
Unique Evolution of SARS-CoV-2 in the Second Large Cruise Ship Cluster in Japan

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Clinical Samples
2.3. Diagnostic Tests
2.4. Whole-Genome Sequencing of SARS-CoV-2
2.5. Phylogenetic Analysis
2.6. Mutation Site Identification
2.7. Haplotype Network Construction
2.8. Pairwise Distance Calculation and Genetic Diversity Analysis
3. Results
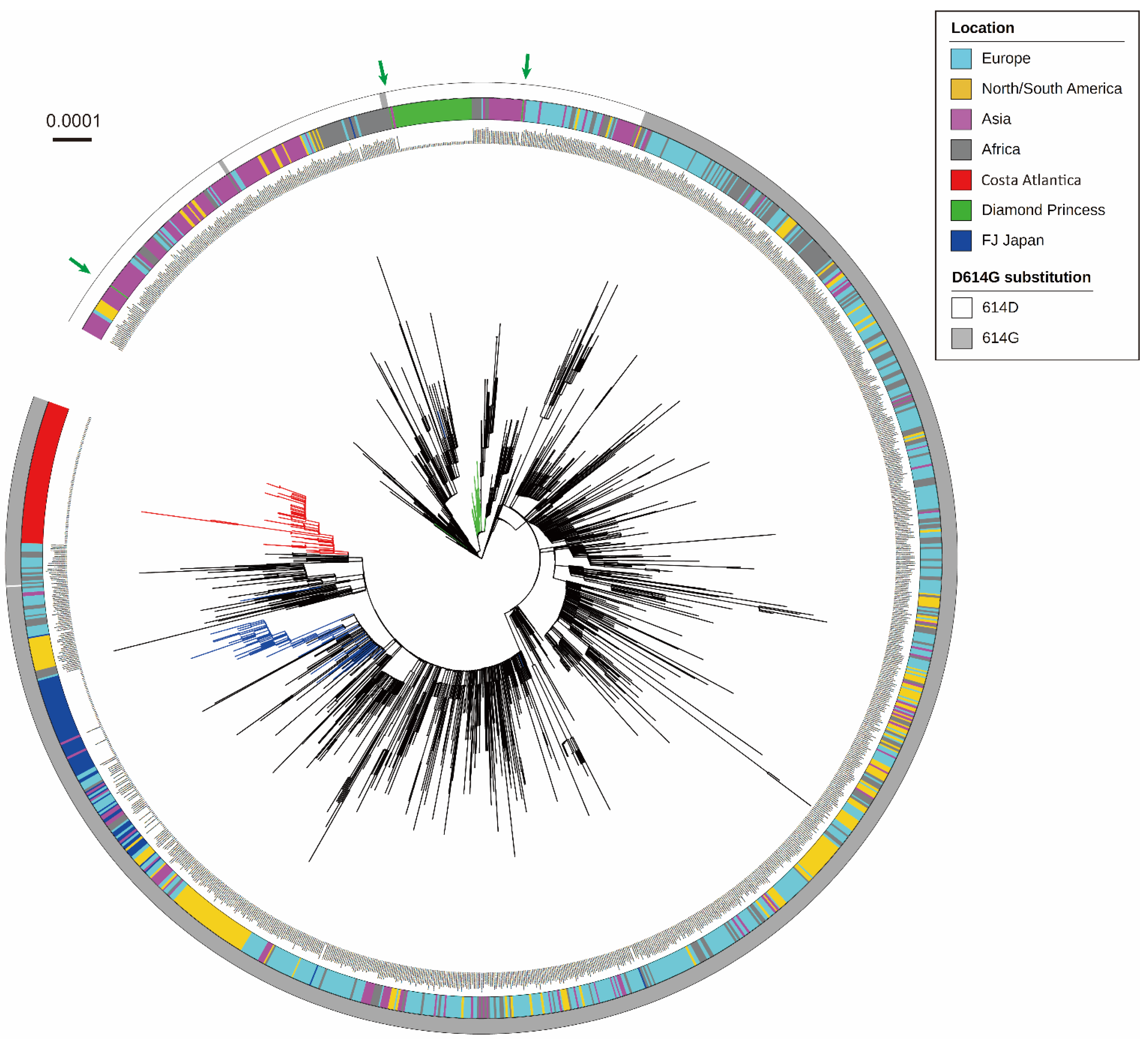
3.1. Phylogenetic Analysis of SARS-CoV-2 Detected in a Cruise Ship Cluster
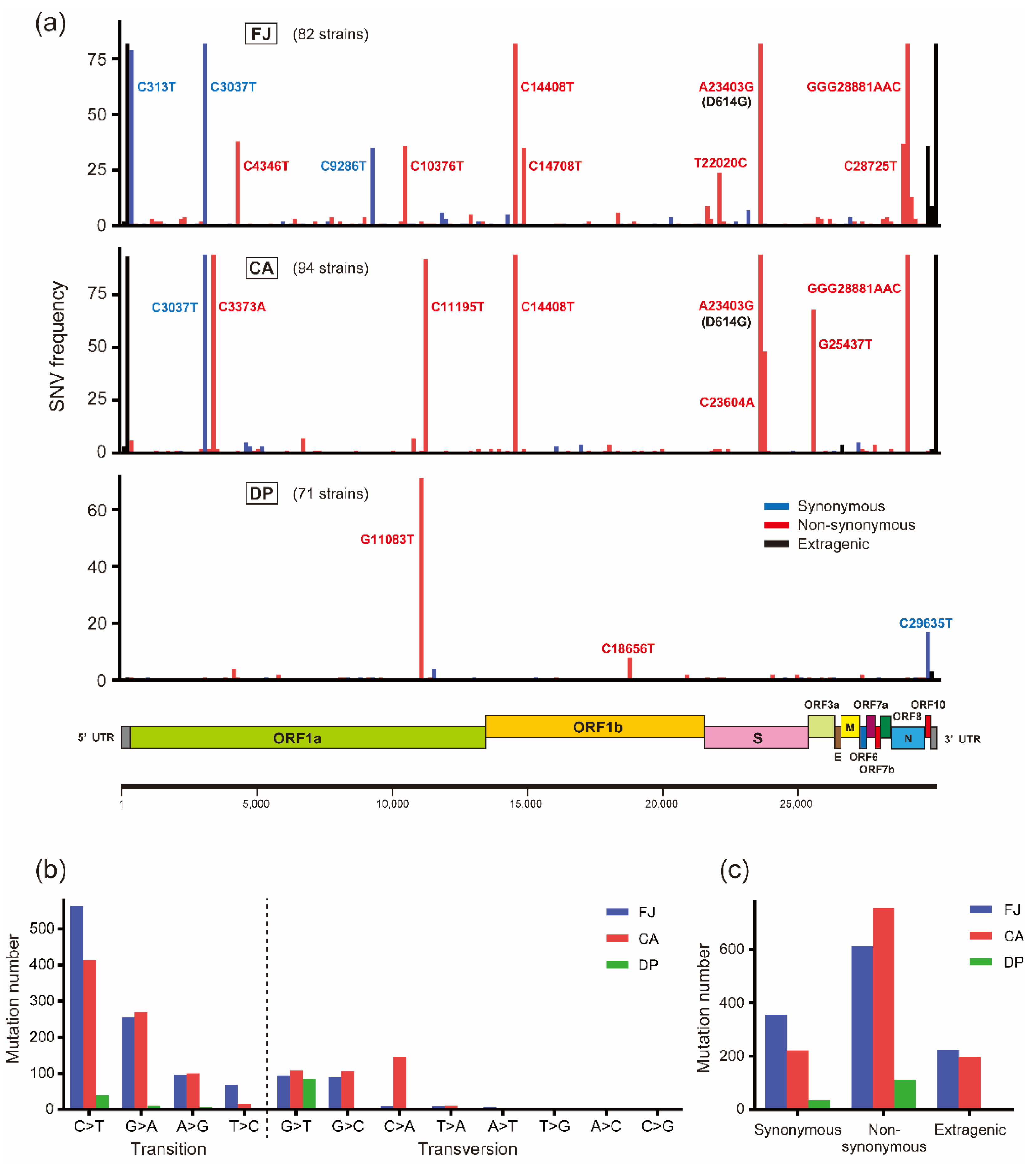
3.2. Characterization of Mutation Sites and Substitution Patterns in CA Strains
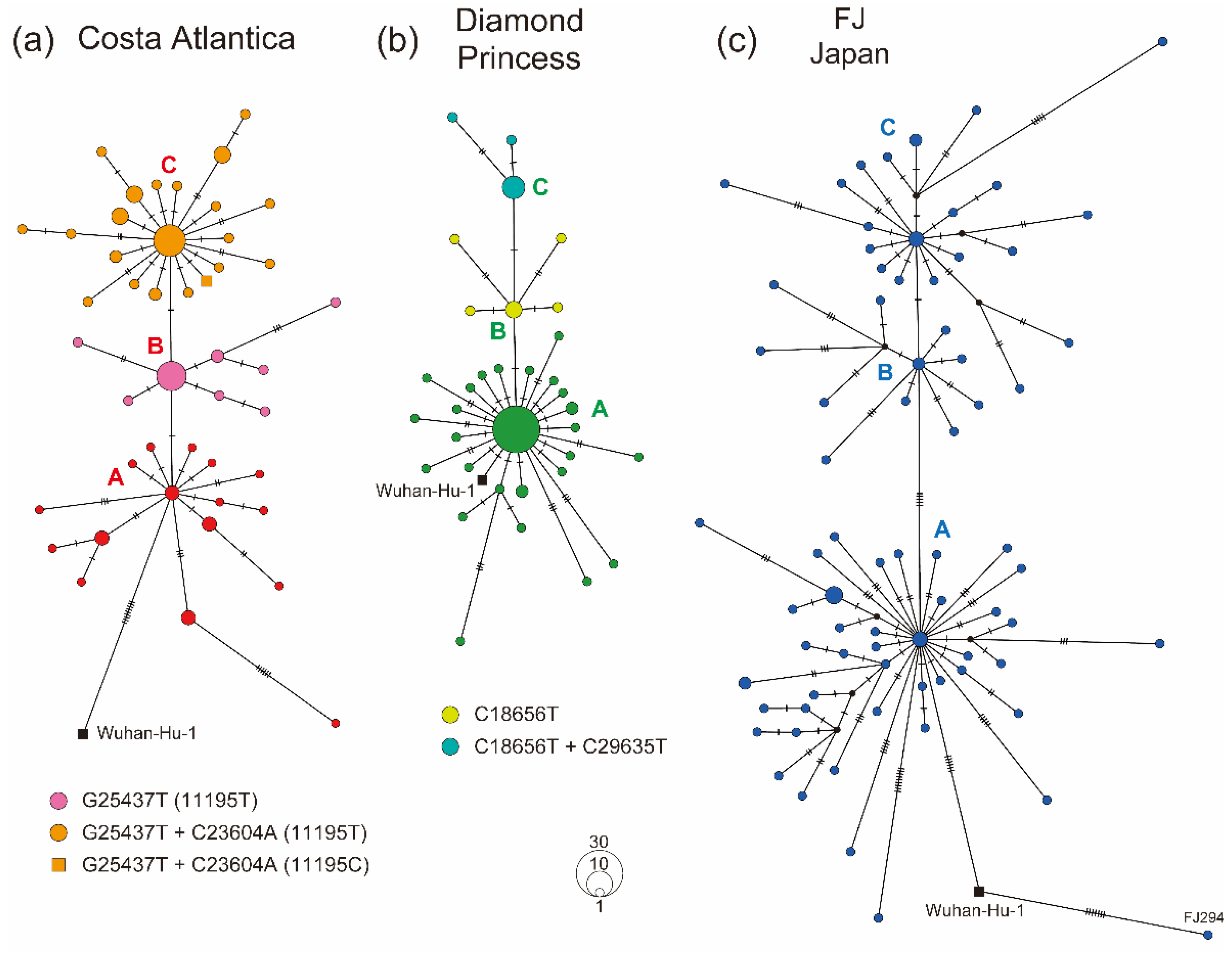
3.3. Haplotype Diversity in SARS-CoV-2 Strains Detected in Cruise Ship Clusters
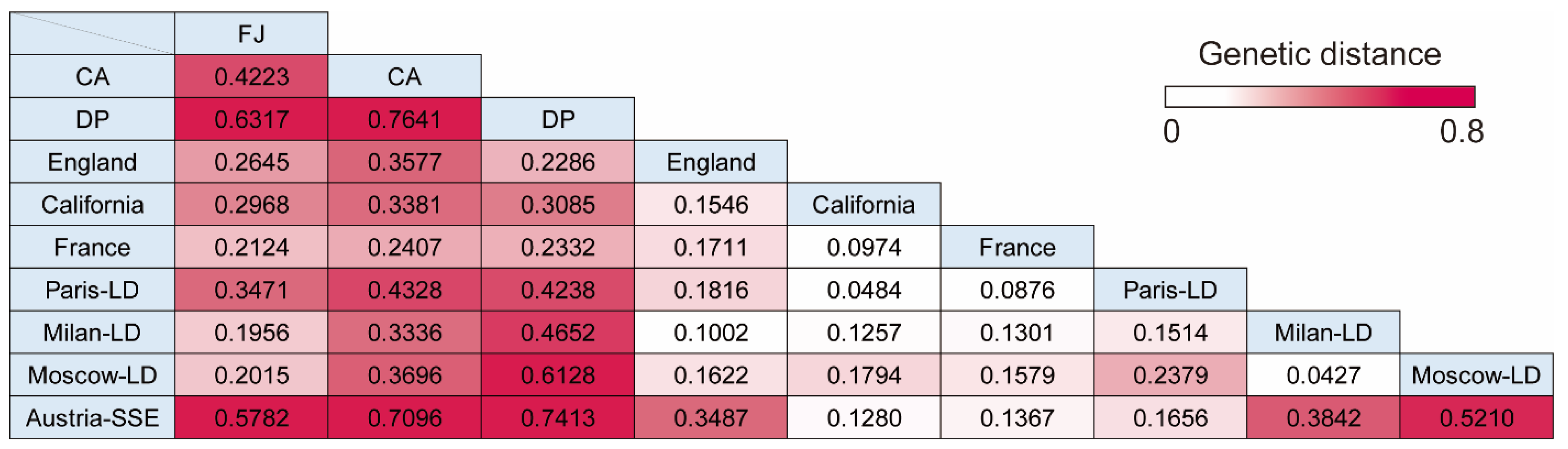
3.4. Pairwise Genetic Distance between Cruise Ship Strains and Isolates from Lockdown Cities or Superspreading Events
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lo, S.W.; Jamrozy, D. Genomics and Epidemiological Surveillance. Nat. Rev. Microbiol. 2020, 18, 478. [Google Scholar] [CrossRef]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing Global and Regional Adaptive Evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2104241118. [Google Scholar] [CrossRef]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 3 September 2021).
- Li, T.; Liu, D.; Yang, Y.; Guo, J.; Feng, Y.; Zhang, X.; Cheng, S.; Feng, J. Phylogenetic Supertree Reveals Detailed Evolution of SARS-CoV-2. Sci. Rep. 2020, 10, 22366. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Barnes, C.O.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; et al. mRNA Vaccine-Elicited Antibodies to SARS-CoV-2 and Circulating Variants. Nature 2021, 592, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, F.; Preibisch, G.; Giziński, S.; Kochańczyk, M.; Lipniacki, T. SARS-CoV-2 Variant of Concern 202012/01 Has about 2-Fold Replicative Advantage and Acquires Concerning Mutations. Viruses 2021, 13, 392. [Google Scholar] [CrossRef] [PubMed]
- du Plessis, L.; McCrone, J.T.; Zarebski, A.E.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.R.; et al. Establishment and Lineage Dynamics of the SARS-CoV-2 Epidemic in the UK. Science 2021, 371, 708–712. [Google Scholar] [CrossRef] [PubMed]
- da Silva Filipe, A.; Shepherd, J.G.; Williams, T.; Hughes, J.; Aranday-Cortes, E.; Asamaphan, P.; Ashraf, S.; Balcazar, C.; Brunker, K.; Campbell, A.; et al. Genomic Epidemiology Reveals Multiple Introductions of SARS-CoV-2 from Mainland Europe into Scotland. Nat. Microbiol. 2021, 6, 112–122. [Google Scholar] [CrossRef]
- Alteri, C.; Cento, V.; Piralla, A.; Costabile, V.; Tallarita, M.; Colagrossi, L.; Renica, S.; Giardina, F.; Novazzi, F.; Gaiarsa, S.; et al. Genomic Epidemiology of SARS-CoV-2 Reveals Multiple Lineages and Early Spread of SARS-CoV-2 Infections in Lombardy, Italy. Nat. Commun. 2021, 12, 434. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Gu, H.; Chang, L.D.J.; Cheuk, S.S.Y.; Gurung, S.; Krishnan, P.; Ng, D.Y.M.; Liu, G.Y.Z.; Wan, C.K.C.; Tsang, D.N.C.; et al. SARS-CoV-2 Superspread in Fitness Center, Hong Kong, China, March 2021. Emerg. Infect. Dis. 2021, 27, 2230–2232. [Google Scholar] [CrossRef]
- Mizumoto, K.; Kagaya, K.; Zarebski, A.; Chowell, G. Estimating the Asymptomatic Proportion of Coronavirus disease 2019 (COVID-19) Cases on Board the Diamond Princess Cruise Ship, Yokohama, Japan, 2020. Eurosurveillance 2020, 25, 32183930. [Google Scholar] [CrossRef] [Green Version]
- Tabata, S.; Imai, K.; Kawano, S.; Ikeda, M.; Kodama, T.; Miyoshi, K.; Obinata, H.; Mimura, S.; Kodera, T.; Kitagaki, M.; et al. Clinical Characteristics of COVID-19 in 104 People with SARS-CoV-2 Infection on the Diamond Princess Cruise Ship: A Retrospective Analysis. Lancet Infect. Dis. 2020, 20, 1043–1050. [Google Scholar] [CrossRef]
- Zhang, S.; Diao, M.; Yu, W.; Pei, L.; Lin, Z.; Chen, D. Estimation of the Reproductive Number of Novel Coronavirus (COVID-19) and the Probable Outbreak Size on the Diamond Princess Cruise Ship: A Data-Driven Analysis. Int. J. Infect. Dis. 2020, 93, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Emery, J.C.; Russell, T.W.; Liu, Y.; Hellewell, J.; Pearson, C.A.; CMMID COVID-19 Working Group; Knight, G.M.; Eggo, R.M.; Kucharski, A.J.; Funk, S.; et al. The contribution of asymptomatic SARS-CoV-2 infections to transmission on the Diamond Princess cruise ship. eLife 2020, 9, e58699. [Google Scholar] [CrossRef] [PubMed]
- Sekizuka, T.; Itokawa, K.; Kageyama, T.; Saito, S.; Takayama, I.; Asanuma, H.; Nao, N.; Tanaka, R.; Hashino, M.; Takahashi, T.; et al. Haplotype Networks of SARS-CoV-2 Infections in the Diamond Princess Cruise Ship Outbreak. Proc. Natl. Acad. Sci. USA 2020, 117, 20198–20201. [Google Scholar] [CrossRef]
- Maeda, H.; Sando, E.; Toizumi, M.; Arima, Y.; Shimada, T.; Tanaka, T.; Tashiro, M.; Fujita, A.; Yanagihara, K.; Takayama, H.; et al. Epidemiology of Coronavirus Disease Outbreak among Crewmembers on Cruise Ship, Nagasaki City, Japan, April 2020. Emerg. Infect. Dis. 2021, 27, 2251–2260. [Google Scholar] [CrossRef]
- Yoshikawa, R.; Abe, H.; Igasaki, Y.; Negishi, S.; Goto, H.; Yasuda, J. Development and Evaluation of a Rapid and Simple Diagnostic Assay for COVID-19 Based on Loop-Mediated Isothermal Amplification. PLoS Negl. Trop. Dis. 2020, 14, e0008855. [Google Scholar] [CrossRef]
- Abe, H.; Ushijima, Y.; Bikangui, R.; Ondo, G.N.; Zadeh, V.R.; Pemba, C.M.; Mpingabo, P.I.; Igasaki, Y.; de Vries, S.G.; Grobusch, M.P.; et al. First Evidence for Continuous Circulation of Hepatitis A Virus Subgenotype IIA in Central Africa. J. Viral Hepat. 2020, 27, 1234–1242. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR Method for MinION and Illumina Sequencing of Zika and Other Virus Genomes Directly from Clinical Samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [Green Version]
- Abe, H.; Ushijima, Y.; Bikangui, R.; Zoa-Assoumou, S.; Ondo, G.N.; Manouana, G.P.; More, A.; Skarwan, E.; Yali-Assy-Oyamli, Y.; Ndeboko, B.; et al. Unrecognized Introduction of SARS-CoV-2 Variants of Concern to Central Africa: Import and Local Transmission of B.1.1.7 in Gabon in the Very Early Stage of the Variant Spread to the African Continent. J. Med. Virol. 2021, 93, 6054–6058. [Google Scholar] [CrossRef]
- Giandhari, J.; Pillay, S.; Wilkinson, E.; Tegally, H.; Sinayskiy, I.; Schuld, M.; Lourenço, J.; Chimukangara, B.; Lessells, R.; Moosa, Y.; et al. Early transmission of SARS-CoV-2 in South Africa: An epidemiological and phylogenetic report. Int. J. Infect. Dis. 2021, 103, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Collivignarelli, M.C.; De Rose, C.; Abbà, A.; Baldi, M.; Bertanza, G.; Pedrazzani, R.; Sorlini, S.; Carnevale Miino, M. Analysis of Lockdown for CoViD-19 Impact on NO2 in London, Milan and Paris: What Lesson Can Be Learnt? Process Saf. Environ. Prot. 2021, 146, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Popa, A.; Genger, J.W.; Nicholson, M.D.; Penz, T.; Schmid, D.; Aberle, S.W.; Agerer, B.; Lercher, A.; Endler, L.; Colaço, H.; et al. Genomic Epidemiology of Superspreading Events in Austria Reveals Mutational Dynamics and Transmission Properties of SARS-CoV-2. Sci. Transl. Med. 2020, 12, eabe2555. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, L.F.; Plucinski, M.M.; Marston, B.J.; Kurbatova, E.V.; Knust, B.; Murray, E.L.; Pesik, N.; Rose, D.; Fitter, D.; Kobayashi, M.; et al. Public Health Responses to COVID-19 Outbreaks on Cruise Ships—Worldwide, February–March 2020. Morb. Mortal. Wkly. Rep. 2020, 69, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal Signal and the Phylodynamic Threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef]
- Nie, Q.; Li, X.; Chen, W.; Liu, D.; Chen, Y.; Li, H.; Li, D.; Tian, M.; Tan, W.; Zai, J. Phylogenetic and Phylodynamic Analyses of SARS-CoV-2. Virus Res. 2020, 287, 198098. [Google Scholar] [CrossRef]
- Li, X.; Zai, J.; Zhao, Q.; Nie, Q.; Li, Y.; Foley, B.T.; Chaillon, A. Evolutionary History, Potential Intermediate Animal Host, and Cross-Species Analyses of SARS-CoV-2. J. Med. Virol. 2020, 92, 602–611. [Google Scholar] [CrossRef]
- Subramanian, R.; He, Q.; Pascual, M. Quantifying Asymptomatic Infection and Transmission of COVID-19 in New York City Using Observed Cases, Serology, and Testing Capacity. Proc. Natl. Acad. Sci. USA 2021, 118, e2019716118. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Ramanathan, M.; Ferguson, I.D.; Miao, W.; Khavari, P.A. SARS-CoV-2 B.1.1.7 and B.1.351 Spike Variants Bind Human ACE2 with Increased Affinity. Lancet Infect. Dis. 2021, 21, 1070. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784. [Google Scholar] [CrossRef]
- Fehr, A.R.; Athmer, J.; Channappanavar, R.; Phillips, J.M.; Meyerholz, D.K.; Perlman, S. The nsp3 macrodomain promotes virulence in mice with coronavirus-induced encephalitis. J. Virol. 2015, 89, 1523–1536. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, M.E.; Chen, Y.; Kuny, C.; Maejima, T.; Lease, R.; Ferraris, D.; Aikawa, M.; Sullivan, C.S.; Perlman, S.; Fehr, A.R. The coronavirus macrodomain is required to prevent PARP-mediated inhibition of virus replication and enhancement of IFN expression. PLoS Pathog. 2019, 15, e1007756. [Google Scholar] [CrossRef] [Green Version]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.K.; Ahel, I.; Perlman, S. The Conserved Coronavirus Macrodomain Promotes Virulence and Suppresses the Innate Immune Response during Severe Acute Respiratory Syndrome Coronavirus Infection. MBio 2016, 7, e01721-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallapaty, S. What the Cruise-Ship Outbreaks Reveal about COVID-19. Nature 2020, 580, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, H.; Hanaoka, S.; Kawasaki, T. The Cruise Industry and the COVID-19 Outbreak. Transp. Res. Interdiscip. Perspect. 2020, 5, 100136. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sample | Strain | Mutations | Amino Acid Substitutions | ||
|---|---|---|---|---|---|---|
| Total Number | Number /Strain | Total Number | Number/Strain | |||
| FJ Japan | Clinical tests | 82 | 1191 | 14.52 | 612 | 7.46 |
| Costa Atlantica | Cruise ship | 94 | 1175 | 12.50 | 755 | 8.03 |
| Diamond Princess | Cruise ship | 71 | 149 | 2.10 | 110 | 1.55 |
| FJ Japan (n = 82) | Costa Atlantica (n = 94) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Position 1 | Ref 2 | Mut 3 | Protein | Substitution | n | Position | Ref | Mut | Protein | Substitution | n |
| 14408 | C | T | ORF1b NSP12b | P314L | 82 | 3373 | C | A | ORF1a NSP3 | D218E | 94 |
| 23403 | A | G | S | D614G | 82 | 14408 | C | T | ORF1b NSP12b | P314L | 94 |
| 28881 | GGG | AAC | N | RG203KR | 82 | 23403 | A | G | S | D614G | 94 |
| 4346 | T | C | ORF1a NSP3 | S543P | 35 | 28881 | GGG | AAC | N | RG203KR | 94 |
| 10376 | C | T | ORF1a NSP5 | P108S | 35 | 11195 | C | T | ORF1a NSP6 | L75F | 92 |
| 14708 | C | T | ORF1b NSP12b | A414V | 35 | 25437 | G | T | ORF3a | L15F | 64 |
| 28725 | C | T | N | P151L | 35 | 23604 | C | A | S | P681H | 44 |
| 22020 | T | C | S | M153T | 23 | 10755 | C | T | ORF1a NSP5 | A234V | 7 |
| 28975 | G | T | N | M234I | 13 | 376 | G | C | ORF1a NSP1 | E37D | 6 |
| 21518 | G | T | ORF1b NSP16 | R287I | 7 | 6660 | G | A | ORF1a NSP3 | S1314N | 5 |
| 18167 | C | T | ORF1b NSP14 | P43L | 6 | 17876 | C | T | ORF1b NSP13 | T547I | 4 |
| 7728 | C | T | ORF1a NSP3 | S1670F | 4 | 25445 | G | T | ORF3a | G18V | 4 |
| 12869 | A | T | ORF1a NSP9 | T62S | 3 | 2901 | T | C | ORF1a NSP3 | V61A | 2 |
| 2167 | G | T | ORF1a NSP2 | K454N | 2 | 3230 | G | T | ORF1a NSP3 | G171C | 2 |
| 2910 | C | T | ORF1a NSP3 | T64I | 2 | 3514 | G | T | ORF1a NSP3 | M265I | 2 |
| 4309 | G | T | ORF1a NSP3 | K530N | 2 | 5018 | G | T | ORF1a NSP3 | D767Y | 2 |
| 11083 | G | T | ORF1a NSP6 | L37F | 2 | 13080 | T | C | ORF1a NSP10 | F19S | 2 |
| 21575 | C | T | S | L5F | 2 | 13624 | G | T | ORF1b NSP12b | D53Y | 2 |
| 21614 | C | T | S | L18F | 2 | 13922 | A | G | ORF1b NSP12b | D152G | 2 |
| 23481 | C | T | S | S640F | 2 | 19885 | AA | CG | ORF1b NSP15 | K89R | 2 |
| 26966 | T | A | M | H148Q | 2 | 27688 | C | T | ORF7a | P99S | 2 |
| 27925 | C | A | ORF8 | T11K | 2 | 28302 | G | T | N | R10L | 2 |
| Diamond Princess (n = 71) | |||||||||||
| Position | Ref | Mut | Protein | Substitution | n | ||||||
| 11083 | G | T | ORF1a NSP6 | L37F | 71 | ||||||
| 18656 | C | T | ORF1b NSP14 | T206I | 8 | ||||||
| 4127 | G | A | ORF1a NSP3 | G470S | 2 | ||||||
| 5845 | A | T | ORF1a NSP3 | K1042N | 2 | ||||||
| FJ Japan (n = 82) | Costa Atlantica (n = 94) | Diamond Princess (n = 71) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pos 1 | Ref 2 | Mut 3 | Sub 4 | n | Pos | Ref | Mut | Sub | n | Pos | Ref | Mut | Sub | n |
| 21575 | C | T | L5F | 2 | 21721 | C | A | D53E | 1 | 21575 | C | T | L5F | 1 |
| 21614 | C | T | L18F | 2 | 21765 | T | G | I68R | 1 | 21917 | A | G | I119V | 1 |
| 21707 | C | T | H49Y | 1 | 22021 | G | T | M153I | 1 | 22104 | G | T | G181V | 1 |
| 22020 | T | C | M153T | 23 | 22275 | T | A | F238Y | 1 | 23856 | G | T | R765L | 1 |
| 22199 | G | C | V213L | 1 | 22289 | G | T | A243S | 1 | 24797 | C | A | P1079T | 1 |
| 22317 | G | T | G252V | 1 | 23403 | A | G | D614G | 94 | 24819 | A | G | K1086R | 1 |
| 23403 | A | G | D614G | 82 | 23604 | C | A | P681H | 44 | 25244 | G | T | V1228L | 1 |
| 23481 | C | T | S640F | 2 | 23705 | C | A | P715T | 1 | |||||
| 24328 | G | C | L922F | 1 | ||||||||||
| 25088 | G | T | V1176F | 1 | ||||||||||
| 25317 | C | A | S1252Y | 1 | ||||||||||
| Group | FJ | CA | DP | England | California | France | Paris-LD | Milan-LD | Moscow-LD | Austria-SSE |
|---|---|---|---|---|---|---|---|---|---|---|
| Fu’s Fs | −24.7827 | −10.2815 | −9.5685 | −23.9885 | −23.7186 | −23.4760 | −24.4464 | −24.5018 | −25.1710 | −13.5053 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abe, H.; Ushijima, Y.; Amano, M.; Sakurai, Y.; Yoshikawa, R.; Kinoshita, T.; Kurosaki, Y.; Yanagihara, K.; Izumikawa, K.; Morita, K.; et al. Unique Evolution of SARS-CoV-2 in the Second Large Cruise Ship Cluster in Japan. Microorganisms 2022, 10, 99. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10010099
Abe H, Ushijima Y, Amano M, Sakurai Y, Yoshikawa R, Kinoshita T, Kurosaki Y, Yanagihara K, Izumikawa K, Morita K, et al. Unique Evolution of SARS-CoV-2 in the Second Large Cruise Ship Cluster in Japan. Microorganisms. 2022; 10(1):99. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10010099
Chicago/Turabian StyleAbe, Haruka, Yuri Ushijima, Murasaki Amano, Yasuteru Sakurai, Rokusuke Yoshikawa, Takaaki Kinoshita, Yohei Kurosaki, Katsunori Yanagihara, Koichi Izumikawa, Kouichi Morita, and et al. 2022. "Unique Evolution of SARS-CoV-2 in the Second Large Cruise Ship Cluster in Japan" Microorganisms 10, no. 1: 99. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10010099