| 7WBQ | ACE2-RBD (L452R, T478K) | SARS-CoV-2 Delta | ||||
| 7ZF7 | ACE2-RBD (G339D, S371L, S373P, S375F, T376A, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H) | SARS-CoV-2 BA.2 | ||||
| 2NY7 | B12 | gp120 | Monoclonal | HIV | Cricetulus griseus | |
| 1HZH | B12 IgG | N/A | ||||
| 5Y2L | AF4H1K1 | Hemagglutinin | Monoclonal | Influenza H3N2 | Homo sapiens | |
| 5Y2M | Influenza H4N6 | |||||
| 5Y2K | N/A | No ligand | ||||
| 6J9O | AF4H1K1 scFv | Escherichia coli | ||||
| 4O5I | F045-092 | Monoclonal | A/Victoria/361/2011 (H3N2) | Trichoplusia ni | ||
| 4O58 | A/Victoria/3/1975 (H3N2) | |||||
| 5KEL | c2G4 and c13C6 | EBOV GP | Cocktail | Ebola | Nicotiana benthamiana | Electron microscopy |
| 5KEN | c4G7 and c13C6 | |||||
| 6DFJ | Z021 | Envelope protein DIII | Monoclonal | DENV-1 | Homo sapiens | X-ray diffraction |
| 6DFI | Zika |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malisheni, M.M.; Bates, M.; Rizvanov, A.A.; MacAry, P.A. SARS-CoV-2 Antibody Effectiveness Is Influenced by Non-Epitope Mutation/Binding-Induced Denaturation of the Epitope 3D Architecture. Pathogens 2022, 11, 1437. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11121437
Malisheni MM, Bates M, Rizvanov AA, MacAry PA. SARS-CoV-2 Antibody Effectiveness Is Influenced by Non-Epitope Mutation/Binding-Induced Denaturation of the Epitope 3D Architecture. Pathogens. 2022; 11(12):1437. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11121437
Chicago/Turabian StyleMalisheni, Moffat M., Matthew Bates, Albert A. Rizvanov, and Paul A. MacAry. 2022. "SARS-CoV-2 Antibody Effectiveness Is Influenced by Non-Epitope Mutation/Binding-Induced Denaturation of the Epitope 3D Architecture" Pathogens 11, no. 12: 1437. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11121437