
First Report and Comparative Genomic Analysis of a Mycoplasma mycoides Subspecies capri HN-A in Hainan Island
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Identification of Mmc HN-A
2.2. Culture and DNA Extraction of Mmc HN-A
2.3. PacBio Sequencing
2.4. Illumina Sequencing
2.5. Genome Assembly
2.6. Genome Annotation and Bioinformatic Analysis
2.7. Gene Function Analysis
2.8. Phylogenetic Tree Construction
3. Results and Discussion
3.1. Results of Isolation and Identification of Mmc HN-A
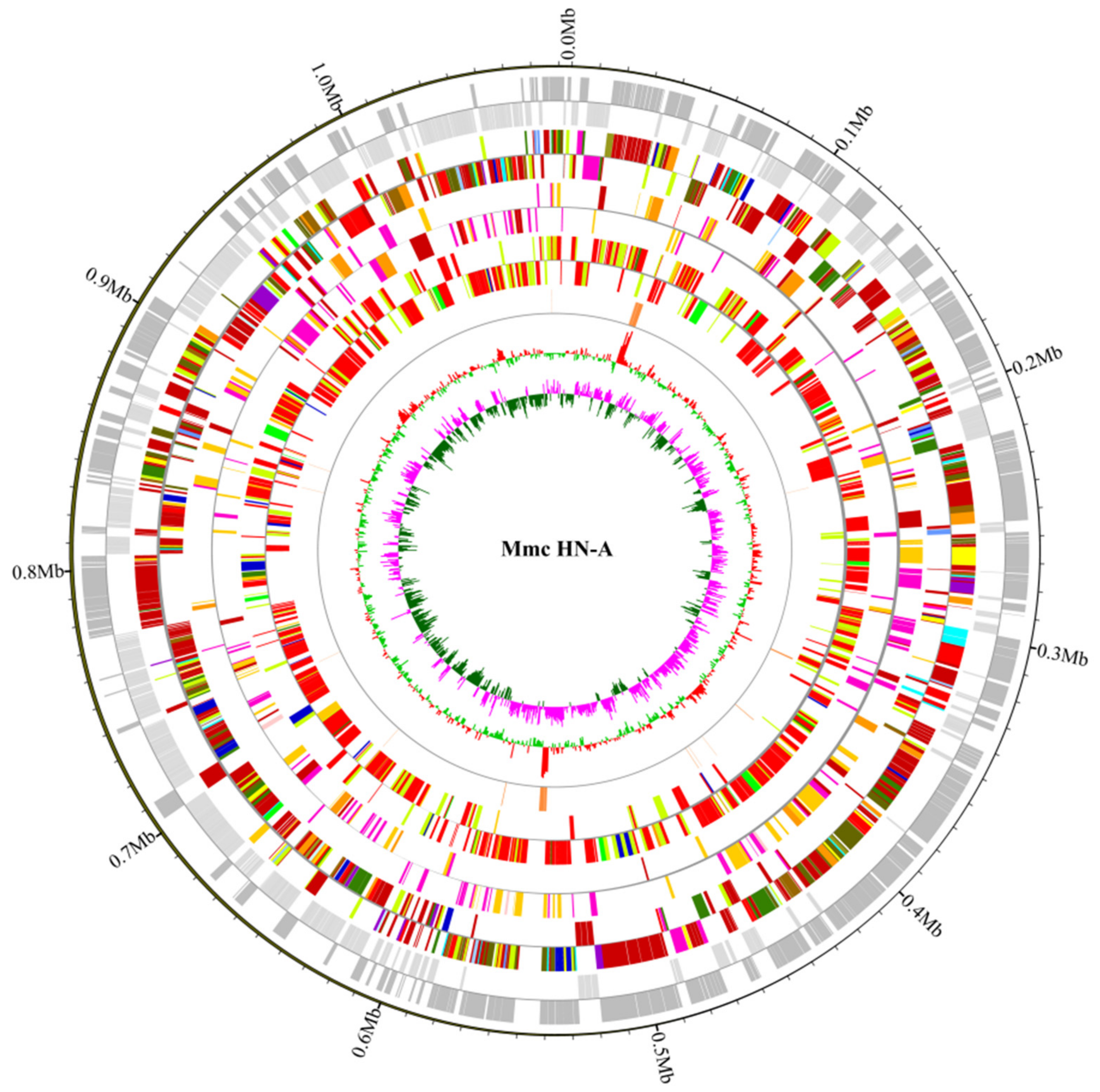
3.2. General Features of Mmc HN-A
3.3. Functional Annotation of Mmc HN-A
3.3.1. Virulence Factors
3.3.2. Metabolism
3.4. Results of Comparative Genomic Analysis
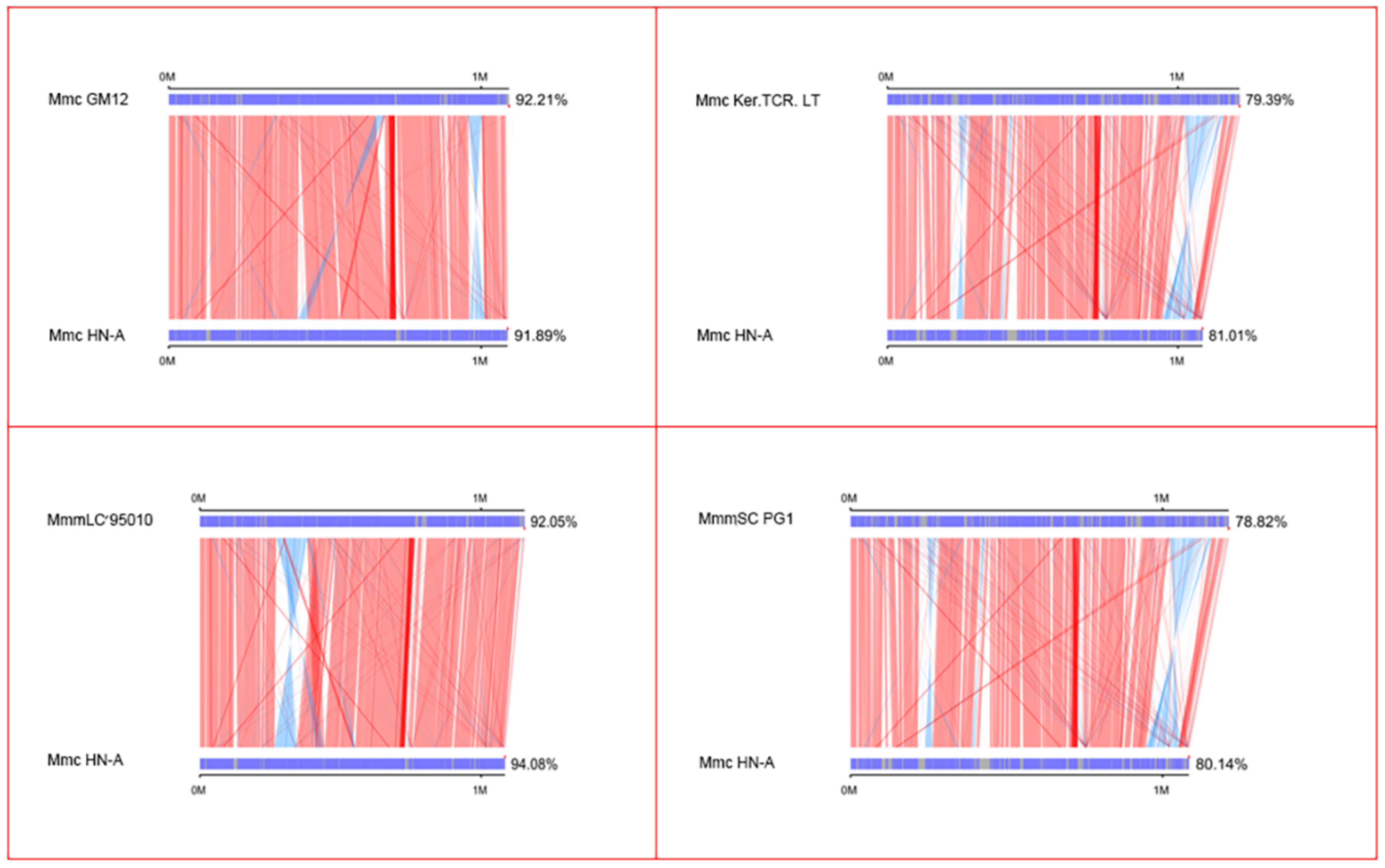
3.4.1. Collinearity Analysis Results of The Five Strains
3.4.2. Statistical Results of SNP, InDel, and SV
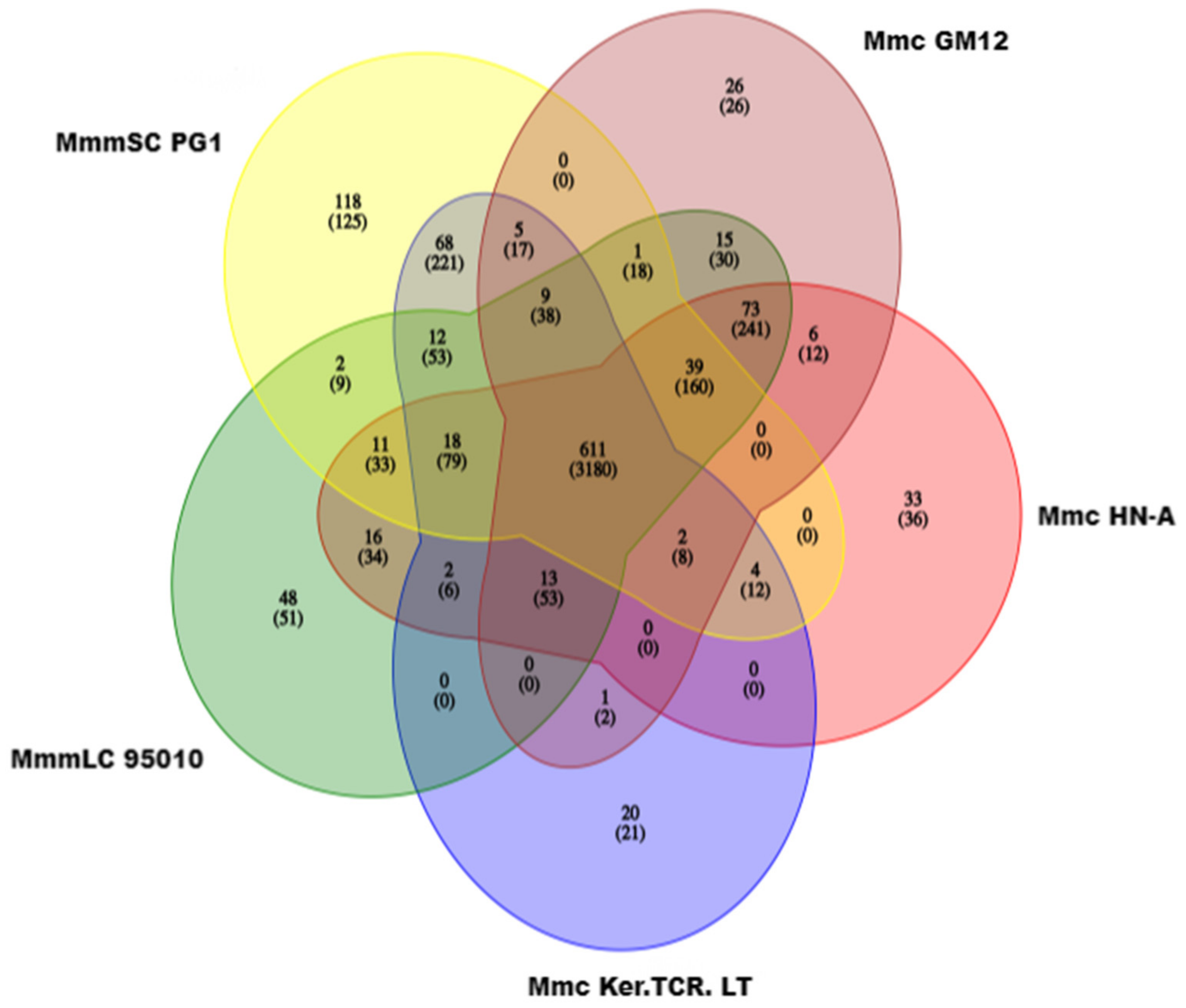
3.5. Gene Family Analysis and Pan-Genome Analysis Results of Five Strains
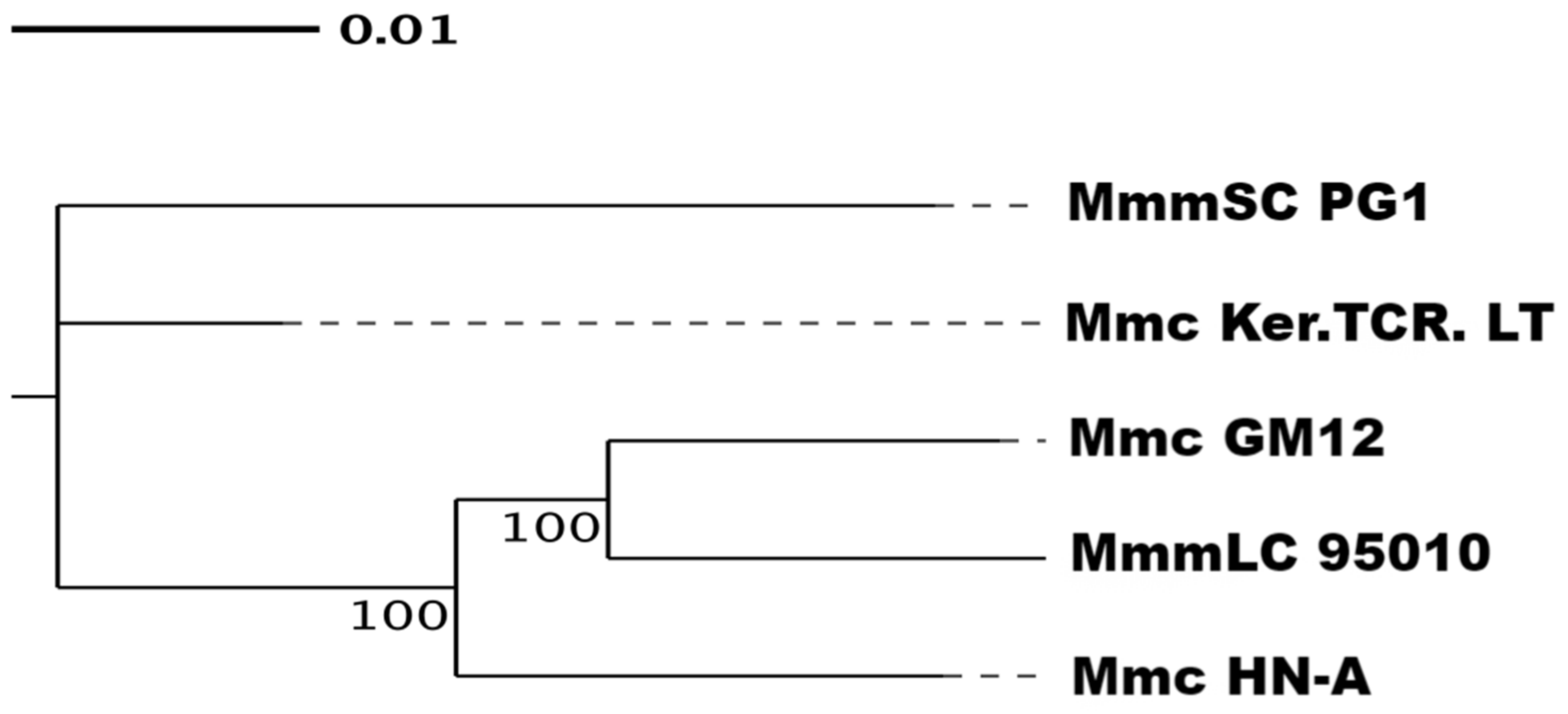
3.6. Phylogenetic Tree Analysis and ANI Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahmad, F.; Khan, H.; Khan, F.A.; Carson, B.D.; Sadique, U.; Ahmad, I.; Saeed, M.; Rehman, F.U.; Rehman, H.U. The First Isolation and Molecular Characterization of Mycoplasma capricolum subsp. capripneumoniae Pakistan Strain: A Causative Agent of Contagious Caprine Pleuropneumonia. J. Microbiol. Immunol. Infect. 2021, 54, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Weldearegay, Y.B.; Müller, S.; Hänske, J.; Schulze, A.; Kostka, A.; Rüger, N.; Hewicker-Trautwein, M.; Brehm, R.; Valentin-Weigand, P.; Kammerer, R.; et al. Host-Pathogen Interactions of Mycoplasma mycoides in Caprine and Bovine Precision-Cut Lung Slices (PCLS) Models. Pathogens 2019, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- OIE. Contagious Bovine Pleuropneumonia. Aetiology, Epidemiology, Diagnosis Prevention and Control References. Technical Disease Cards. Available online: http://www.oie.int/en/animal-health-in-the-world/technical-disease-cards/ (accessed on 6 March 2018).
- Tambi, N.E.; Maina, W.O.; Ndi, C. An Estimation of the Economic Impact of Contagious Bovine Pleuropneumonia in Africa. Rev. Sci. Tech. 2006, 25, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Thiaucourt, F.; Bölske, G.; Leneguersh, B.; Smith, D.; Wesonga, H. Diagnosis and Control of Contagious Caprine Pleuropneumonia. Rev. Sci. Tech. 1996, 15, 1415–1429. [Google Scholar] [CrossRef]
- World Organization for Animal Health. Contagious Caprine pleu-ropneumonia. In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals (Mammals, Birds and Bees), 5th ed.; Office International Des Epizooties: Paris, France, 2004; Volume 2, Chapter 2.4.6; pp. 623–634. [Google Scholar]
- Nicholas, R.A.J. Improvements in the Diagnosis and Control of Diseases of Small Ruminants Caused by Mycoplasmas. Small Rumin. Res. 2002, 45, 145–149. [Google Scholar] [CrossRef]
- Xu, C.G.; Hao, Y.Q.; Zhang, L.; Hao, R.X.; Liu, X.L.; Huang, Z.Y. Molecular cloning and immune response analysis of putative variable lipoproteins from Mycoplasma mycoides subsp capri. Genet. Mol. Res. 2014, 13, 1527–1539. [Google Scholar] [CrossRef]
- Thiaucourt, F.; Bölske, G. Contagious Caprine Pleuropneumonia and Other Pulmonary Mycoplasmoses of Sheep and Goats. Rev. Sci. Tech. 1996, 15, 1397–1414. [Google Scholar] [CrossRef]
- Hernandez, L.; Lopez, J.; St-Jacques, M.; Ontiveros, L.; Acosta, J.; Handel, K. Mycoplasma mycoides subsp. capri Associated With Goat Respiratory Disease and High Flock Mortality. Can. Vet. J. 2006, 47, 366–369. [Google Scholar]
- Shah, M.K.; Saddique, U.; Ahmad, S.; Hayat, Y.; Rahman, S.; Hassan, M.F.; Ali, T. Isolation Rate and Antimicrobial Susceptibility Profiles of Mycoplasma mycoides Subspecies capri Field Isolates from Sheep and Goats in Pakistan. Small Rumin. Res. 2017, 153, 118–122. [Google Scholar] [CrossRef]
- Solana, P.; Rivera, E. Infection of Goats in Mexico by Mycoplasma mycoides var. capri. Ann. N. Y. Acad. Sci. 1967, 143, 357–365. [Google Scholar] [CrossRef]
- Kinde, H.; DaMassa, A.J.; Wakenell, P.S.; Petty, R. Mycoplasma Infection in a Commercial Goat Dairy Caused by Mycoplasma agalactiae and Mycoplasma mycoides subsp. mycoides (Caprine Biotype). J. Vet. Diagn. Invest. 1994, 6, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Corona, L.; Cillara, G.; Tola, S. Proteomic Approach for Identification of Immunogenic Proteins of Mycoplasma mycoides subsp. capri. Vet. Microbiol. 2013, 167, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Bertin, C.; Pau-Roblot, C.; Courtois, J.; Manso-Silván, L.; Tardy, F.; Poumarat, F.; Citti, C.; Sirand-Pugnet, P.; Gaurivaud, P.; Thiaucourt, F. Highly Dynamic Genomic Loci Drive the Synthesis of Two Types of Capsular or Secreted Polysaccharides Within the Mycoplasma mycoides Cluster. Appl. Environ. Microbiol. 2015, 81, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Eden, P.A.; Schmidt, T.M.; Blakemore, R.P.; Pace, N.R. Phylogenetic analysis of Aquaspirillum magnetotacticum using polymerase chain reaction-amplified 16S rRNA-specific DNA. Int. J. Syst. Bacteriol. 1991, 41, 324–325. [Google Scholar] [CrossRef]
- Zhang, J.P.; Jiang, J.X.; Lin, Y.S.; You, W.; Hu, Q.L. Cloning and Expression of Specific Protein Gene, Mmc-3740, of Mycoplasma mycoides. Fujian J. Agric. Sci. 2019, 34, 1124–1128. [Google Scholar] [CrossRef]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased Diploid Genome Assembly with Single-Molecule Real-Time Sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. bioRxiv 2018, 274100. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI Prokaryotic Genome Annotation Pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. TRNAscan-SE On-Line: Integrating Search and Context for Analysis of Transfer RNA Genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and Rapid Annotation of Ribosomal RNA Genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-Fold Faster RNA Homology Searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Brinkman, F.S.L. Improved Genomic Island Predictions with IslandPath-DIMOB. Bioinformatics 2018, 34, 2161–2167. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E. Phage_Finder: Automated Identification and Classification of Prophage Regions in Complete Bacterial Genome Sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A Web Tool to Identify Clustered Regularly Interspaced Short Palindromic Repeats. Nucleic Acids Res. 2007, 35 (Suppl. S2), W52–W57. [Google Scholar] [CrossRef]
- Haas, B. TransposonPSI: An Application of PSI-Blast to Mine (Retro-) Transposon ORF Homologies. Available online: http://transposonpsi.sourceforge.net/ (accessed on 1 August 2022).
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From Genomics to Chemical Genomics: New Developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded Microbial Genome Coverage and Improved Protein Family Annotation in the COG Database. Nucleic Acids Res. 2014, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating Some Redundancy Significantly Speeds up Clustering of Large Protein Databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating Signal Peptides from Transmembrane Regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Arnold, R.; Brandmaier, S.; Kleine, F.; Tischler, P.; Heinz, E.; Behrens, S.; Niinikoski, A.; Mewes, H.W.; Horn, M.; Rattei, T. Sequence-Based Prediction of Type III Secreted Proteins. PLoS Pathog. 2009, 5, e1000376. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-Base: The Pathogen–Host Interactions Database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiong, Z.; Sun, L.; Yang, J.; Jin, Q. VFDB 2012 Update: Toward the Genetic Diversity and Molecular Evolution of Bacterial Virulence Factors. Nucleic Acids Res. 2012, 40, D641–D645. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The Protein Families Database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Blin, K.; Wolf, T.; Chevrette, M.G.; Lu, X.; Schwalen, C.J.; Kautsar, S.A.; Suarez Duran, H.G.; de Los Santos, E.L.C.; Kim, H.U.; Nave, M.; et al. AntiSMASH 4.0—Improvements in Chemistry Rrediction and Gene Cluster Boundary Identification. Nucleic Acids Res. 2017, 45, W36–W41. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and Open Software for Comparing Large Genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Goel, M.; Sun, H.; Jiao, W.B.; Schneeberger, K. SyRI: Finding Genomic Rearrangements and Local Sequence Differences from Whole-Genome Assemblies. Genome Biol. 2019, 20, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive Protein Alignments at Tree-of-Life Scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert Jr, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and Taxonomy in Diagnostics for Food Security: Soft-Rotting Enterobacterial Plant Pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Schieck, E.; Lartigue, C.; Frey, J.; Vozza, N.; Hegermann, J.; Miller, R.A.; Valguarnera, E.; Muriuki, C.; Meens, J.; Nene, V.; et al. Galactofuranose in Mycoplasma mycoides is Important for Membrane Integrity and Conceals Adhesins but does not Contribute to Serum Resistance. Mol. Microbiol. 2016, 99, 55–70. [Google Scholar] [CrossRef]
- Goebel, W.; Chakraborty, T.; Kreft, J. Bacterial Hemolysins as Virulence Factors. Antonie Van Leeuwenhoek 1988, 54, 453–463. [Google Scholar] [CrossRef]
- Li, Y.; Wang, R.; Sun, W.; Song, Z.; Bai, F.; Zheng, H.; Xin, J. Comparative Genomics Analysis of Mycoplasma capricolum subsp. capripneumoniae 87001. Genomics 2020, 112, 615–620. [Google Scholar] [CrossRef]
- Deutscher, J.; Francke, C.; Postma, P.W. How Phosphotransferase System-Related Protein Phosphorylation Regulates Carbohydrate Metabolism in Bacteria. Microbiol. Mol. Biol. Rev. 2006, 70, 939–1031. [Google Scholar] [CrossRef]
- Vasu, K.; Nagaraja, V. Diverse Functions of Restriction-Modification Systems in addition to Cellular Defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef]
- Doberenz, S.; Eckweiler, D.; Reichert, O.; Jensen, V.; Bunk, B.; Spröer, C.; Kordes, A.; Frangipani, E.; Luong, K.; Korlach, J.; et al. Identification of a Pseudomonas aeruginosa PAO1 DNA Methyltransferase, Its Targets, and Physiological Roles. mBio 2017, 8, e02312–e02316. [Google Scholar] [CrossRef] [PubMed]
- Nye, T.M.; Jacob, K.M.; Holley, E.K.; Nevarez, J.M.; Dawid, S.; Simmons, L.A.; Watson, M.E., Jr. DNA Methylation from a Type I Restriction Modification System Influences Gene Expression and Virulence in Streptococcus pyogenes. PLoS Pathog. 2019, 15, e1007841. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for their use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Detection Object | Primer Name | Primer Sequence (5′-3′) | Product Size (bp) | Annealing Temperature (°C) |
|---|---|---|---|---|
| Bacterial 16S rRNA | 16S rRNA | F: AGAGTTTGATCCTGGCTCAG | 1465 | 56 |
| R: GGTTACCTTGTTACGACTT | ||||
| Mmc | Mmc 3740 | F: GGATCCGTGATAAAGTTATT GACTTTATTAG | 435 | 50 |
| R:AAGCTTTTATTGTACTGTATTGTGTTCTTC |
| Item | Number | Item | Number |
|---|---|---|---|
| Genome size (bp) | 1,084,691 | Number of genes | 848 |
| Total length of gene (bp) | 966,987 | Average length of gene (bp) | 1,140.31 |
| Genome GC content (%) | 23.76 | Total length of gene/genome (%) | 89.15 |
| GC content of gene region (%) | 23.89 | Total length of intergenic region/genome (%) | 10.85 |
| The length of gene (bp) | 75–9840 | Tandem repeat length/genome (%) | 0.62 |
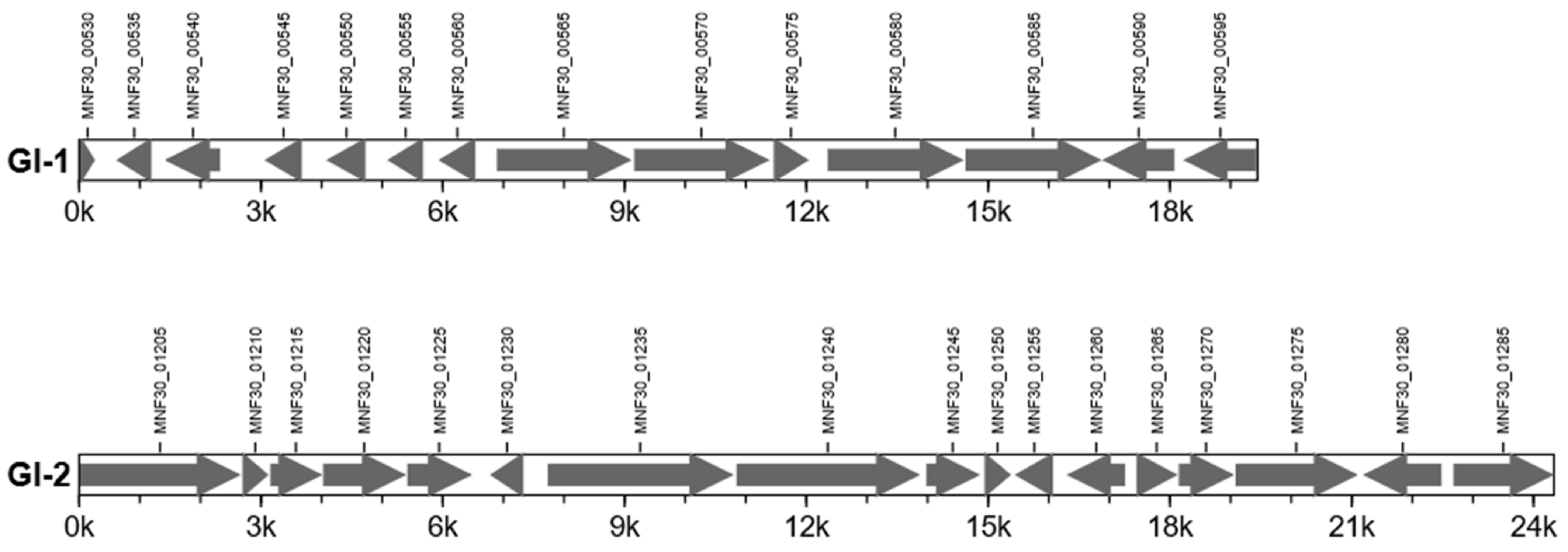
| Number of tandem repeats | 146 | Number of gene islands | 2 |
| Total bases in tandem repeats | 6747 | Total length of gene island (bp) | 43,779 |
| Number of interspersed repeats | 8 | Number of short-interspersed elements | 5 |
| Number of rRNA genes | 6 | Number of DNA elements | 3 |
| Number of tRNA genes | 30 | Total potential CRISPRs | 5 |
| Number of sRNA genes | 2 | Number of Prophages | 0 |
| Mmc HN-A | Mmc GM12 | Mmc Ker.TCR. LT | MmmLC 95010 | MmmSC PG1 | |
|---|---|---|---|---|---|
| Accession number | CP093215 | CP001621.1 | CP068548.1 | FQ377874.1 | BX293980.2 |
| Place of isolation | China | USA | India | France | Sweden |
| Collection date | 2021 | 2009 | 2019 | 1995 | 2003 |
| Size (bp) | 1,084,691 | 1,089,202 | 1,211,756 | 1,153,998 | 1,211,703 |
| G + C (%) | 23.76 | 23.9 | 24.0 | 23.81 | 24.0 |
| SNPs | Insertion | Deletion | SV | |
|---|---|---|---|---|
| Mmc GM12 | 25,297 | 566 | 540 | 51 |
| Mmc Ker.TCR. LT | 19,624 | 605 | 676 | 98 |
| MmmLC 95010 | 26,003 | 520 | 527 | 68 |
| MmmSC PG1 | 27,246 | 574 | 657 | 101 |
| Species | Total Genes | Gene in Families | Unclustered Genes | Families | Unique Families |
|---|---|---|---|---|---|
| Mmc Ker.TCR. LT | 847 | 828 | 19 | 746 | 20 |
| MmmLC 95010 | 922 | 877 | 45 | 825 | 48 |
| MmmSC PG1 | 1016 | 905 | 111 | 789 | 118 |
| Mmc GM12 | 832 | 806 | 26 | 775 | 26 |
| Mmc HN-A | 848 | 818 | 30 | 798 | 33 |
| Species | Total Genes | Shared Gene Number | Specific Gene Number |
|---|---|---|---|
| Mmc Ker.TCR. LT | 847 | 826 | 21 |
| MmmLC 95010 | 922 | 871 | 51 |
| MmmSC PG1 | 1016 | 891 | 125 |
| Mmc GM12 | 832 | 806 | 26 |
| Mmc HN-A | 848 | 812 | 36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Jiang, J.; He, M.; Li, H.; Cheng, Y.; An, Q.; Chen, S.; Du, L.; Man, C.; Chen, Q.; et al. First Report and Comparative Genomic Analysis of a Mycoplasma mycoides Subspecies capri HN-A in Hainan Island. Microorganisms 2022, 10, 1908. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10101908
Zhang Z, Jiang J, He M, Li H, Cheng Y, An Q, Chen S, Du L, Man C, Chen Q, et al. First Report and Comparative Genomic Analysis of a Mycoplasma mycoides Subspecies capri HN-A in Hainan Island. Microorganisms. 2022; 10(10):1908. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10101908
Chicago/Turabian StyleZhang, Zhenxing, Junming Jiang, Meirong He, Haoyang Li, Yiwen Cheng, Qi An, Si Chen, Li Du, Churiga Man, Qiaoling Chen, and et al. 2022. "First Report and Comparative Genomic Analysis of a Mycoplasma mycoides Subspecies capri HN-A in Hainan Island" Microorganisms 10, no. 10: 1908. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10101908