
Unraveling the Tropaeolum majus L. (Nasturtium) Root-Associated Bacterial Community in Search of Potential Biofertilizers



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Description and Experimental Design
2.2. Isolation of Endophytic Bacteria
2.3. DNA Extraction of Bulk Soil, Rhizosphere and Endosphere
2.4. Amplicon Metagenomic Sequencing of 16S rRNA Genes from Bulk Soil, Rhizosphere and Endosphere
2.5. Bioinformatic Analysis
2.6. Statistical Analyses
2.7. Endosphere PGPB Selection
2.7.1. Production of Indole-Related Compounds
2.7.2. Organic Phosphate Mineralization and Inorganic Phosphate Solubilization
2.7.3. Siderophore Production
2.7.4. Production of Antimicrobial Substances
2.7.5. Amplification of the Nitrogenase Encoding Gene
2.8. DNA Extraction of the Selected PGPB
2.9. Molecular Identification of Selected PGPB
2.10. Phylogenetic Analyses
2.11. Comparison between the Total Endophytic Bacterial Community and the Selected PGPB
3. Results
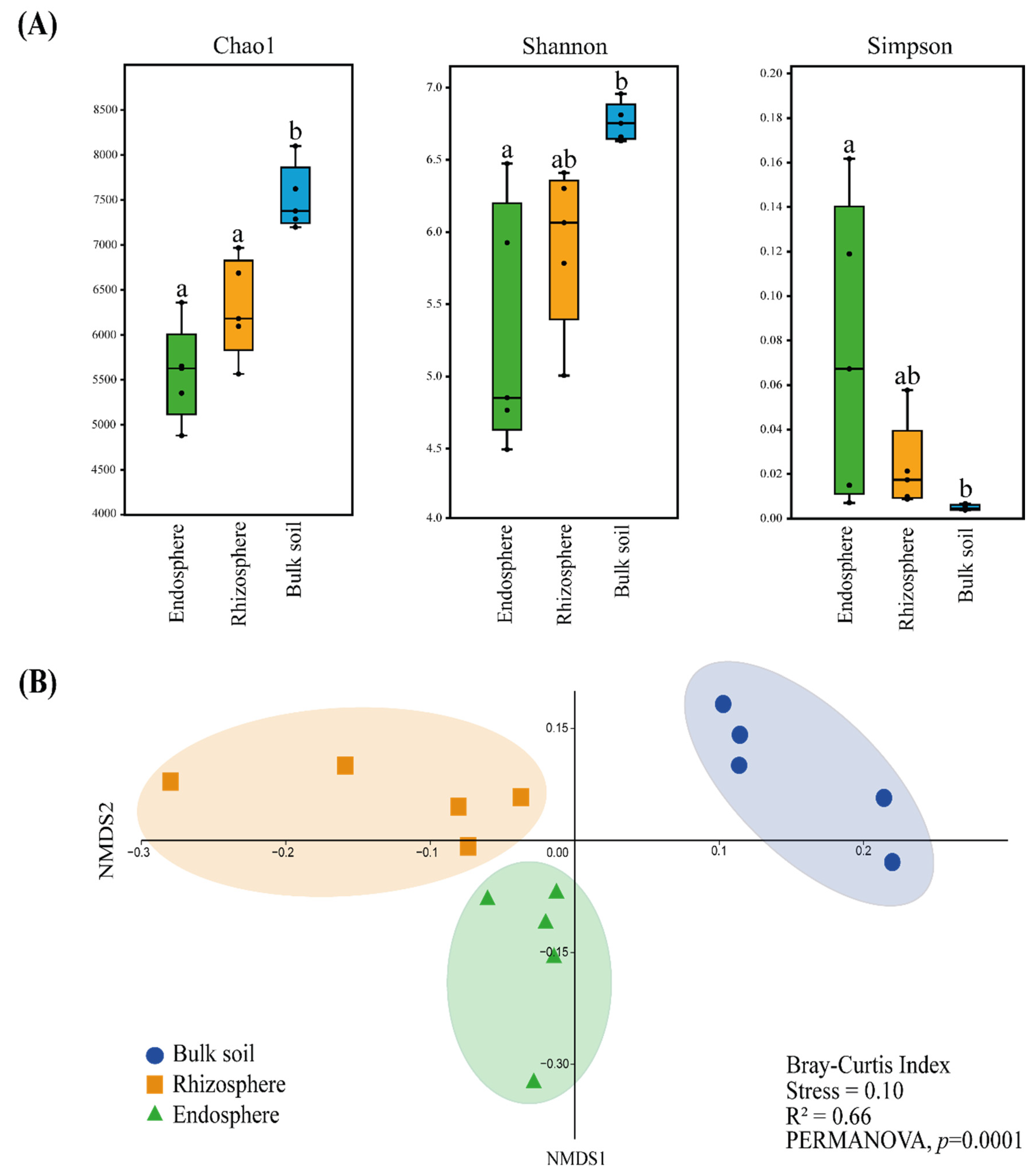
3.1. Endosphere, Rhizosphere and Bulk Soil Bacteriome Analyses through 16S rRNA Amplicon Metagenomic Sequencing
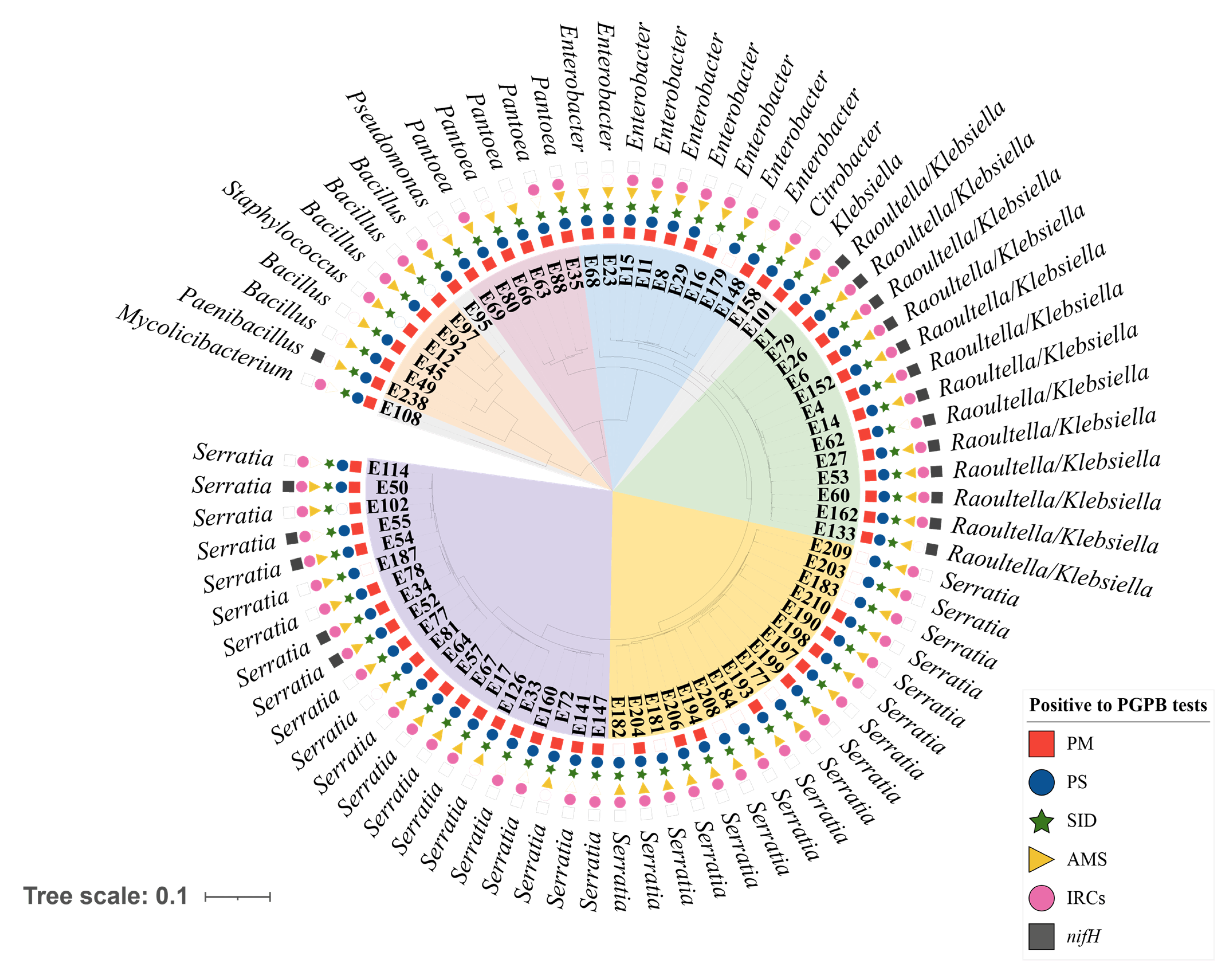
3.2. Selection and Identification of Plant Growth-Promoting Bacteria (PGPB)
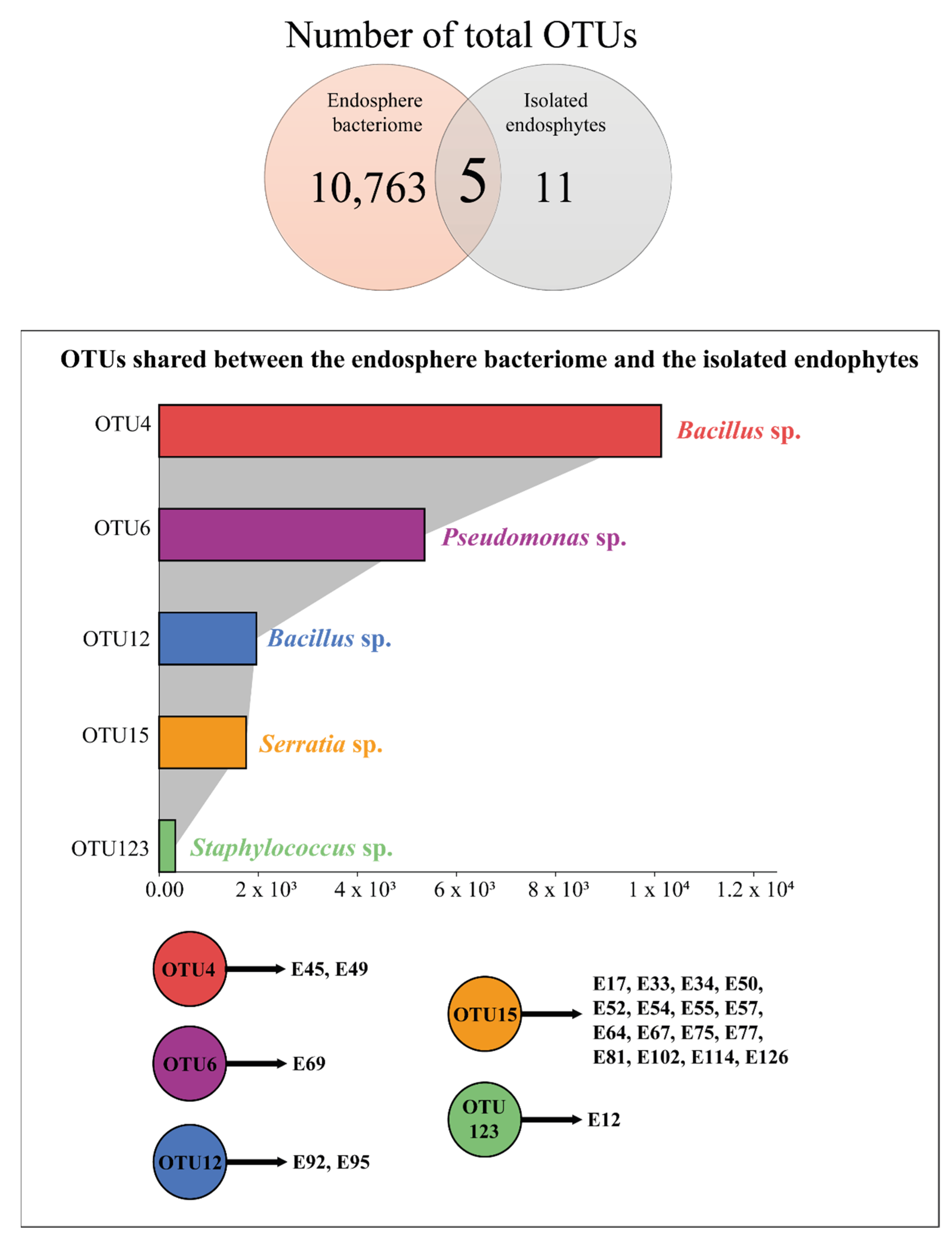
3.3. Comparison between the Endosphere Bacteriome and the Bacterial Isolates
4. Discussion
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Okoye, T.C.; Uzor, P.F.; Onyeto, C.A.; Okereke, E.K. Safe African Medicinal Plants for Clinical Studies. In Toxicological Survey of African Medicinal Plants; Kuete, V., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 535–555. [Google Scholar]
- Jakubczyk, K.; Janda, K.; Watychowicz, K.; Łukasiak, J.; Wolska, J. Garden nasturtium (Tropaeolum majus L.)—A source of mineral elements and bioactive compounds. Rocz. Panstw. Zakl. Hig. 2018, 69, 119–126. [Google Scholar] [PubMed]
- da Silva, O.B.; Goelzer, A.; Moreira dos Santos, F.H.; Carnevali, T.O.; Vieira, M.C.; Zárate, N.A.H. Increased yield and nutrient content of Tropaeolum majus L. with use of chicken manure. Rev. Ceres 2021, 68, 379–389. [Google Scholar] [CrossRef]
- Biswas, K.R.; Rahmatullah, M.A. Survey of Nonconventional Plants Consumed during Times of Food Scarcity in Three Adjoining Villages of Narail and Jessore Districts, Bangladesh. Am.-Eurasian J. Sustain. Agric. 2011, 5, 1–5. [Google Scholar]
- Pintão, A.M.; Pais, M.S.; Coley, H.; Kelland, L.R.; Judson, I.R. In vitro and in vivo antitumor activity of benzyl isothiocyanate: A natural product from Tropaeolum majus. Planta Med. 1995, 61, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Lim, D.Y.; Kwon, G.T.; Kim, J.H.; Huang, Z.; Song, H.; Oh, Y.S.; Kang, Y.H.; Lee, K.W.; Dong, Z.; et al. Benzyl isothiocyanate inhibits prostate cancer development in the transgenic adenocarcinoma mouse prostate (TRAMP) model, which is associated with the induction of cell cycle G1 arrest. Int. J. Mol. Sci. 2016, 17, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butnariu, M.; Bostan, C. Antimicrobial and anti-inflammatory activities of the volatile oil compounds from Tropaeolum majus L. (Nasturtium). J. Biotechnol. 2010, 10, 5900–5909. [Google Scholar]
- Brondani, J.C.; Cuelho, C.H.F.; Marangoni, L.D.; Lima, R.; Guex, C.G.; Bonilha, I.F.; Manfron, M.P. Traditional usages, botany, phytochemistry, biological activity and toxicology of Tropaeolum majus L.—A review. Bol. Latinoam. Caribe Plantas Med. Aromát. 2016, 15, 264–273. [Google Scholar]
- Gasparotto Junior, A.; Prando, T.B.; Leme, T.S.V.; Gasparotto, F.M.; Lourenço, E.L.B.; Rattmann, Y.D.; Silva-Santos, J.E.; Kassuya, C.A.L.; Marques, M.C.A. Mechanisms underlying the diuretic effects of Tropaeolum majus L. extracts and its main component isoquercitrin. J. Ethnopharmacol. 2012, 141, 501–509. [Google Scholar] [CrossRef]
- Schulz, V.S.; Schumann, C.; Weisenburger, S.; Müller-Lindenlauf, M.; Stolzenburg, K.; Möller, K. Row-Intercropping Maize (Zea mays L.) with Biodiversity-Enhancing Flowering-Partners—Effect on Plant Growth, Silage Yield, and Composition of Harvest Material. Agriculture 2020, 10, 524. [Google Scholar] [CrossRef]
- Béliveau, A.; Lucotte, M.; Davidson, R.; Paquet, S.; Mertens, F.; Passos, C.J.; Romana, C.A. Reduction of soil erosion and mercury losses in agroforestry systems compared to forests and cultivated fields in the Brazilian Amazon. J. Environ. Manag. 2017, 203, 522–532. [Google Scholar] [CrossRef]
- Wei, M.S.; Li, G.F. First Report of Broad bean wilt virus 2 and Turnip mosaic virus in Tropaeolum majus in China. Plant Dis. 2017, 101, 1332. [Google Scholar] [CrossRef]
- Schulze, G.A.; Roberts, R.; Pietersen, G. First report of the detection of Bean yellow mosaic virus (BYMV) on Tropaeolum majus; Hippeastrum spp., and Liatris spp. in South Africa. Plant Dis. 2017, 101, 846. [Google Scholar] [CrossRef] [Green Version]
- Russomanno, O.M.R.; Coutinho, L.N.; Kruppa, P.C.; Fabri, E.G. Anthracnose (Colletotrichum gloeosporioides) in medicinal plants. Biológico 2014, 76, 25–28. [Google Scholar]
- Olsson, K. Experience of brown rot caused by Pseudomonas solanacearum in Sweden. Bull. OEPP/EPPO Bull. 1976, 6, 199–207. [Google Scholar] [CrossRef]
- Pradhanang, P.M.; Elphinstone, J.G.; Fov, R.T.V. Identification of crop and weed hosts of Ralstonia solanacearum biovar 2 in the hills of Nepal. Plant Pathol. 2000, 49, 403–413. [Google Scholar] [CrossRef]
- Backer, R.; Rokem, J.S.; Ilangumaran, G.; Lamont, J.; Praslickova, D.; Ricci, E.; Subramanian, S.; Smith, D.L. Plant Growth-Promoting Rhizobacteria: Context, mechanisms of action, and roadmap to commercialization of biostimulants for sustainable agriculture. Front. Plant Sci. 2018, 9, 1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.; Mishra, J.; Arora, N.K. Plant growth promoting bacteria for combating salinity stress in plants—Recent developments and prospects: A review. Microbiol. Res. 2021, 252, 126861. [Google Scholar] [CrossRef]
- Ngalimat, M.S.; Mohd Hata, E.; Zulperi, D.; Ismail, S.I.; Ismail, M.R.; Mohd Zainudin, N.A.I.; Saidi, N.B.; Yusof, M.T. Plant growth-promoting bacteria as an emerging tool to manage bacterial rice pathogens. Microorganisms 2021, 9, 682. [Google Scholar] [CrossRef]
- Orozco-Mosqueda, M.d.C.; Flores, A.; Rojas-Sánchez, B.; Urtis-Flores, C.A.; Morales-Cedeño, L.R.; Valencia-Marin, M.F.; Chávez-Avila, S.; Rojas-Solis, D.; Santoyo, G. Plant Growth-Promoting Bacteria as bioinoculants: Attributes and challenges for sustainable crop improvement. Agronomy 2021, 11, 1167. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning—A Laboratory Manual; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1989; 412p. [Google Scholar]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef] [Green Version]
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. Past: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 1, 9. [Google Scholar]
- Legendre, P.; Borcard, D. Box–Cox-chord transformations for community composition data prior to beta diversity analysis. Ecography 2018, 41, 1820–1824. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- King, E.O.; Ward, M.K.; Raney, D.E. Two simple media for the demonstration of pyocyanin and fluorescin. J. Lab. Clin. Med. 1954, 44, 301–307. [Google Scholar]
- Tang, Y.W.; Bonner, J. The enzymatic inactivation of indoleacetic acid; some characteristics of the enzyme contained in pea seedlings. Arch. Biochem. 1947, 13, 11–25. [Google Scholar]
- Nautiyal, C.S. An efficient microbiological growth medium for screening phosphate solubilizing microorganisms. FEMS Microbiol. Lett. 1999, 170, 265–270. [Google Scholar] [CrossRef]
- Rosado, A.S.; de Azevedo, F.S.; da Cruz, D.W.; van Elsas, J.D.; Seldin, L. Phenotypic and genetic diversity of Paenibacillus azotofixans strains isolated from the rhizoplane or rhizosphere soil of different grasses. J. Appl. Microbiol. 1998, 84, 216–226. [Google Scholar] [CrossRef]
- Schwyn, B.; Neilands, J.B. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 1987, 160, 47–56. [Google Scholar] [CrossRef]
- Rosado, A.S.; Seldin, L. Production of a potentially novel antimicrobial substance by Bacillus polymyxa. World J. Microbiol. Biotechnol. 1993, 9, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Cotta, S.R.; da Mota, F.F.; Tupinambá, G.; Ishida, K.; Rozental, S.; Silva, D.O.; da Silva, A.J.; Bizzo, H.R.; Alviano, D.S.; Alviano, C.S.; et al. Antimicrobial activity of Paenibacillus kribbensis POC 115 against the dermatophyte Trichophyton rubrum. World J. Microbiol. Biotechnol. 2012, 28, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Woodman, M.E. Direct PCR of intact bacteria (colony PCR). Curr. Protoc. Microbiol. 2008, 9, A.3D.1–A.3D.6. [Google Scholar] [CrossRef]
- Angel, R.; Nepel, M.; Panhölzl, C.; Schmidt, H.; Herbold, C.W.; Eichorst, S.A.; Woebken, D. Evaluation of Primers Targeting the Diazotroph Functional Gene and Development of NifMAP—A Bioinformatics Pipeline for Analyzing nifH Amplicon Data. Front. Microbiol. 2018, 9, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massol-Deya, A.A.; Odelson, D.A.; Hickey, R.F.; Tiedje, J.M. Bacterial community fingerprinting of amplified 16S and 16-23S ribosomal DNA gene sequences and restriction endonuclease analysis (ARDRA). In Molecular Microbial Ecology Manual; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995; pp. 1–8. [Google Scholar]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley and Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Hall, T. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Yashiro, E.; Spear, R.N.; McManus, P.S. Culture-dependent and culture-independent assessment of bacteria in the apple phyllosphere. J. Appl. Microbiol. 2011, 110, 1284–1296. [Google Scholar] [CrossRef]
- Anguita-Maeso, M.; Olivares-García, C.; Haro, C.; Imperial, J.; Navas-Cortés, J.A.; Landa, B.B. Culture-dependent and culture-independent characterization of the Olive xylem microbiota: Effect of Sap extraction methods. Front. Plant Sci. 2020, 10, 1708. [Google Scholar] [CrossRef] [Green Version]
- Pirttilä, A.M.; Mohammad Parast Tabas, H.; Baruah, N.; Koskimäki, J.J. Biofertilizers and biocontrol agents for agriculture: How to identify and develop new potent microbial strains and traits. Microorganisms 2021, 9, 817. [Google Scholar] [CrossRef] [PubMed]
- Marilley, L.; Aragno, M. Phylogenetic diversity of bacterial communities differing in degree of proximity of Lolium perenne and Trifolium repens roots. Appl. Soil Ecol. 1999, 13, 127–136. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Shiwa, Y.; Ishige, T.; Sakamoto, H.; Tanaka, K.; Uchino, M.; Tanaka, N.; Oguri, S.; Saitoh, H.; Tsushima, S. Bacterial diversity associated with the rhizosphere and endosphere of two halophytes: Glaux maritima and Salicornia europaea. Front. Microbiol. 2018, 9, 2878. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Zhao, L.; Xu, X.; Feng, H.; Shi, Y.; Deakin, G.; Feng, Z.; Zhu, H. Cultivar-dependent variation of the cotton rhizosphere and endosphere microbiome under field conditions. Front. Plant Sci. 2019, 10, 1659. [Google Scholar] [CrossRef] [PubMed]
- Bruto, M.; Prigent-Combaret, C.; Muller, D.; Moënne-Loccoz, Y. Analysis of genes contributing to plant-beneficial functions in plant growth-promoting rhizobacteria and related Proteobacteria. Sci. Rep. 2014, 4, 6261. [Google Scholar] [CrossRef] [Green Version]
- Raio, A.; Puopolo, G. Pseudomonas chlororaphis metabolites as biocontrol promoters of plant health and improved crop yield. World J. Microbiol. Biotechnol. 2021, 37, 99. [Google Scholar] [CrossRef]
- Ryan, M.P.; Adley, C.C. Ralstonia spp.: Emerging global opportunistic pathogens. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Tan, L.; Gu, S.; Xiao, Y.; Xiong, X.; Zeng, W.A.; Feng, K.; Wei, Z.; Deng, Y. Network analysis infers the wilt pathogen invasion associated with nondetrimental bacteria. NPJ Biofilms Microbiomes 2020, 6, 8. [Google Scholar] [CrossRef]
- Elsayed, T.R.; Jacquiod, S.; Nour, E.H.; Sørensen, S.J.; Smalla, K. Biocontrol of bacterial wilt disease through complex interaction between tomato plant, antagonists, the indigenous rhizosphere microbiota, and Ralstonia solanacearum. Front. Microbiol. 2020, 10, 2835. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, T.; Biddle, J.F.; Teske, A.; Aitken, M.D. Cultivation-dependent and cultivation-independent characterization of hydrocarbon-degrading bacteria in Guaymas Basin sediments. Front. Microbiol. 2015, 6, 695. [Google Scholar] [CrossRef]
- Hajjar, R.; Ambaraghassi, G.; Sebajang, H.; Schwenter, F.; Su, S.H. Raoultella ornithinolytica: Emergence and resistance. Infect. Drug Resist. 2020, 13, 1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Wu, X.; Li, S.; Tang, L.; Chen, M.; An, Q. Proposal for reunification of the genus Raoultella with the genus Klebsiella and reclassification of Raoultella electrica as Klebsiella electrica comb. nov. Res. Microbiol. 2021, 172, 103851. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jia, J.; Atkinson, S.; Cámara, M.; Gao, K.; Li, H.; Cao, J. Biocontrol potential of an endophytic Serratia sp. G3 and its mode of action. World J. Microbiol. Biotechnol. 2010, 26, 1465–1471. [Google Scholar] [CrossRef]
- Lin, L.; Wei, C.; Chen, M.; Wang, H.; Li, Y.; Li, Y.; Litao, Y.; An, Q. Complete genome sequence of endophytic nitrogen-fixing Klebsiella variicola strain DX120E. Stand. Genom. Sci. 2015, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Prasad, V.; Charu Lata, C. Chapter 3—Bacillus: Plant Growth Promoting Bacteria for Sustainable Agriculture and Environment. In New and Future Developments in Microbial Biotechnology and Bioengineering; Singh, J.S., Singh, D.P., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 43–55. [Google Scholar]
- Bokhari, A.; Essack, M.; Lafi, F.F.; Andres-Barrao, C.; Jalal, R.; Alamoudi, S.; Razali, R.; Alzubaidy, H.; Shah, K.H.; Siddique, S.; et al. Bioprospecting desert plant Bacillus endophytic strains for their potential to enhance plant stress tolerance. Sci. Rep. 2019, 9, 18154. [Google Scholar] [CrossRef] [PubMed]
- Daud, N.S.; Mohd Din, A.J.; Rosli, M.A.; Azam, Z.M.; Othman, N.; Sarmidi, M.R. Paenibacillus polymyxa bioactive compounds for agricultural and biotechnological applications. Biocatal. Agric. Biotechnol. 2019, 18, 101092. [Google Scholar] [CrossRef]
- Duca, D.; Lorv, J.; Patten, C.L.; Rose, D.; Glick, B.R. Indole-3-acetic acid in plant–microbe interactions. Antonie Van Leeuwenhoek 2014, 106, 85–125. [Google Scholar] [CrossRef] [PubMed]
- Vílchez, J.I.; Navas, A.; González-López, J.; Arcos, S.C.; Manzanera, M. Biosafety test for Plant Growth-Promoting Bacteria: Proposed Environmental and Human Safety Index (EHSI) protocol. Front. Microbiol. 2016, 6, 1514. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dal’Rio, I.; Mateus, J.R.; Seldin, L. Unraveling the Tropaeolum majus L. (Nasturtium) Root-Associated Bacterial Community in Search of Potential Biofertilizers. Microorganisms 2022, 10, 638. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10030638
Dal’Rio I, Mateus JR, Seldin L. Unraveling the Tropaeolum majus L. (Nasturtium) Root-Associated Bacterial Community in Search of Potential Biofertilizers. Microorganisms. 2022; 10(3):638. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10030638
Chicago/Turabian StyleDal’Rio, Isabella, Jackeline Rossetti Mateus, and Lucy Seldin. 2022. "Unraveling the Tropaeolum majus L. (Nasturtium) Root-Associated Bacterial Community in Search of Potential Biofertilizers" Microorganisms 10, no. 3: 638. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10030638