
Mexican Strains of Anaplasma marginale: A First Comparative Genomics and Phylogeographic Analysis

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. General Features of Anaplasma marginale Genomes
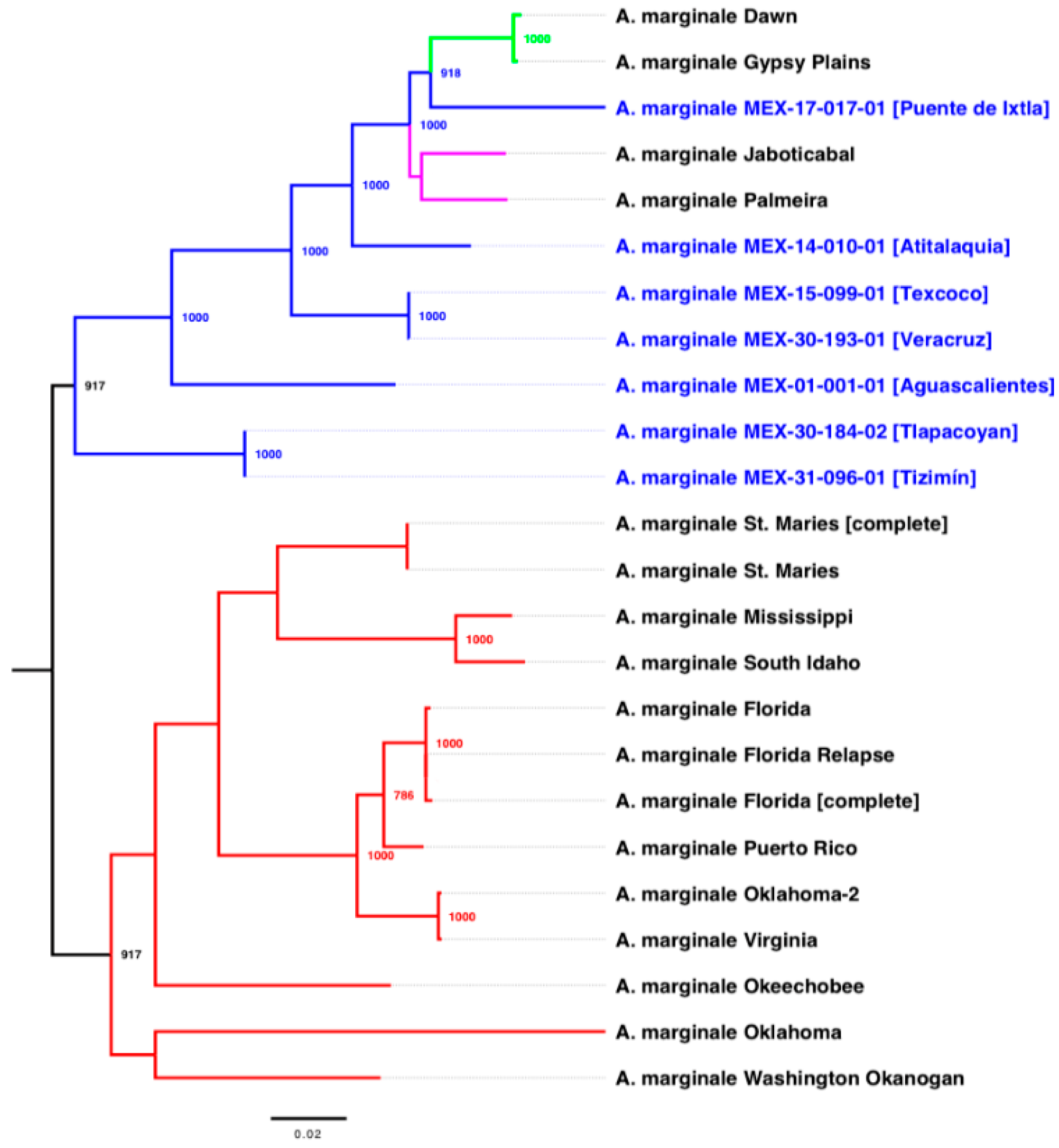
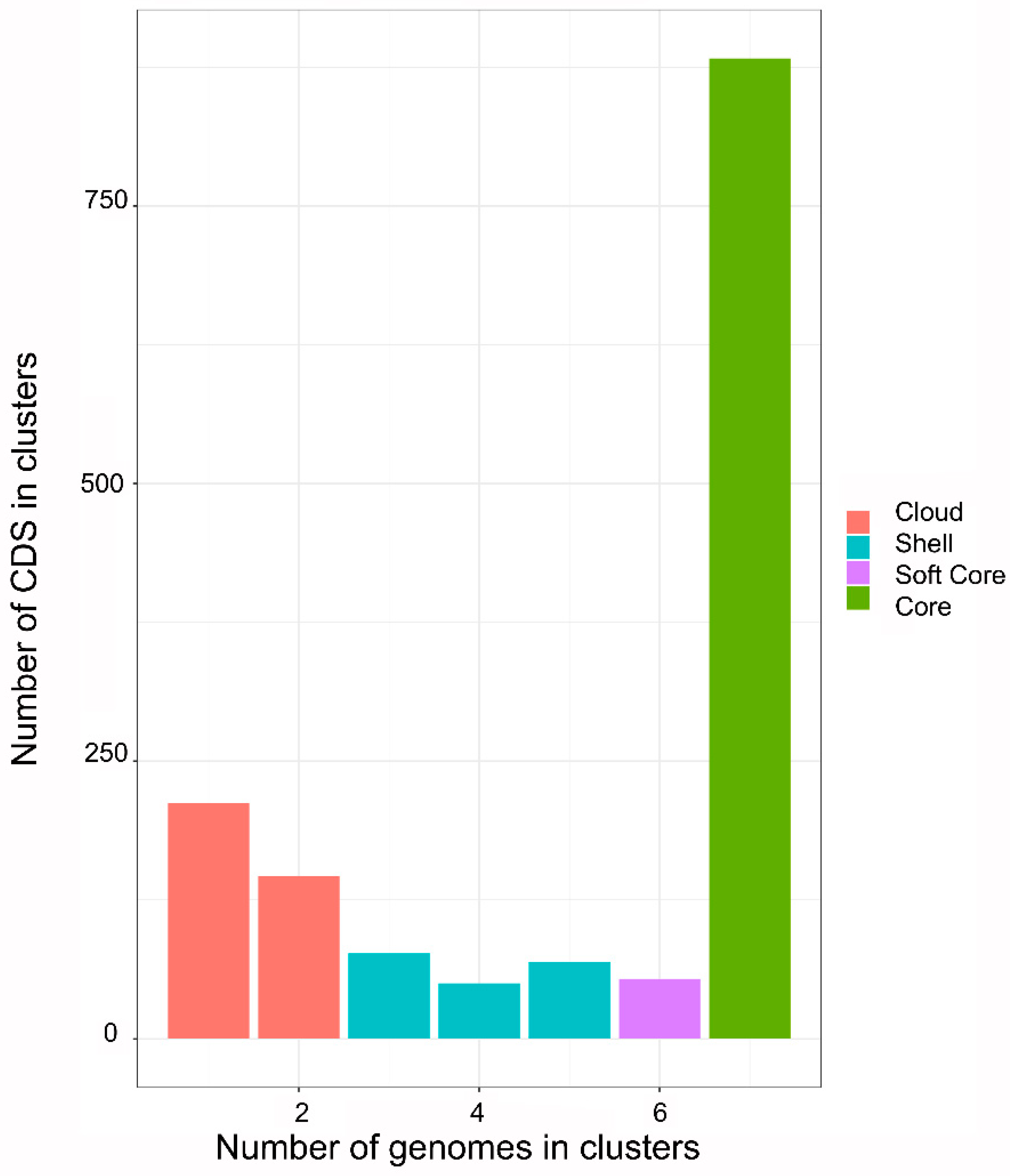
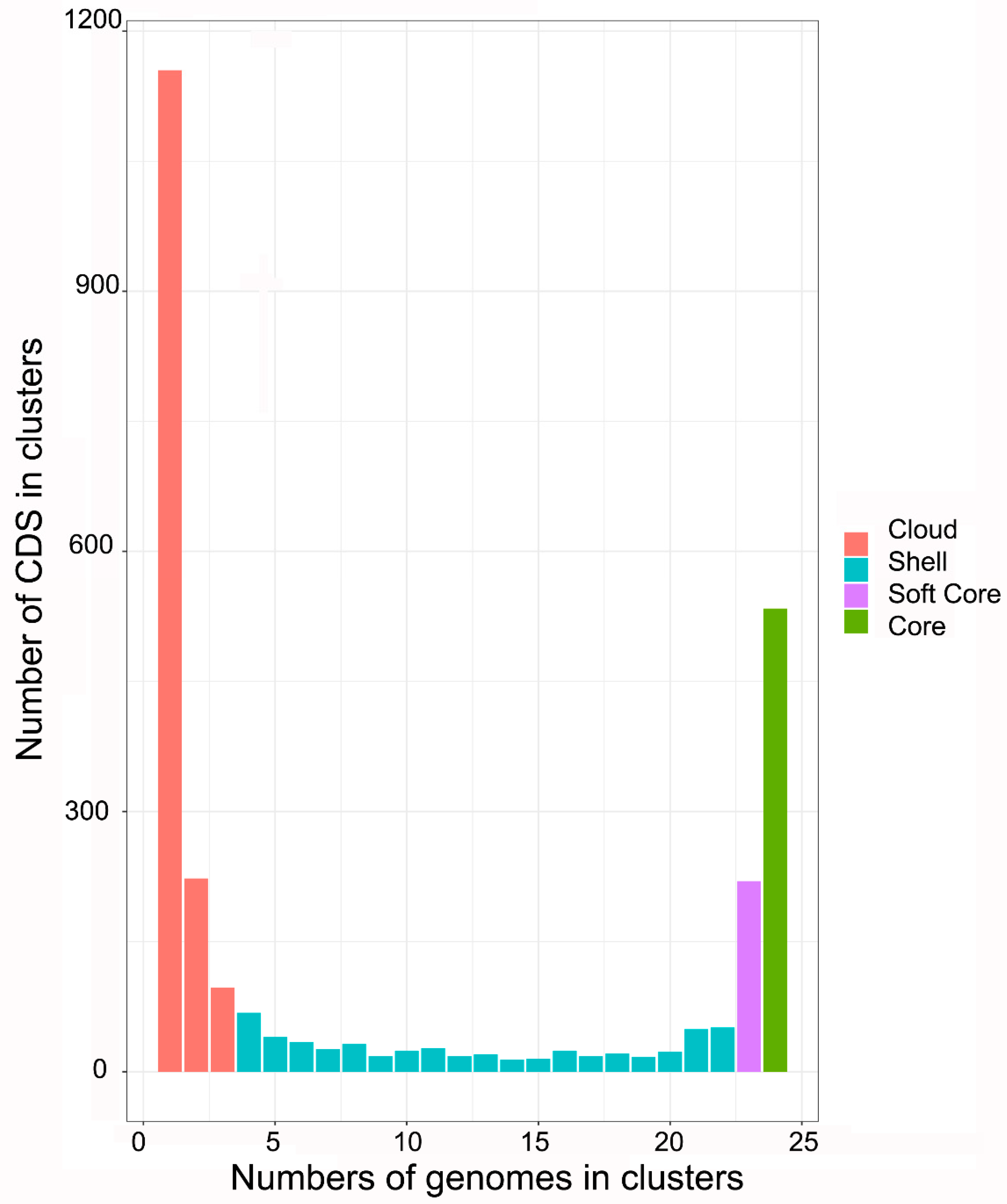
2.2. Phylogenetic and Pan-Genomic Analysis
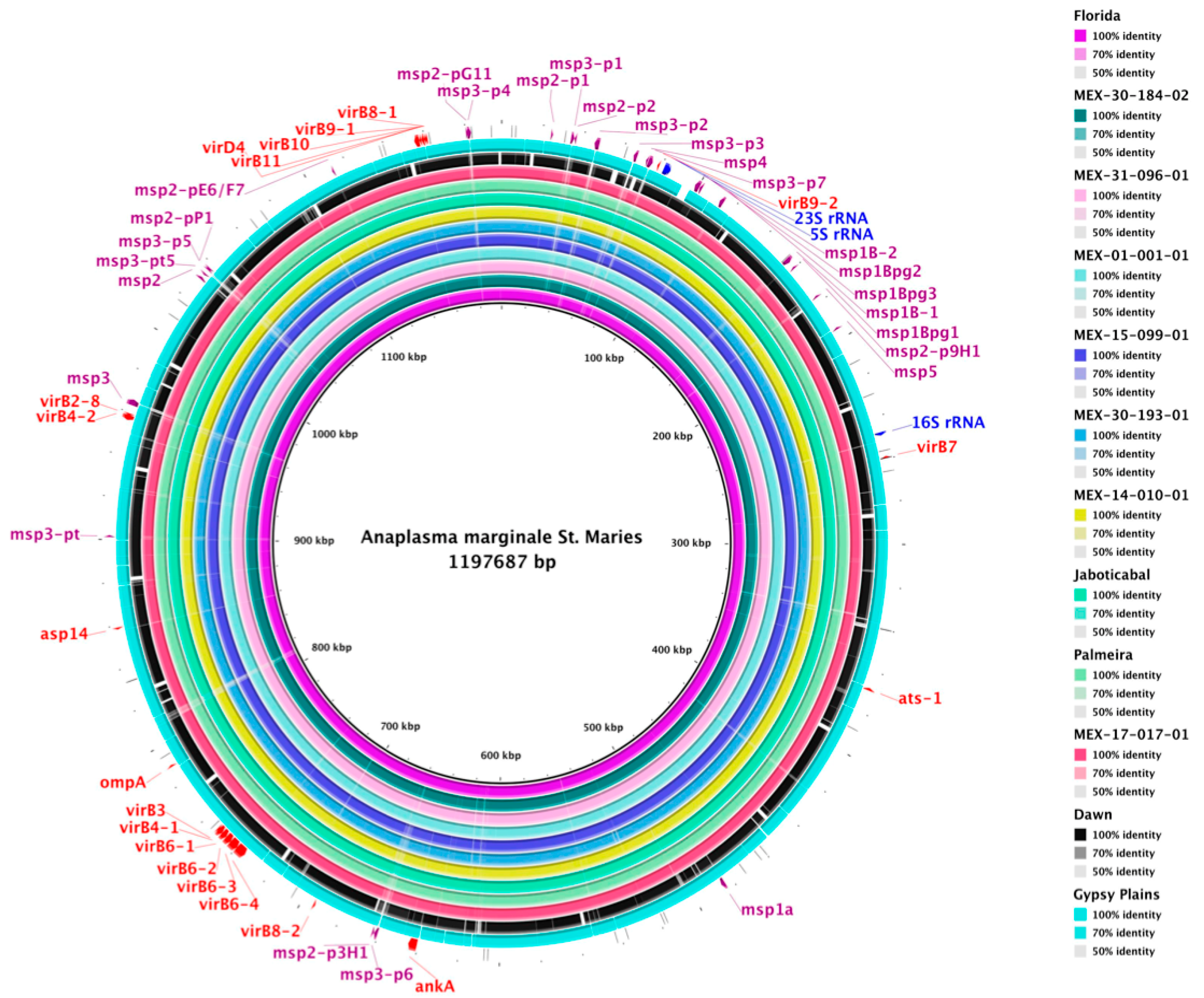
2.3. Genome Comparison
3. Discussion
4. Materials and Methods
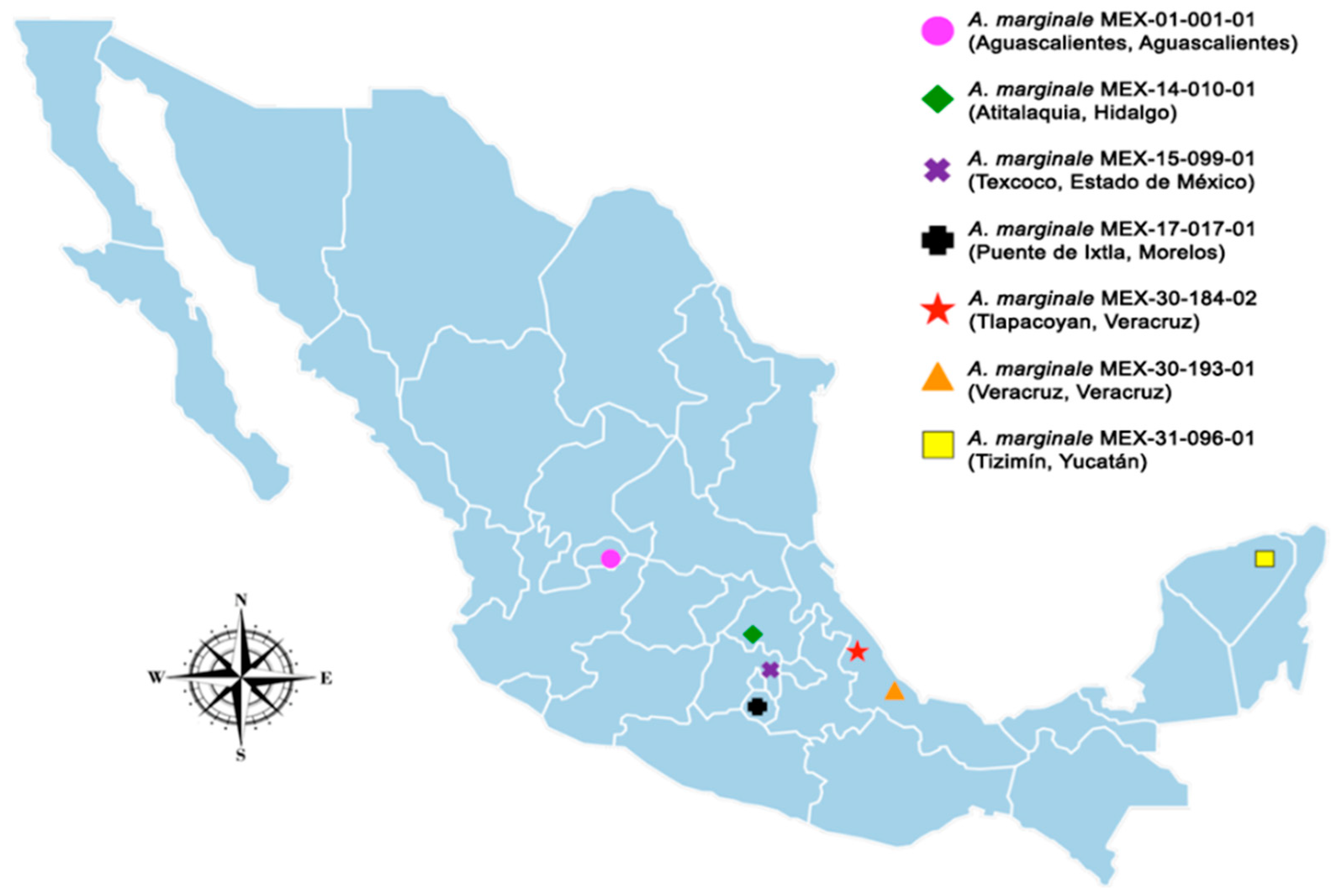
4.1. Sample Collection and Nomenclature
4.2. Genome Sequences and Annotation
4.3. Analysis of MSP1a
4.4. Phylogenetic and Pan-Genomic Analysis
4.5. Comparative Genomics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aubry, P.; Geale, D.W. A review of Bovine anaplasmosis. Transbound. Emerg. Dis. 2011, 58, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Ristic, M. Anaplasmosis. In Diseases of Cattle in the Tropics Economic and Zoonotic Relevance, 1st ed.; Ristic, I.M., Ed.; Martinus Nijhoff: The Hague, The Netherlands, 1981; pp. 327–344. [Google Scholar]
- Theiler, A. Gall-sickness of South Africa. (Anaplasmosis of Cattle.). J. Comp. Pathol. Ther. 1910, 23, 98–115. [Google Scholar] [CrossRef]
- Brayton, K.A.; Dark, M.J.; Palmer, G.H. Anaplasma. In Genome Mapping and Genomics in Animal-Associated Microbes; Springer: Berlin/Heidelberg, Germany, 2009; pp. 85–116. [Google Scholar]
- Dark, M.J.; Al-Khedery, B.; Barbet, A.F. Multistrain genome analysis identifies candidate vaccine antigens of Anaplasma marginale. Vaccine 2011, 29, 4923–4932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocan, K.M.; de la Fuente, J.; Guglielmone, A.A.; Meléndez, R.D. Antigens and alternatives for control of Anaplasma marginale infection in Cattle. Clin. Microbiol. Rev. 2003, 16, 698–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez Ocampo, R.; Vega y Murguía, C.A.; Oviedo Ortega, N.; Rojas Ramírez, E.E.; García Ortiz M, Á.; Preciado de la Torre, J.F.; Cruz, R.R.; García, D.I.D.; Camarillo, S.D.R. Diversidad genética de la región variable de los genes Msp1a y Msp4 en cepas de Anaplasma marginale de México. Rev. Mex. Cienc. Pecu. 2012, 3, 373–387. [Google Scholar]
- Abdala, A.A.; Pipano, E.; Aguirre, D.H.; Gaido, A.B.; Zurbriggen, M.A.; Mangold, A.J.; Guglielmone, A.A. Frozen and fresh Anaplasma centrale vaccines in the protection of cattle against Anaplasma marginale infection. Rev. Elev. Med. Vet. Pays Trop. 1990, 43, 155–158. [Google Scholar] [CrossRef]
- Van Borm, S.; Belak, S.; Freimanis, G.; Fusaro, A.; Granberg, F.; Hoper, D.; King, D.P.; Monne, I.; Orton, R.; Rosseel, T. Next-generation sequencing in veterinary medicine: How can the massive amount of information arising from high-throughput technologies improve diagnosis, control, and management of infectious diseases? Methods Mol. Biol. 2015, 1247, 415–436. [Google Scholar]
- Perea Razo, C.A.; Rodriguez Hernandez, E.; Ponce, S.I.R.; Milian Suazo, F.; Robbe-Austerman, S.; Stuber, T.; Cantó Alarcón, G.J. Molecular epidemiology of cattle tuberculosis in Mexico through whole-genome sequencing and spoligotyping. PLoS ONE 2018, 13, e0201981. [Google Scholar] [CrossRef] [PubMed]
- Cockle, P.J.; Gordon, S.V.; Lalvani, A.; Buddle, B.M.; Hewinson, R.G.; Vordermeier, H.M. Identification of Novel Mycobacterium tuberculosis Antigens with Potential as Diagnostic Reagents or Subunit Vaccine Candidates by Comparative Genomics. Infect. Immun. 2002, 70, 6996–7003. [Google Scholar] [CrossRef] [Green Version]
- Quiroz Castañeda, R.E.; Amaro-Estrada, I.; Martínez-Ocampo, F.; Rodríguez-Camarillo, S.; Dantán González, E.; Cobaxin-Cárdenas, M.; Preciado-de la Torre, J.F. Draft Genome Sequence of Anaplasma marginale Strain Mex-01-001-01, a Mexican Strain That Causes Bovine Anaplasmosis. Microbiol. Resour. Announc. 2018, 7, e01101-18. Available online: http://mra.asm.org/content/7/16/e01101-18.abstract (accessed on 15 May 2022). [CrossRef] [Green Version]
- Martínez-Ocampo, F.; Quiroz-Castañeda, R.E.; Amaro-Estrada, I.; Dantán-González, E.; De La Torre, J.F.P.; Rodríguez-Camarillo, S. Whole-Genome Sequencing of Mexican Strains of Anaplasma marginale: An Approach to the Causal Agent of Bovine Anaplasmosis. Int. J. Genom. 2020, 2020, 5902029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buermans, H.P.J.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1932–1941. Available online: http://0-www-sciencedirect-com.brum.beds.ac.uk/science/article/pii/S092544391400180X (accessed on 16 May 2022). [CrossRef] [PubMed] [Green Version]
- Besser, J.; Carleton, H.A.; Gerner-Smidt, P.; Lindsey, R.L.; Trees, E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin. Microbiol. Infect. 2018, 24, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Fang, R.; Pan, T.; Li, T.; Li, N.; He, Q.; Wu, R.; Peng, Y.; Zhou, Z. Comparative Genomics Analysis of Two Different Virulent Bovine Pasteurella multocida Isolates. Int. J. Genom. 2016, 2016, 4512493. [Google Scholar] [CrossRef] [Green Version]
- Sachman-Ruiz, B.; Lozano, L.; Lira, J.J.; Martínez, G.; Rojas, C.; Álvarez, J.A.; Figueroa, J.V. A Comparative Genomic Study of Attenuated and Virulent Strains of Babesia bigemina. Pathogens 2021, 10, 318. Available online: https://0-www-mdpi-com.brum.beds.ac.uk/2076-0817/10/3/318 (accessed on 17 May 2022). [CrossRef]
- Lebov, J.; Grieger, K.; Womack, D.; Zaccaro, D.; Whitehead, N.; Kowalcyk, B.; MacDonald, P.D. A framework for One Health research. One Health 2017, 3, 44–50. Available online: https://pubmed.ncbi.nlm.nih.gov/28616503 (accessed on 18 May 2022). [CrossRef] [PubMed]
- Estrada-Peña, A.; Naranjo, V.; Acevedo-Whitehouse, K.; Mangold, A.J.; Kocan, K.M.; de la Fuente, J. Phylogeographic analysis reveals association of tick-borne pathogen, Anaplasma marginale, MSP1a sequences with ecological traits affecting tick vector performance. BMC Biol. 2009, 7, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacciani-Mori, L.; Giometto, A.; Suweis, S.; Maritan, A. Dynamic metabolic adaptation can promote species coexistence in competitive microbial communities. PLoS Comput. Biol. 2020, 16, e1007896. Available online: https://pubmed.ncbi.nlm.nih.gov/32379752 (accessed on 19 May 2022). [CrossRef]
- Peel, D.S.; Johnson, R.J.; Mathews, K.H. Cow-calf beef production in Mexico. Beef Sel. Res. Glob. Beef Prod. Trade 2012, 1, 153–172. [Google Scholar]
- Aguilar-Díaz, H.; Quiroz-Castañeda, R.; Cobaxin-Cárdenas, M.; Salinas-Estrella, E.; Amaro-Estrada, I. Advances in the Study of the Tick Cattle Microbiota and the Influence on Vectorial Capacity. Front. Vet. Sci. 2021, 8, 710352. Available online: https://www.frontiersin.org/article/10.3389/fvets.2021.710352 (accessed on 19 May 2022). [CrossRef]
- dos Santos, P.N.; de Almeida-Valim, J.R.; Matos, P.C.M.; da Silva, J.B.; da Fonseca, A.H. Molecular characterization of the msp1 α AmRio1 strain of Anaplasma marginale in calves and experimentally infected ticks. Vet. Parasitol. Reg. Stud. Rep. 2019, 16, 100268. [Google Scholar] [CrossRef] [PubMed]
- Hove, P.; Khumalo, Z.T.H.; Chaisi, M.E.; Oosthuizen, M.C.; Brayton, K.A.; Collins, N.E. Detection and characterisation of Anaplasma marginale and A. centrale in South Africa. Vet. Sci. 2018, 5, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lew, A.E.; Bock, R.E.; Minchin, C.M.; Masaka, S. A msp1alpha polymerase chain reaction assay for specific detection and differentiation of Anaplasma marginale isolates. Vet. Microbiol. 2002, 4, 325–335. [Google Scholar] [CrossRef]
- Peona, V.; Weissensteiner, M.H.; Suh, A. How complete are “complete” genome assemblies?-An avian perspective. Mol. Ecol. Resour. 2018, 18, 1188–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Ocampo, F.; Quiroz-Castañeda, R.E.; Amaro-Estrada, I.; Cobaxin Cárdenas, M.; Dantán-González, E.; Rodríguez-Camarillo, S. Draft Genome Sequences of Anaplasma marginale Strains MEX-15-099-01 and MEX-31-096-01, Two Mexican Isolates with Different Degrees of Virulence. Microbiol. Resour. Announc. 2019, 8, e01184-19. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pmc/articles/PMC3624806/ (accessed on 13 May 2022). [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- NCBI BLAST Download. Available online: http://www.ncbi.nih.gov/BLAST/download.shtml (accessed on 17 May 2022).
- Catanese, H.N.; Brayton, K.A.; Gebremedhin, A.H. RepeatAnalyzer: A tool for analysing and managing short-sequence repeat data. BMC Genom. 2016, 17, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Fuente, J.; Ruybal, P.; Mtshali, M.S.; Naranjo, V.; Shuqing, L.; Mangold, A.J. Analysis of world strains of Anaplasma marginale using major surface protein 1a repeat sequences. Vet. Microbiol. 2007, 119, 382–390. Available online: http://0-www-sciencedirect-com.brum.beds.ac.uk/science/article/pii/S0378113506003828 (accessed on 10 May 2022). [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high-performance computing Europe PMC Funders Group. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A Simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [Green Version]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Assembly Level | Total Length (bp) * | G + C Content (%) * | Genes ** | CDS ** | rRNAs # | tRNAs ## |
|---|---|---|---|---|---|---|---|
| A. marginale Dawn | Incomplete chromosome | 1,196,760 | 49.73 | 1145 | 1107 | 3 | 35 |
| A. marginale Gypsy Plains | Incomplete chromosome | 1,198,622 | 49.72 | 1189 | 1149 | 3 | 37 |
| A. marginale Jaboticabal | Incomplete chromosome | 1,195,321 | 49.77 | 1238 | 1198 | 3 | 37 |
| A. marginale Palmeira | Incomplete chromosome | 1,195,200 | 49.75 | 1219 | 1179 | 3 | 37 |
| A. marginale MEX-01-001-01 | Draft (34 contigs) | 1,179,425 | 49.79 | 1218 | 1178 | 3 | 37 |
| A. marginale MEX-14-010-01 | Draft (46 contigs) | 1,172,327 | 49.79 | 1190 | 1150 | 3 | 37 |
| A. marginale MEX-15-099-01 | Draft (32 contigs) | 1,169,440 | 49.79 | 1185 | 1145 | 3 | 37 |
| A. marginale MEX-17-017-01 | Draft (41 contigs) | 1,172,716 | 49.79 | 1203 | 1163 | 3 | 37 |
| A. marginale MEX-30-184-02 | Draft (40 contigs) | 1,176,681 | 49.79 | 1205 | 1165 | 3 | 37 |
| A. marginale MEX-30-193-01 | Draft (41 contigs) | 1,167,111 | 49.80 | 1178 | 1138 | 3 | 37 |
| A. marginale MEX-31-096-01 | Draft (43 contigs) | 1,176,579 | 49.79 | 1204 | 1164 | 3 | 37 |
| A. marginale Puerto Rico | Draft (59 contigs) | 1,158,530 | 49.80 | 1220 | 1180 | 3 | 37 |
| A. marginale Florida | Complete chromosome | 1,202,435 | 49.77 | 1227 | 1187 | 3 | 37 |
| A. marginale Florida | Draft (204 contigs) | 1,136,981 | 49.84 | 1186 | 1147 | 3 | 35 |
| A. marginale Florida Relapse | Draft (61 contigs) | 1,154,411 | 49.81 | 1189 | 1149 | 3 | 37 |
| A. marginale Mississippi | Draft (82 contigs) | 1,141,520 | 49.79 | 1208 | 1168 | 3 | 37 |
| A. marginale Okeechobee | Draft (403 contigs) | 1,390,987 | 47.49 | 1267 | 1225 | 3 | 38 |
| A. marginale Oklahoma | Draft (57 contigs) | 1,156,921 | 49.82 | 1167 | 1127 | 3 | 37 |
| A. marginale Oklahoma-2 | Draft (44 scaffolds) | 1,160,766 | 49.79 | 1188 | 1148 | 3 | 37 |
| A. marginale South Idaho | Draft (358 contigs) | 1,409,432 | 46.73 | 1316 | 1262 | 3 | 47 |
| A. marginale St. Maries | Complete chromosome | 1,197,687 | 49.76 | 1250 | 1210 | 3 | 37 |
| A. marginale St. Maries | Draft (60 contigs) | 1,155,236 | 49.79 | 1194 | 1154 | 3 | 37 |
| A. marginale Virginia | Draft (70 contigs) | 1,153,875 | 49.79 | 1241 | 1201 | 3 | 37 |
| A. marginale Washington Okanogan | Draft (332 contigs) | 1,383,255 | 46.94 | 1331 | 1274 | 3 | 52 |
| Organism | Geographic Region | Repeats Structure Previously Reported * | Repeats Structure Reported in This Work ** | m | n | SD-ATG Distance | Genotype *** | Genotype Frequency Per Ecoregion Cluster *** | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||||||||
| A. marginale MEX-01-001-01 | Aguascalientes, Aguascalientes | 4 9 10 11 9 | 4 9 10 11 9 | 2 | 7 | 23 | E | 0.75 | 0.00 | 0.25 | 0.00 |
| A. marginale MEX-14-010-01 | Atitalaquia, Hidalgo | τ 57 13 18 | τ 22-2 13 18 | 2 | 7 | 23 | E | 0.75 | 0.00 | 0.25 | 0.00 |
| A. marginale MEX-15-099-01 | Texcoco, Estado de México | α β β Γ | α β Γ | 2 | 7 | 23 | E | 0.75 | 0.00 | 0.25 | 0.00 |
| A. marginale MEX-17-017-01 | Puente de Ixtla, Morelos | 12 13 14 | 12 14 | 3 | 5 | 23 | G | 0.15 | 0.14 | 0.56 | 0.14 |
| A. marginale MEX-30-184-02 | Tlapacoyan, Veracruz | 73 β β β Γ | T C | 3 | 5 | 23 | G | 0.15 | 0.14 | 0.56 | 0.14 |
| A. marginale MEX-30-193-01 | Veracruz, Veracruz | α β β Γ | α β β Γ | 2 | 7 | 23 | E | 0.75 | 0.00 | 0.25 | 0.00 |
| A. marginale MEX-31-096-01 | Tizimín, Yucatán | T C B B C B π | T C | 3 | 5 | 23 | G | 0.15 | 0.14 | 0.56 | 0.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dantán-González, E.; Quiroz-Castañeda, R.E.; Aguilar-Díaz, H.; Amaro-Estrada, I.; Martínez-Ocampo, F.; Rodríguez-Camarillo, S. Mexican Strains of Anaplasma marginale: A First Comparative Genomics and Phylogeographic Analysis. Pathogens 2022, 11, 873. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11080873
Dantán-González E, Quiroz-Castañeda RE, Aguilar-Díaz H, Amaro-Estrada I, Martínez-Ocampo F, Rodríguez-Camarillo S. Mexican Strains of Anaplasma marginale: A First Comparative Genomics and Phylogeographic Analysis. Pathogens. 2022; 11(8):873. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11080873
Chicago/Turabian StyleDantán-González, Edgar, Rosa Estela Quiroz-Castañeda, Hugo Aguilar-Díaz, Itzel Amaro-Estrada, Fernando Martínez-Ocampo, and Sergio Rodríguez-Camarillo. 2022. "Mexican Strains of Anaplasma marginale: A First Comparative Genomics and Phylogeographic Analysis" Pathogens 11, no. 8: 873. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11080873