
An Update of Bovine Hemoplasmas Based on Phylogenetic and Genomics Analysis
 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Sequences and Annotation
2.2. Phylogenetic and Pangenome Analysis
2.3. Comparative Genomics
2.4. Prediction of Antigenic Proteins
2.5. Prediction of Subcellular Localization and Stability of Proteins
2.6. Linear B-Cell Epitope Prediction and Three-Dimensional Modeling
3. Results
3.1. General Features of Genomes
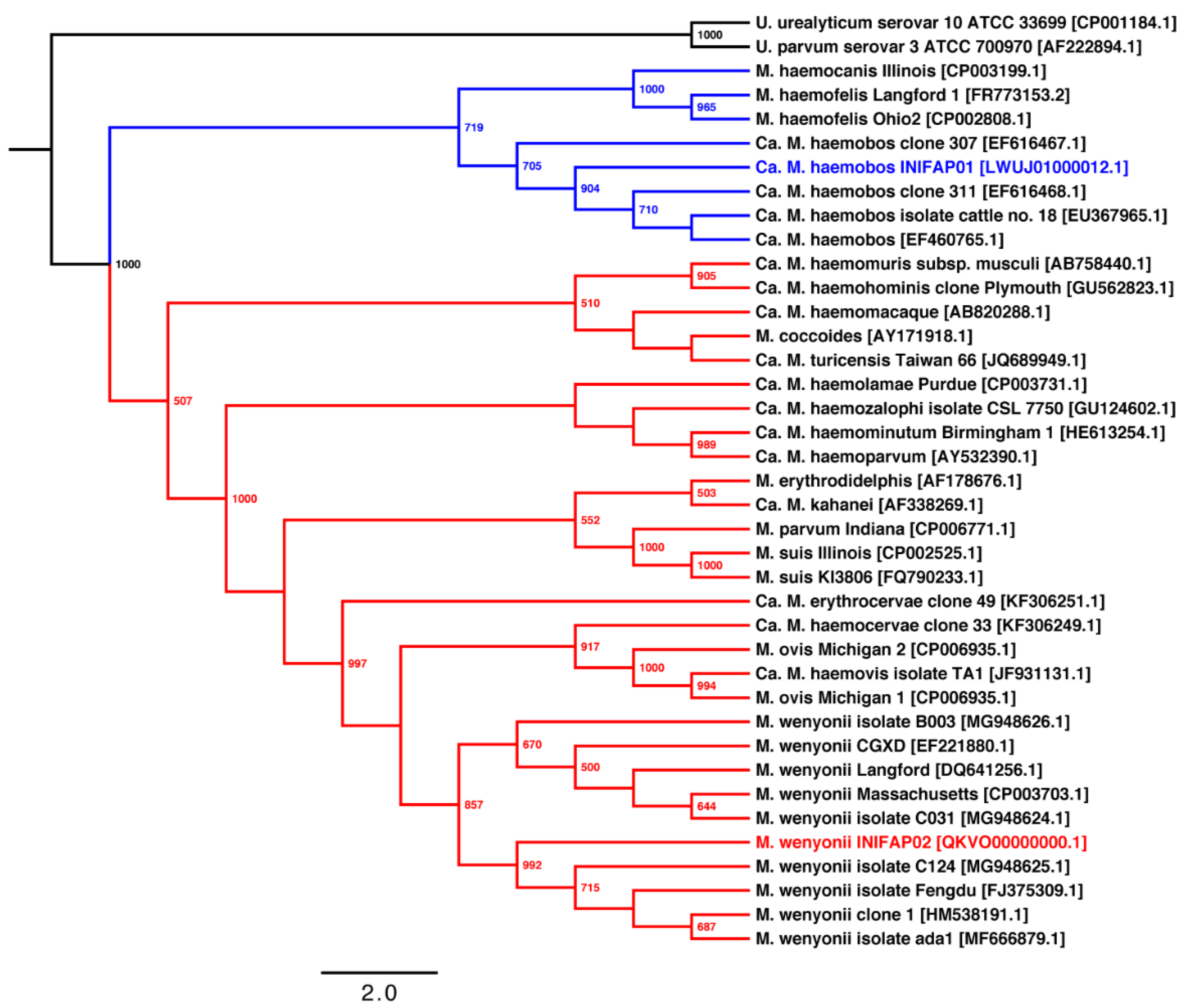
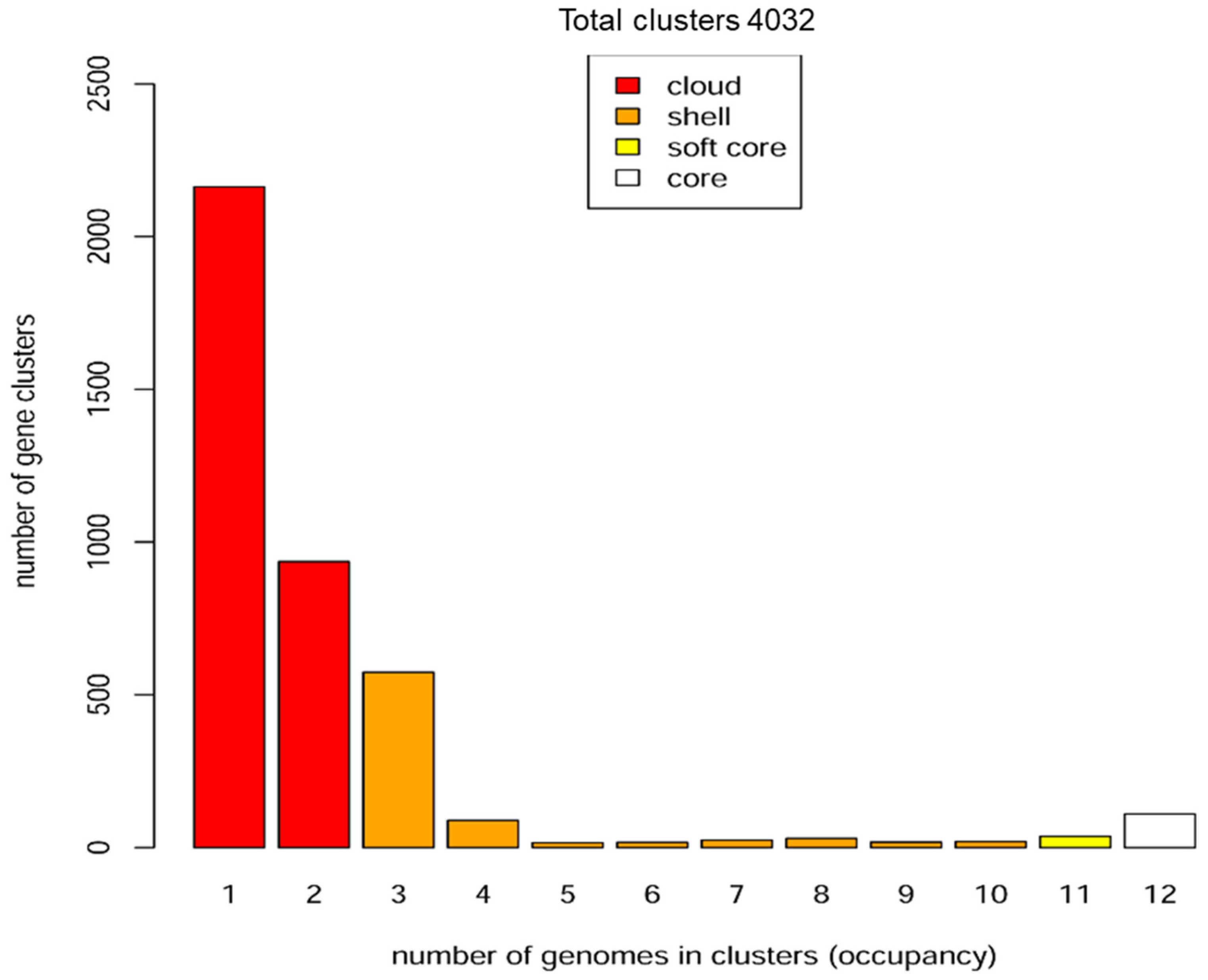
3.2. Phylogenetic and Pangenome Analyzes
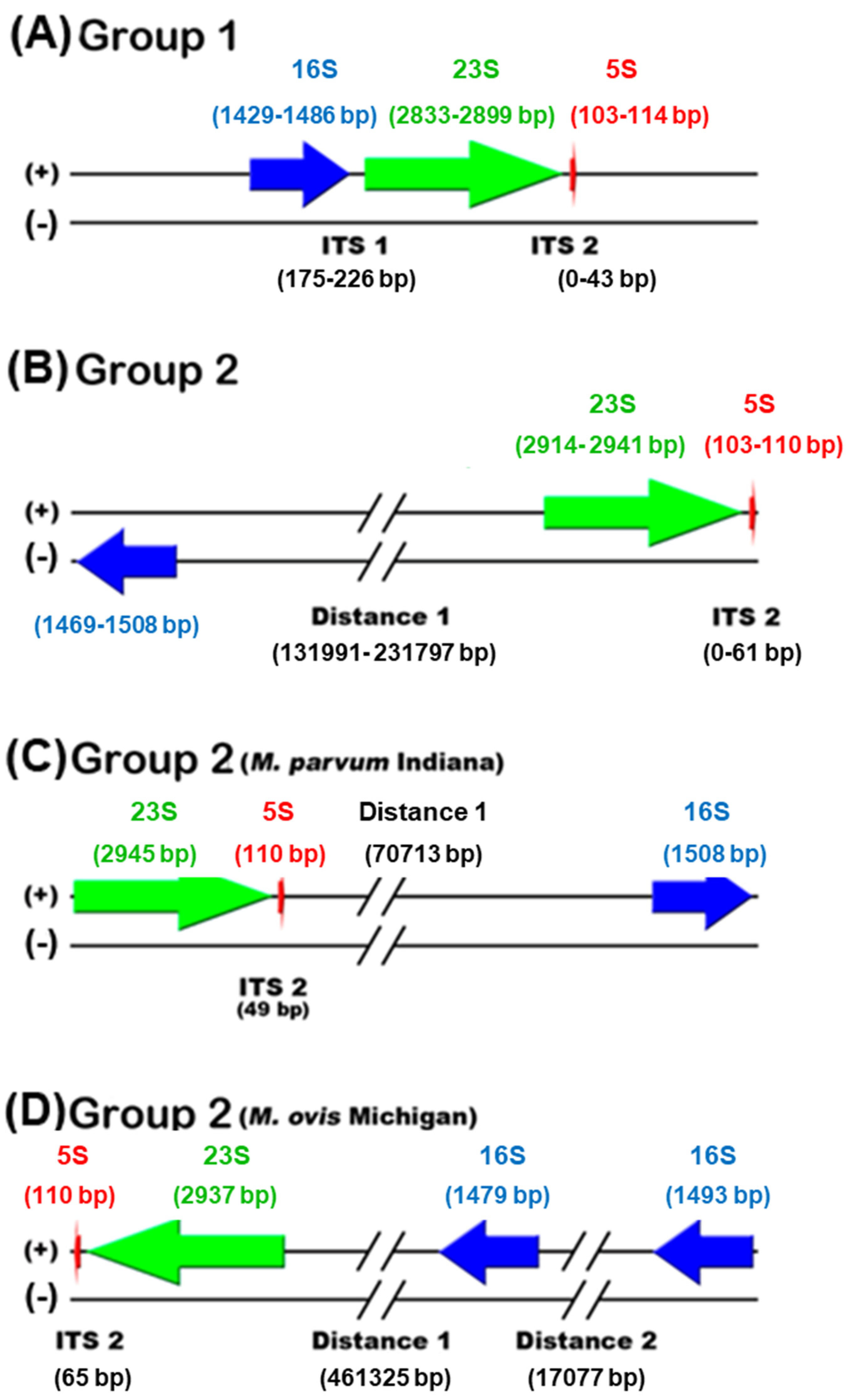
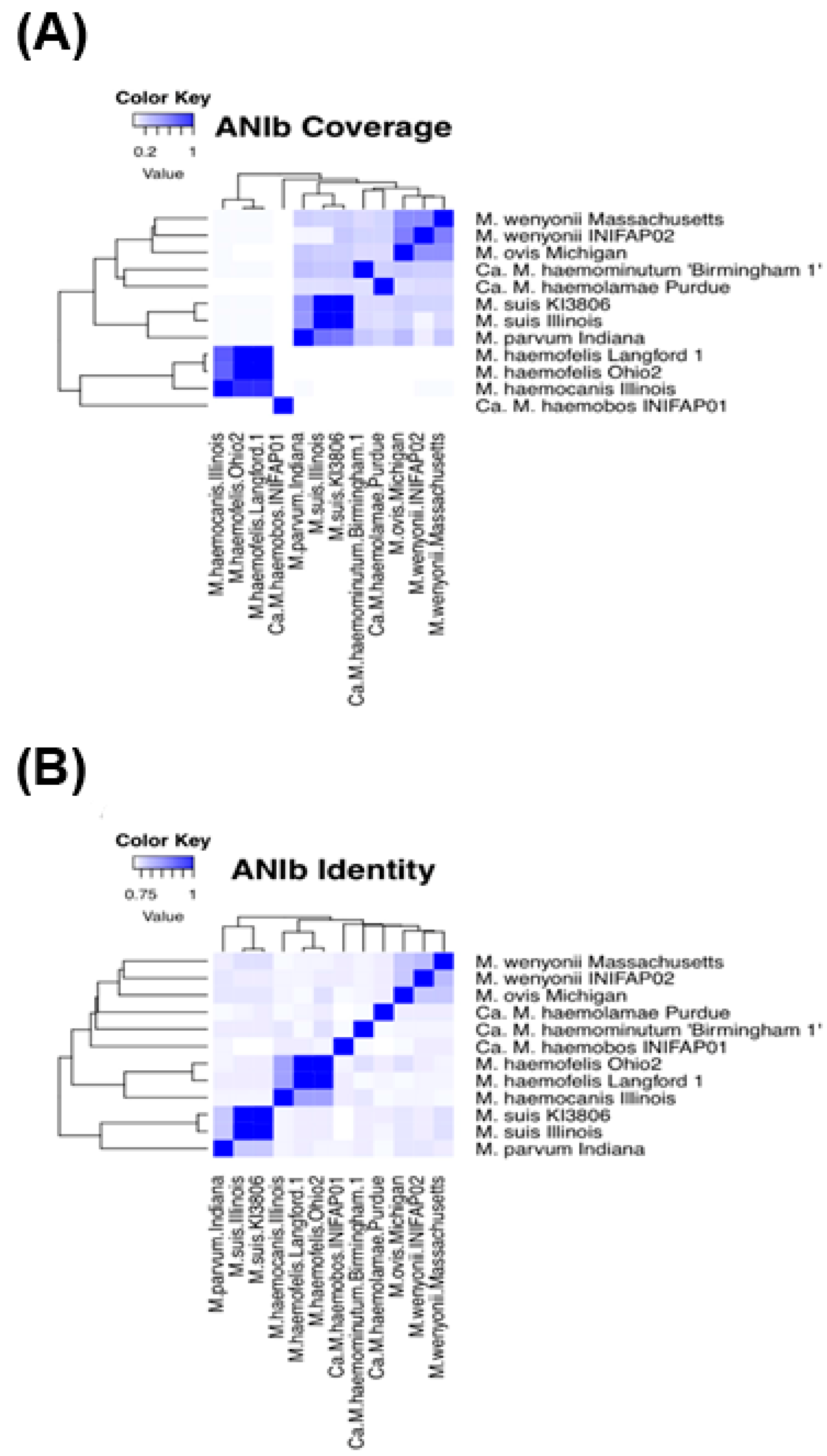
3.3. Comparative Genomics
3.4. Selection and Prediction of B-Cell Epitopes in Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guimaraes, A.M.S.; Santos, A.P.; do Nascimento, N.C.; Timenetsky, J.; Messick, J.B. Comparative genomics and phylogenomics of hemotrophic mycoplasmas. PLoS ONE 2014, 9, e91445. [Google Scholar] [CrossRef] [PubMed]
- Messick, J.B. Hemotrophic Mycoplasmas (Hemoplasmas): A review and new insights into pathogenic potential. Vet. Clin. Pathol. 2004, 33, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Rikihisa, Y.; Kawahara, M.; Wen, B.; Kociba, G.; Fuerst, P.; Kawamori, F.; Suto, C.; Shibata, S.; Futohashi, M. Western immunoblot analysis of Haemobartonella muris and comparison of 16S rRNA gene sequences of H. muris, H. felis, and Eperythrozoon suis. J. Clin. Microbiol. 1997, 35, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Neimark, H.; Johansson, K.-E.; Rikihisa, Y.; Tully, J.G. Proposal to transfer some members of the genera Haemobartonella and Eperythrozoon to the genus Mycoplasma with descriptions of Candidatus Mycoplasma haemofelis, Candidatus mycoplasma haemomuris, Candidatus mycoplasma haemosui’ and Candidatus mycoplasma. Int. J. Syst. Evol. Microbiol. 2001, 51, 891–899. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, A.P.; Guimaraes, A.M.S.; do Nascimento, N.C.; SanMiguel, P.J.; Messick, J.B. Complete genome sequence of Mycoplasma wenyonii strain Massachusetts. J. Bacteriol. 2012, 194, 5458–5459. [Google Scholar] [CrossRef]
- Martínez-Ocampo, F.; Rodríguez-Camarillo, S.D.; Amaro-Estrada, I.; Quiroz-Castañeda, R.E. Draft genome sequence of “Candidatus mycoplasma haemobos,” a hemotropic mycoplasma identified in cattle in Mexico. Genome Announc. 2016, 4, e00656-16. [Google Scholar] [CrossRef]
- Quiroz-Castañeda, R.E.; Martínez-Ocampo, F.; Dantán-González, E. Draft genome sequence of Mycoplasma wenyonii, a second hemotropic mycoplasma species identified in Mexican bovine cattle. Microbiol. Resour. Announc. 2018, 7, e00875-18. [Google Scholar] [CrossRef]
- Hoelzle, K.; Winkler, M.; Kramer, M.M.; Wittenbrink, M.M.; Dieckmann, S.M.; Hoelzle, L.E. Detection of Candidatus mycoplasma haemobos in cattle with anaemia. Vet. J. 2011, 187, 408–410. [Google Scholar] [CrossRef]
- Tagawa, M.; Matsumoto, K.; Inokuma, H. Molecular Detection of Mycoplasma wenyonii and “Candidatus mycoplasma haemobos” in cattle in Hokkaido, Japan. Vet. Microbiol. 2008, 132, 177–180. [Google Scholar] [CrossRef]
- Niethammer, F.M.; Ade, J.; Hoelzle, L.E.; Schade, B. Hemotrophic Mycoplasma in simmental cattle in Bavaria: Prevalence, blood parameters, and transplacental transmission of “Candidatus Mycoplasma haemobos” and Mycoplasma wenyonii. Acta Vet. Scand. 2018, 60, 74. [Google Scholar] [CrossRef]
- Ade, J.; Niethammer, F.; Schade, B.; Schilling, T.; Hoelzle, K.; Hoelzle, L.E. Quantitative analysis of Mycoplasma wenyonii and “Candidatus mycoplasma haemobos” infections in cattle using novel gapn-based realtime PCR assays. Vet. Microbiol. 2018, 220, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Genova, S.G.; Streeter, R.N.; Velguth, K.E.; Snider, T.A.; Kocan, K.M.; Simpson, K.M. Severe anemia associated with Mycoplasma wenyonii infection in a mature cow. Can. Vet. J. 2011, 52, 1018–1021. [Google Scholar] [PubMed]
- McFadden, A.; Ha, H.J.; Donald, J.J.; Bueno, I.M.; van Andel, M.; Thompson, J.C.; Tisdall, D.J.; Pulford, D.J. Investigation of bovine haemoplasmas and their association with anaemia in New Zealand cattle. N. Z. Vet. J. 2016, 64, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, M.; Yamakawa, K.; Aoki, T.; Matsumoto, K.; Ishii, M.; Inokuma, H. Effect of chronic hemoplasma infection on cattle productivity. J. Vet. Med. Sci. 2013, 75, 1271–1275. [Google Scholar] [CrossRef]
- Fujihara, Y.; Sasaoka, F.; Suzuki, J.; Watanabe, Y.; Fujihara, M.; OOSHITA, K.; Ano, H.; Harasawa, R. Prevalence of hemoplasma infection among cattle in the Western part of Japan. J. Vet. Med. Sci. 2011, 73, 1653–1655. [Google Scholar] [CrossRef] [PubMed]
- Girotto, A.; Zangirólamo, A.F.; Bogado, A.L.G.; Souza, A.S.L.; Silva, G.C.F.; Garcia, J.L. Molecular Detection and occurrence of ‘Candidatus mycoplasma haemobos’ in dairy cattle of Southern Brazil. Rev. Bras. De Parasitol. Vet. 2012, 21, 342–344. [Google Scholar] [CrossRef]
- Tagawa, M.; Ybanez, A.P.; Matsumoto, K.; Yokoyama, N.; Inokuma, H. interference between Theileria orientalis and hemotropic Mycoplasma spp. (hemoplasmas) in grazing cattle. Vet. Parasitol. 2013, 195, 165–168. [Google Scholar] [CrossRef]
- Ahmad, T.A.; Eweida, A.E.; Sheweita, S.A. B-Cell Epitope mapping for the design of vaccines and effective diagnostics. Trials Vaccinol. 2016, 5, 71–83. [Google Scholar] [CrossRef]
- Parvizpour, S.; Pourseif, M.M.; Razmara, J.; Rafi, M.A.; Omidi, Y. Epitope-based vaccine design: A comprehensive overview of bioinformatics approaches. Drug Discov. Today 2020, 25, 1034–1042. [Google Scholar] [CrossRef]
- Ranjbar, M.H.; Ebrahimi, M.M.; Shahsavandi, M.M.; Farhadi, S.; Mirjalili, T.; Tebianian, A.; Motedayen, M. Novel applications of immuno-bioinformatics in vaccine and bio-product developments at research institutes. Arch. Razi Inst. 2019, 74, 219–233. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect trna genes and tmrna genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Connors, J.; Krzywinski, M.; Schein, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Díaz-Sánchez, A.A.; Corona-González, B.; Meli, M.L.; Álvarez, D.O.; Cañizares, E.V.; Rodríguez, O.F.; Rivero, E.L.; Hofmann-lehmann, R. First molecular evidence of bovine hemoplasma species (Mycoplasma spp.) in water buffalo and dairy cattle herds in Cuba. Parasites Vectors 2019, 12, 78. [Google Scholar] [CrossRef] [PubMed]
- Quiroz Castañeda, R.E.; Aragón, K.M.; Diaz, H.A.; Preciado de la Torre, J.F. Molecular Detection of bovine hemotrophic mycoplasmas in Mexico. Rev. Cent. Investig. Univ. Salle 2020, 13, 67–82. [Google Scholar] [CrossRef]
- Schambow, R.A.; Poulsen, K.; Bolin, S.; Krahn, D.; Norby, B.; Sockett, D.; Ruegg, P.L. Apparent prevalence of Mycoplasma wenyonii, Candidatus mycoplasma haemobos, and bovine leukemia virus in Wisconsin and Michigan dairy cattle herds. JDS Commun. 2021, 2, 61–66. [Google Scholar] [CrossRef]
- Stevanović, O.; Jurković, D.; Polkinghorne, A.; Ćeleš, A.; Ilić, T.; Dimitrijević, S.; Nedić, D.; Beck, R. Molecular detection of Babesia divergens and Mycoplasma wenyonii infection in cattle from Bosnia And Herzegovina. Parasitol. Res. 2020, 119, 1423–1427. [Google Scholar] [CrossRef]
- Tatsukawa, F.; Nohara, R.; Taniguchi, T.; Goto, A.; Misawa, N.; Katamoto, H. Detection of Mycoplasma wenyonii and “Candidatus mycoplasma haemobos” from japanese black breeding cows in Kyushu and Okinawa Region, Southern Part of Japan. J. Vet. Med. Sci. 2021, 83, 9–16. [Google Scholar] [CrossRef]
- Ayling, R.; Bisgaard-Frantzen, S.; Adler, A.; Blowey, R.; Barlow, A.; Millar, M.; van der Burgt, G. Detection of Candidatus Mycoplasma haemobos, Mycoplasma wenyonii and Anaplasma phagocytophilum from cattle in England. Vet. Rec. 2012, 170, 543. [Google Scholar] [CrossRef]
- Hornok, S.; Micsutka, a.; Meli, M.L.; Lutz, H.; Hofmann-Lehmann, R. Molecular investigation of transplacental and vector-borne transmission of bovine haemoplasmas. Vet. Microbiol. 2011, 152, 411–414. [Google Scholar] [CrossRef]
- Peters, I.R.; Helps, C.R.; McAuliffe, L.; Neimark, H.; Lappin, M.R.; Gruffydd-Jones, T.J.; Day, M.J.; Hoelzle, L.E.; Willi, B.; Meli, M.; et al. RNase P RNA Gene (rnpB) Phylogeny of hemoplasmas and other mycoplasma species. J. Clin. Microbiol. 2008, 46, 1873–1877. [Google Scholar] [CrossRef]
- Andersson, J.O.; Andersson, S.G. Genome degradation is an ongoing process in Rickettsia. Mol. Biol. Evol. 1999, 16, 1178–1191. [Google Scholar] [CrossRef]
- Merhej, V.; El Karkouri, K.; Raoult, D. Whole genome-based phylogenetic analysis of Rickettsiae. Clin. Microbiol. Infect. 2009, 15, 336–337. [Google Scholar] [CrossRef] [PubMed]
- Rurangirwa, F.R.; Brayton, K.A.; McGuire, T.C.; Knowles, D.P.; Palmer, G.H. Conservation of the unique Rickettsial rRNA gene arrangement in Anaplasma. Int. J. Syst. Evol. Microbiol. 2002, 52, 1405–1409. [Google Scholar] [CrossRef] [PubMed]
- Figueras, M.J.; Beaz-Hidalgo, R.; Hossain, M.J.; Liles, M.R. Taxonomic Affiliation of new genomes should be verified using average nucleotide identity and multilocus phylogenetic analysis. Genome Announc. 2014, 2, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA Hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Liu, X.; Marrakchi, M.; Xu, D.; Dong, H.; Andreescu, S. Biosensors Based on modularly designed synthetic peptides for recognition, detection and live/dead differentiation of pathogenic bacteria. Biosens. Bioelectron. 2016, 80, 9–16. [Google Scholar] [CrossRef]
- Quiroz-Castañeda, R.E.; Tapia-Uriza, T.R.; Mujica, C.V.; Rodríguez-Camarillo, S.D.; Preciado-De la Torre, J.F.; Amaro-Estrada, I.; Cobaxin-Cárdenas, M. Synthetic peptides-based indirect ELISA for the diagnosis of bovine anaplasmosis. Int. J. Appl. Res. Vet. Med. 2019, 17, 65–70. [Google Scholar]
- Molina, A.I.B.; Bayúgar, R.C.; Gutiérrez-Pabello, J.A.; López, A.T.T.; De La Torre, J.F.P.; Camarillo, S.D.R. Immunolocalization of VirB11 protein in the Anaplasma marginale outer membrane and its reaction with bovine immune Sera. Rev. Mex. Cienc. Pecu. 2018, 9, 769–791. [Google Scholar] [CrossRef]
- Bashir, S.; Abd-elrahman, K.A.; Hassan, M.A.; Almofti, Y.A. Multi Epitope based peptide vaccine against Mare’s disease virus serotype 1 glycoprotein H and B. Am. J. Microbiol. Res. 2018, 6, 124–139. [Google Scholar]
- Droppa-Almeida, D.; Franceschi, E.; Padilha, F.F. Immune-informatic analysis and design of peptide vaccine from multi-epitopes against Corynebacterium pseudotuberculosis. Bioinform. Biol. Insights 2018, 12, 1177932218755337. [Google Scholar] [CrossRef]
- Wang, C.Y.; Chang, T.Y.; Walfield, A.M.; Ye, J.; Shen, M.; Chen, S.P.; Li, M.C.; Lin, Y.L.; Jong, M.H.; Yang, P.C.; et al. Effective synthetic peptide vaccine for foot-and-mouth disease in swine. Vaccine 2002, 20, 2603–2610. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Organism | Assembly Level | Length (bp) * | G + C Content (%) * | CDS ** | rRNAs # | tRNAs ## |
|---|---|---|---|---|---|---|---|
| Group 1 | ‘Ca. M. haemobos’ INIFAP01 | 18 contigs | 935,638 | 30.46 | 1180 | 3 | 31 |
| Group 1 | M. haemocanis Illinois | Chromosome | 919,992 | 35.33 | 1234 | 3 | 31 |
| Group 1 | M. haemofelis Langford 1 | Chromosome | 1,147,259 | 38.85 | 1595 | 3 | 31 |
| Group 1 | M. haemofelis Ohio2 | Chromosome | 1,155,937 | 38.81 | 1650 | 3 | 31 |
| Group 2 | ‘Ca. M. haemolamae’ Purdue | Chromosome | 756,845 | 39.27 | 1045 | 3 | 33 |
| Group 2 | ‘Ca. M. haemominutum’ Birmingham 1 | Chromosome | 513,880 | 35.52 | 587 | 3 | 32 |
| Group 2 | M. ovis Michigan | Chromosome | 702,511 | 31.69 | 918 | 4 | 32 |
| Group 2 | M. parvum Indiana | Chromosome | 564,395 | 26.98 | 578 | 3 | 32 |
| Group 2 | M. suis Illinois | Chromosome | 742,431 | 31.08 | 914 | 3 | 32 |
| Group 2 | M. suis KI3806 | Chromosome | 709,270 | 31.08 | 856 | 3 | 32 |
| Group 2 | M. wenyonii INIFAP02 | 37 contigs | 596,665 | 33.43 | 678 | 3 | 32 |
| Group 2 | M. wenyonii Massachusetts | Chromosome | 650,228 | 33.92 | 727 | 3 | 32 |
| Candidatus Mycoplasma Haemobos INIFAP01 | |
|---|---|
| Classification (NCBI Accession Number) | Prediction Score as Antigen (VaxiJen) |
| RAST Category: Virulence, disease, and defense | |
| DNA gyrase subunit B (OAL10308.1) | 0.5352 (antigen) |
| DNA gyrase subunit A (OAL10309.1) | 0.4373 (non-antigen) |
| SSU ribosomal protein S7p (WP_187150158.1) | 0.5180 (antigen) |
| Translation elongation factor G (WP_187150159.1) | 0.5399 (antigen) |
| Translation elongation factor thermo unstable (Tu) (WP_187150070.1) | 0.4268 (non-antigen) |
| SSU ribosomal protein S12p (WP_187150157.1) | 0.7537 (antigen) |
| DNA-directed RNA polymerase beta subunit (WP_187150197.1) | 0.3781 (non-antigen) |
| DNA-directed RNA polymerase (WP_187150196.1) | 0.4488 (non-antigen) |
| RAST Category: Division and cell cycle | |
| ProteinTsaD/Kae1/Qri7 (WP_187150270.1) | 0.3846 (non-antigen) |
| RNA polymerase sigma factor RpoD (WP_187150278.19 | 0.3857 (non-antigen) |
| DNA primase (WP_187150493.1) | 0.3009 (non-antigen) |
| RAST Category: Fatty acids, lipids and isoprenoids | |
| Cardiolipin synthase (WP_187150134.1) | 0.3463 (non-antigen) |
| RAST Category: Stress Response | |
| Manganese superoxide dismutase (WP_187150149.1) | 0.3487 (non-antigen) |
| Mycoplasma wenyonii INIFAP02 | |
| RAST Category: Virulence, disease, and defense | |
| Ribosomal protein SSU S7p (RAO94848.1) | 0.5435 (antigen) |
| Translation elongation factor G (RAO94847.1) | 0.5650 (antigen) |
| Translation elongation factor thermo unstable (Tu) (RAO95121.1) | 0.4359 (non-antigen) |
| Ribosomal protein SSU S12p (RAO94849.1) | 0.7774 (antigen) |
| Ribosomal protein LSU L35p (RAO95358.1) | 0.5491 (antigen) |
| Translation initiation factor 3 (RAO95223.1) | 0.4488 (non-antigen) |
| Ribosomal protein LSU L20p (RAO95357.1) | 0.3668 (non-antigen) |
| RAST Category: Division and cell cycle | |
| Protein TsaD/Kae1/Qri7 (RAO95106.1) | 0.3954 (non-antigen) |
| RNA polymerase sigma factor RpoD (RAO94807.1) | 0.3979 (non-antigen) |
| DNA primase (RAO95339.1) | 0.3929 (non-antigen) |
| RAST Category: Fatty acids, lipids and isoprenoids | |
| Cardiolipin synthase (RAO95377.1) | 0.3305 (non-antigen) |
| Ca. M. Haemobos INIFAP01 | M. wenyonii INIFAP02 | ||||
|---|---|---|---|---|---|
| RAST Category: Virulence, disease, and defense | Predicted epitopes (SVMTrip, recommend score 1.0) | Predicted epitopes (BCEPred) | RAST Category: Virulence, disease, and defense | Predicted epitopes (SVMTrip, recommend score 1.0) | Predicted epitopes (BCEPred) |
| DNA gyrase subunit B | RKLALEGFMSFAGKLADCTT | AGGDSSDSGGQYTDS GGKFDNNSYKTSGG * EVNVYRNGEEHY ENGGKIKDEPKMVSKCEEDKTG IESRLTKLAYLNKGKKFV VNEITKEEKEFFYEEGIKDW FIHSEGKVKNRRAPE FGRFLEENPEQRKVILQRVDQERNFRLK VVEGDSAGGSAKSARNREYQAI NVWKRSKYTAILENEEVKSL NKEVVYLFDDKKKDEFLKNLSNP | Ribosomal protein SSU S7p | MWEGKKQLARRIVYNALEKI | NALEKIREKTEKNPVEV YQVPVESSKERREALA LIKYSRKRN |
| SSU ribosomal protein S7p | PLEVFMEALKNIAPTIELKT | Translation elongation factor G | IPKEYIKSIREGLVDAMKAG VPRIIFCNKMDKVGASFQSS | DAGKTTTSERDWMEQEREKGITDEEFEEIPI PEDQQEEVKTLR KAFTRSGEELTIENKDESN | |
| Translation elongation factor G | EFVDKIVGGKIPKEYIKSIK AKVIKSKIPLKEMFGYATAL | DWMEQEKEKGIT TKKAYEFDGKQEEEYKEIPI ETPAFDKEQNPISIKNSPDNDF QMHSNHRTEIES AIEPKTKVDQEKMSM FRETFTQEAEVEGKYIKQSGGRG HVWIKYEPNKDKGFEF LSLKDASKKCASILLEPI SRRGTIEGDEQVENAKVIKSKIPLKEM | Ribosomal protein SSU S12p | RVKDLPGVKYHIIRGKLDAA | VEKRKKERSKYGVKKEKKS |
| SSU ribosomal protein S12p | RVKDLPGVKYHIVRGKLDTV | Ribosomal protein LSU L35p | SHRSHCASAKTTKRKRQLRK | KKIKHKTKKSLSKR SGAIKRKRSHRS SGAIKRKRSHRSHCASAKTTKRKRQLRKSA | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-García, D.L.; Aguilar-Díaz, H.; Amaro-Estrada, I.; Martínez-Ocampo, F.; Quiroz-Castañeda, R.E. An Update of Bovine Hemoplasmas Based on Phylogenetic and Genomics Analysis. Microorganisms 2022, 10, 1916. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10101916
Flores-García DL, Aguilar-Díaz H, Amaro-Estrada I, Martínez-Ocampo F, Quiroz-Castañeda RE. An Update of Bovine Hemoplasmas Based on Phylogenetic and Genomics Analysis. Microorganisms. 2022; 10(10):1916. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10101916
Chicago/Turabian StyleFlores-García, Diana Laura, Hugo Aguilar-Díaz, Itzel Amaro-Estrada, Fernando Martínez-Ocampo, and Rosa Estela Quiroz-Castañeda. 2022. "An Update of Bovine Hemoplasmas Based on Phylogenetic and Genomics Analysis" Microorganisms 10, no. 10: 1916. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10101916